

Abstract
Background and purpose:
Voltage-gated potassium (Kv) channels contribute to resting membrane potential in pulmonary artery smooth muscle cells and are down regulated in patients with pulmonary arterial hypertension (PAH) and a contribution from Kv7 channels has been recently proposed. We investigated the effect of the Kv7 channel activator, flupirtine, on PAH in two independent mouse models: PAH induced by hypoxia and spontaneous PAH in mice over-expressing the 5-HT transporter (SERT+ mice).
Experimental approach:
Right ventricular pressure was assessed in vivo in mice chronically treated with flupirtine (30 mg·kg−1·day−1). In separate in vitro experiments, pulmonary arteries from untreated mice were mounted in a wire myograph. Relaxations to acute administration of flupirtine and contractions to Kv channel blocking drugs, including the Kv7 channel blocker linopirdine, were measured.
Key results:
In wild-type (WT) mice, hypoxia increased right ventricular pressure, pulmonary vascular remodelling and right ventricular hypertrophy. These effects were attenuated by flupirtine, which also attenuated these indices of PAH in SERT+ mice. In the in vitro experiments, flupirtine induced a potent relaxant response in arteries from untreated WT and SERT+ mice. The relaxation was fully reversed by linopirdine, which potently contracted mouse pulmonary arteries while other Kv channel blockers did not.
Conclusions and implications:
Flupirtine significantly attenuated development of chronic hypoxia-induced PAH in mice and reversed established PAH in SERT+ mice, apparently via Kv7 channel activation. These results provide the first direct evidence that drugs activating Kv7 channels may be of benefit in the treatment of PAH with different aetiologies.
Keywords: Kv7 channels, pulmonary hypertension, 5-HT
Introduction
Pulmonary arterial hypertension (PAH) is characterized by a progressive increase in pulmonary arterial pressure, concomitant with increased pulmonary vascular resistance, pulmonary vascular remodelling, right ventricle (RV) failure and death (Chin and Rubin, 2008). About 70% of patients with the familial form of the disease (fPAH) have a mutation in the gene encoding bone morphogenetic protein (BMP) receptor type II (BMPR-II), a member of the transforming growth factor-β superfamily, as the primary genetic defect (Lane et al., 2000; Machado et al., 2001). Voltage-gated K+ (Kv) channels, several families of which are expressed in pulmonary artery smooth muscle cells (PASMCs), contribute to resting membrane potential and have been implicated in the regulation of vascular smooth muscle function (Archer et al., 1998; Yuan et al., 1998). BMP can regulate Kv channel expression in PASMCs (Young et al., 2006). Moreover, K+ channel downregulation is observed in PASMCs from patients with PAH (Yuan et al., 1998; Remillard et al., 2007) and in PASMCs from rats with chronic hypoxia-induced PAH (Smirnov et al., 1994; Evans et al., 1996; Platoshyn et al., 2001). Considerable effort has focused on the importance of the Kv1.5 and Kv2.1 channels, where downregulation or dysfunction may be a predisposing factor in the development of pulmonary arterial vasoconstriction and pulmonary vascular remodelling in patients with fPAH (Yuan et al., 1998; Remillard et al., 2007). However, these particular Kv channels may not play a major role in maintaining PASMC resting potential as they have a very low probability of opening at the resting membrane potential (Evans et al., 1996; Gurney et al., 2003).
KCNQ (KCNQ1–5) genes encode a subfamily of voltage-gated K+ channels, denoted Kv7.1-Kv7.5. These channels have been studied primarily in the heart, CNS and auditory pathway (Jentsch, 2000; Robbins, 2001; Jespersen et al., 2005; Jin et al., 2008), but were recently identified in vascular (Ohya et al., 2003; Yeung et al., 2007; Mackie et al., 2008) and uterine (McCallum et al., 2009) smooth muscle and may regulate Na+ flux across pulmonary epithelial cells (Greenwood et al., 2009). KCNQ genes express the well described non-inactivating, outwardly rectifying K+ currents (‘M’ currents) that play a pivotal role in controlling membrane excitability in neurones (Marrion, 1997; Robbins, 2001). The background K+ current giving rise to the resting potential of PASMCs has biophysical properties similar to those of recombinant and native Kv7 channels (Evans et al., 1996). Combined with the finding that Kv7 channel blocking drugs are potent constrictors of rodent pulmonary arteries (Joshi et al., 2006), this suggests the possible involvement of Kv7 channels in the resting potential of PASMCs.
The Kv7 channel activator, flupirtine, is used in Europe as a non-opioid analgesic and in several human trials has proven to be essentially free of cardiovascular side effects (Herrmann et al., 1987; Hummel et al., 1991; Friedel and Fitton, 1993). Flupirtine was recently shown to activate recombinant Kv7.4 or Kv7.5 channels (Yeung et al., 2008). Its more potent analogue, retigabine, which is not yet available for clinical use, selectively activates Kv7.2–Kv7.5 channels while having little effect on the cardiac Kv7.1 channel (Main et al., 2000; Tatulian et al., 2001; Schenzer et al., 2005; Wuttke et al., 2005). Flupirtine and retigabine have been reported to relax vascular smooth muscle (Joshi et al., 2006; Yeung et al., 2007; Mackie et al., 2008). Here we investigated the effects of flupirtine (Wladyka and Kunze, 2006) on pulmonary vascular haemodynamics and indices of PAH in two mouse models of the disease. Mice with pulmonary hypertension, induced by chronic hypoxia, were used to investigate the effect of flupirtine on the development of PAH. Mice that over-express the 5-HT transporter (SERT) have established PAH and were therefore studied to determine the effects of flupirtine in an alternative model of PAH (MacLean et al., 2004). Importantly, we demonstrated that flupirtine treatment attenuated the indices of PAH in these two independent murine models of the disease. Flupirtine prevented the development of hypoxia-induced PAH and reversed PAH in SERT+ mice, where the disease was already established.
Methods
In vivo experiments
All animal procedures and experiments were conducted in accordance with the United Kingdom Animals (Scientific Procedures) Act 1986 and conformed to institutional regulations at the University of Glasgow. The generation and characterization of the SERT+ mice have been described previously (MacLean et al., 2004; Mair et al., 2008). All mice were bred in the University of Glasgow and the genotype of each mouse was confirmed by PCR.
Exposure to hypoxia
Female wild-type (WT) control mice (C57BL/6XCBA strain, 5–6 months old; n= 8–10 mice per group) were maintained in normoxic or hypobaric/hypoxic conditions for 2 weeks as previously described (Keegan et al., 2001; MacLean et al., 2004). The hypobaric chamber was depressurized over the course of 2 days to 550 mbar (equivalent to 10% O2). Temperature was maintained at 21°C to 22°C, and the chamber was ventilated with air at 45 L·min−1. As male SERT+ mice do not develop PAH (MacLean et al., 2004), female mice were used in this study.
Drug administration protocol
Subsets of the WT and SERT+ mice were given orally, either flupirtine (30 mg·kg−1·day−1; n= 8–10 mice per group) or vehicle (1% carboxymethylcellulose, n= 8–10 mice per group) alone on each of the 14 experimental days. In those WT mice also exposed to hypoxia, flupirtine (or vehicle) was administered during the 14 days of hypoxic exposure. The effectiveness of the chosen dose of flupirtine has been demonstrated in models testing for anti-nociceptive and anti-Parkinson activity in rodents (Nickel, 1987; Szelenyi et al., 1989; Schmidt et al., 1997). Age-matched flupirtine and vehicle-treated control groups were maintained in room air. All animals were maintained in cages with a 12 h light/dark cycle and free access to food and water.
Under isoflurane (1.5% in O2) anaesthesia, mean right ventricular pressure (mRVP) was measured via a 25-gauge needle advanced into the RV trans-diaphragmatically (MacLean et al., 2004; Morecroft et al., 2007). RVP and heart rate (HR; derived from the RVP trace) were recorded on a data acquisition system (MP 100, Biopac Systems). Depth of anaesthesia was confirmed by lack of a pinch withdrawal reflex applied to the hind paw.
Indices of PAH
The ratio of right ventricular weight to left ventricular weight plus septum [RV/(LV + S)] was used as an index of right ventricular hypertrophy (RVH) (MacLean et al., 2004; Morecroft et al., 2007) and the ratio of (LV + S) to body weight (bw) was also calculated. Sagittal sections were obtained from left lungs, stained with Elastica van Giessen stain and microscopically assessed for muscularization of pulmonary arteries (<80 µm external diameter) as described previously (MacLean et al., 2004; Morecroft et al., 2007). Lung sections from four to six mice from each group were studied.
Myography
Arteries from SERT+ mice and control WT mice were studied. The animals were killed by an overdose of sodium pentobarbitone (200 mg·kg−1, i.p.) and the lungs removed. Small pulmonary arteries (third order, first intralobar) of ∼350 µm internal diameter and mesenteric arteries (first order branches) were dissected, cut into 2 mm long segments, threaded onto 40 µm stainless steel wires and mounted on an isometric myograph (610 M; Danish Myo Technology, Aarhus, Denmark) as described previously (Keegan et al., 2001; Morecroft et al., 2007). The vessels were maintained at 37°C in Krebs buffer solution (pH7.4) of the following composition (in mM): NaCl 118.4, NaHCO3 25, KCl 4.7, KH2PO4 1.2, MgSO4 0.6, CaCl2 2.5, glucose 11.0 and EDTA 0.023. Pulmonary arteries were aerated with 16% O2/5% CO2 balance N2 and set up at tensions equivalent to their mean in vivo RVP (12–15 mmHg). Mesenteric arteries were gassed with 95% O2/5% CO2 and mounted under their optimal resting tensions, previously calculated to be 170 mg (Daly et al., 2002). Vessel responses were recorded onto a computer using a data acquisition and recording software system (Myodaq and Myodata, Danish Myo Technology). After a 45 min equilibration period, the response to 50 mM KCl, the concentration that produced maximal contraction in these vessels, was measured. On washout and re-equilibration, vessels were contracted with phenylephrine to ∼75% of the maximal phenylephrine-induced tone using a concentration of 10–100 nM and cumulative concentration-response curves to flupirtine (1 nM–0.1 mM) or retigabine (1 nM–30 µM, pulmonary arteries only) constructed. In a separate series of experiments on WT pulmonary and mesenteric arteries, following phenylephrine pre-constriction, concentration-response curves to flupirtine (1 nM–0.1 mM) were constructed immediately followed by constructing cumulative concentration-response curves to linopirdine (1 nM–0.1 mM) to assess if Kv7 channel block could reverse the flupirtine-induced relaxation. In other separate experiments, after challenging mouse pulmonary arteries with 50 mM KCl and subsequent washout, cumulative concentration-response curves were constructed for the Kv7 channel blocking drug linopirdine and several other K+ channel blocking drugs. The EC50 for linopirdine acting at Kv7 channels is well documented in native tissues (Lamas et al., 1997; Schnee and Brown, 1998; Joshi et al., 2006) and expression systems (Robbins, 2001). The other K+ channel blocking drugs included 4-aminopyridine (4-AP) and capsaicin, used as broad spectrum Kv channel inhibitors (Grissmer et al., 1994; Coetzee et al., 1999), tetraethylammonium ions (TEA), which inhibit BKCa, Kv1.1, Kv1.2 and Kv3.1 at low millimolar concentrations (Grissmer et al., 1994; Coetzee et al., 1999), glibenclamide for selective KATP channel block (Clapp and Gurney, 1992) and ZnCl2, which blocks the TASK-like K+ current in rabbit PASMCs (Gurney et al., 2003).
The drug/molecular target nomenclature conforms to the BJP's Guide to Receptors and Channels (Alexander et al., 2008).
Data analysis
Contractile responses to K+ channel blockers were normalized by expressing them as a percentage of the 50 mM KCl response of each arterial segment. Vasodilator responses were measured as a percentage of the pre-constrictor tone induced by phenylephrine. The maximal vasoconstrictor/vasodilator effect (Emax) and the pEC50 or pIC50 were calculated from a sigmoidal nonlinear regression curve fit. Individual comparisons were made using Student's unpaired t-test, as appropriate. Multiple comparisons were made using one-way anova, followed by Neuman Keuls post hoc test. GraphPad Prism (version 4) was used to perform all statistical analyses and values of P < 0.05 were considered to be significant.
Materials and solutions
Flupirtine maleate, linopirdine dihydrochloride, glibenclamide, capsaicin and 4-AP were purchased from Tocris Cookson (Bristol, UK). Retigabine was a gift from Astra Zeneca. Stock solutions were prepared in distilled water (linopirdine), dimethylsulphoxide (other drugs) or 100 mM HEPES buffer (4-AP) with the pH adjusted to 7.3. Stock solutions of flupirtine for oral administration were made up in 1% caboxymethylcellulose in water. Phenylephrine hydrochloride, ZnCl2, TEA chloride and carboxymethylcellulose were from Sigma-Aldrich (Poole, UK) and stock solutions were prepared in distilled water.
Results
In vivo experiments
As previously reported (MacLean et al., 2004), under normoxia SERT+ mice demonstrated markedly elevated mRVP, compared with WT mice (P < 0.001; Figure 1A). Treatment with flupirtine markedly reversed this elevation to levels commensurate with those in WT mice (P < 0.001 vs. vehicle-treated SERT+ mice) (Figure 1A). As in previous studies (MacLean et al., 2004), the SERT+ mice displayed a low proportion of remodelled pulmonary arteries and this was not significantly affected by flupirtine treatment (Figure 1B). RVH, as indicated by an increase in the ratio RV/(LV + S), was observed in the SERT+ mice compared with WT mice (Figure 1C) and this was also attenuated by flupirtine [Figure 1C (P < 0.05)].
Figure 1.
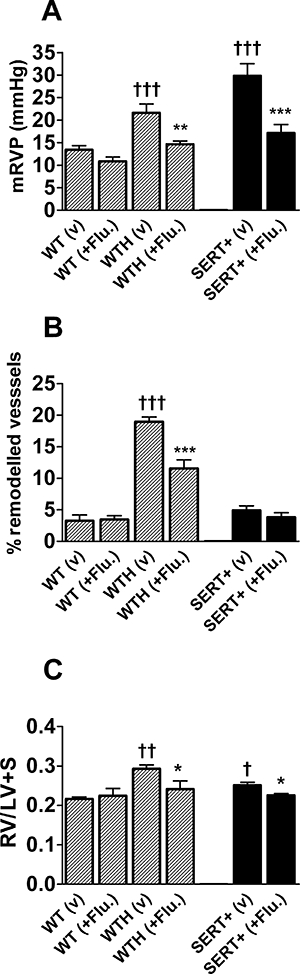
Effect of chronic flupirtine treatment on indices of PAH in anaesthetized mice. (A) Mean RVP (mRVP), (B) pulmonary vascular remodelling and (C) right ventricular hypertrophy in vehicle treated (v) and flupirtine (+Flu.) treated (30 mg·kg−1·day−1 by oral gavage) WT normoxic and hypoxic (WTH) mice and in normoxic SERT+ mice. *Value significantly less than corresponding value in vehicle treated mice (*P < 0.05, **P < 0.01, ***P < 0.001). †Value significantly greater than corresponding value in normoxic WT mice (†P < 0.05, ††P < 0.01, †††P < 0.001). Data are expressed as mean ± SEM from six to eight experiments. LV + S, left ventricle plus septum; mRVP, mean right ventricular pressure; SERT+, mice that over-express the 5-HT transporter; WT, wild-type mice.
Wild-type mice maintained in a hypoxic environment developed pulmonary hypertension within 14 days, with mRVP increased by around 50% compared with normoxic WT animals (Figure 1A). Flupirtine treatment attenuated the development of chronic hypoxia-induced pulmonary hypertension, with mRVP reduced to near normal values (Figure 1A). As a consequence of the increased RVP, the hypoxic WT mice developed RVH, with RV/(LV + S) increasing in the normoxic animals after exposure to hypoxia (Figure 1C; P < 0.001) and treatment with flupirtine reduced the hypertrophy, (Figure 1C; P < 0.05 vs. vehicle-treated hypoxic animals). In hypoxia-induced PAH, the rise in mRVP is associated with remodelling of the pulmonary arteries of diameter <80 µm. In control WT mice, the majority of vessels of this diameter were not remodelled, only a very small percentage being fully muscularized (Figure 1B). In hypoxic WT mice, a significant increase in the percentage of remodelled pulmonary arteries was evident (Figure 1B). Treatment with flupirtine resulted in a significant reduction in the percentage of remodelled arteries, compared with the vehicle-treated hypoxia group (Figure 1B).
The effects of flupirtine were specific for the rise in mRVP seen in SERT+ mice and WT mice exposed to hypoxia for 14 days. The drug had no effect on the indices of PAH measured in normoxic WT mice (Figure 1). Flupirtine also had no significant effect on the HR, bw or (LV + S)/bw ratio in any of the treatment groups (Table 1).
Table 1.
Body weight (bw), heart rate and left heart weight to bw ratio in wild-type (WT) and SERT+ mice after 14 days of chronic hypoxia and treatment with vehicle or flupirtine (30 mg−1·kg−1·day−1)
| Group | Heart rate (beats·min−1) | bw (g) | LV+S/bw (mg·g−1) |
|---|---|---|---|
| WT normoxic vehicle-dosed | 365 ± 10 | 32.1 ± 1.3 | 3.22 ± 0.10 |
| WT normoxic flupirtine-dosed | 397 ± 32 | 28.0 ± 1.2 | 3.17 ± 0.22 |
| WT hypoxic vehicle-dosed | 379 ± 39 | 28.3 ± 1.1 | 3.04 ± 0.15 |
| WT hypoxic flupirtine-dosed | 418 ± 24 | 30.5 ± 1.6 | 3.13 ± 0.08 |
| SERT+ normoxic vehicle-dosed | 469 ± 25 | 27.3 ± 4.8 | 3.43 ± 0.19 |
| SERT+ normoxic flupirtine-dosed | 499 ± 29 | 22.8 ± 2.5 | 3.53 ± 0.11 |
Values shown are mean ± SEM; n= 6–8 animals per group.
LV + S, left ventricle plus septum; SERT+, mice which over-express the 5-HT transporter.
Myography
Flupirtine relaxed pre-constricted pulmonary arteries from WT and SERT+ mice (Figure 2), although it was more potent in the WT vessels where it caused ∼40% and ∼60% relaxation at 10 and 30 µM respectively. Retigabine also relaxed pre-constricted pulmonary arteries from WT mice, with around 60% relaxation produced at 10 µM and 75% relaxation at 30 µM (Figure 3A). The effect of flupirtine was not restricted to the pulmonary circulation, because it also relaxed pre-constricted mesenteric arteries from WT mice (Figure 3A). Flupirtine appeared to be slightly more potent in relaxing mesenteric arteries and, unlike the response in pulmonary arteries, the relaxation response in mesenteric arteries reached a maximum at 30 µM (Figure 3A), allowing us to measure the pIC50 (5.6 ± 0.1) and Emax (94 ± 8%; n= 6 mice). To assess whether the relaxation effect of flupirtine was mediated by the opening of Kv7 channels, the ability of linopirdine to reverse the relaxation was investigated. In the continued presence of phenylephrine and 100 µM flupirtine, linopirdine was applied at increasing concentrations and the recovery of tension measured. Figure 3B shows that the response of pulmonary arteries to flupirtine was completely reversed by linopirdine with pEC50= 6.25 ± 0.06 (n= 5). In contrast, the response to flupirtine was only partially reversed by linopirdine in the mesenteric arteries (Figure 3B; pEC50= 6.47 ± 0.13, n= 5).
Figure 2.
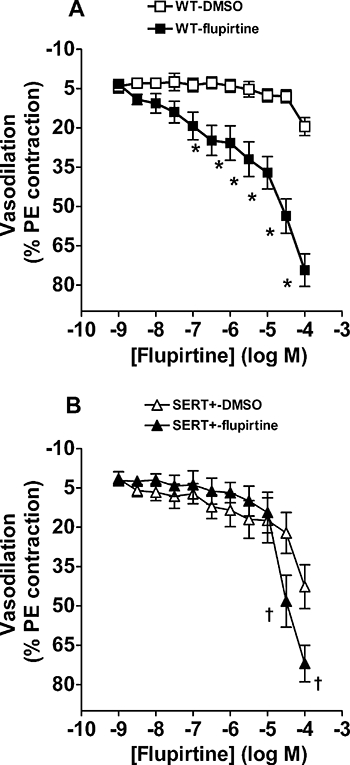
Vasodilator responses to the Kv7 channel activator flupirtine in precontracted pulmonary arteries. Cumulative concentration response curves to flupirtine in (A) WT and (B) SERT+ mouse pulmonary arteries pre-constricted with phenylephrine (PE; 10–100 nM). Vehicle (DMSO) data are also shown and illustrate the fall in vascular tone in vessels set up in parallel with those to which flupirtine was added. *Value significantly greater than corresponding value in vehicle treated WT vessels (*P < 0.001). †Value significantly greater than corresponding value in vehicle treated SERT+ vessels (†P < 0.001). Data are expressed as a percentage of the response to PE-induced preconstriction and shown as mean ± SEM from five to seven experiments. DMSO, dimethylsulphoxide; PE, phenylephrine; SERT+, mice that over-express the 5-HT transporter; WT, wild-type mice.
Figure 3.
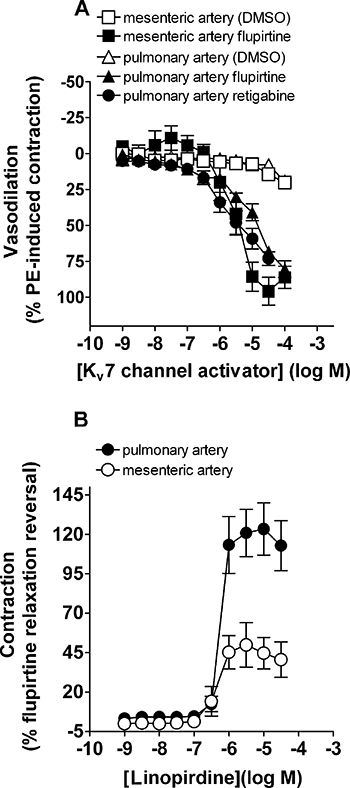
(A) Vasodilator responses to the Kv7 channel activators flupirtine and retigabine in WT mouse pulmonary arteries and flupirtine in mesenteric arteries pre-constricted with phenylephrine (PE; 10–100 nM). Vehicle (DMSO) data are also shown and illustrate the fall in vascular tone in vessels set up in parallel with those to which Kv7 channel activators were added. (B) Reversal by linopirdine of the flupirtine-induced vasodilator response shown in Figure 3A in the same WT mouse pulmonary and mesenteric arteries. Data are expressed as a percentage of the response to PE-induced preconstruction (A) and as a percentage reversal of flupirtine-induced vasodilation (B) and shown as mean ± SEM from five to eight experiments. DMSO, dimethylsulphoxide; PE, phenylephrine; WT, wild-type mice.
The effects of a range of different K+ channel blockers were tested on the baseline tone of WT and SERT+ pulmonary arteries, in order to assess which of the K+ channels might be responsible for maintaining a negative membrane potential in the PASMCs, thereby keeping the vessels relaxed. Only three drugs, linopirdine, 4-AP and TEA, produced sizeable constriction (Figure 4). In vessels from WT mice, linopirdine was around five orders of magnitude more potent than any other drug, producing constriction with pEC50= 6.37 ± 0.18 and Emax= 56 ± 4% (n= 7) of the response to 50 mM KCl (Figure 4A). TEA and 4-AP were very weak vasoconstrictors, eliciting a significant response only at concentrations ≥10 mM (Figure 4A), while glibenclamide (data not shown), capsaicin (data not shown) and ZnCl2 (Figure 4A) all failed to evoke constriction at concentrations up to 1 mM. A similar pattern of effects was observed in pulmonary arteries from the SERT+ mice (Figure 4B). Interestingly although, SERT+ vessels were significantly less responsive to linopirdine than WT vessels, with pEC50= 5.24 ± 0.16 (P < 0.001) and Emax= 33 ± 4 % (Figure 4B; n= 8, P < 0.001). In contrast, SERT+ vessels appeared to have increased sensitivity to 4-AP, with 10 mM 4-AP evoking constriction amounting to 76 ± 8% (n= 5) of the response to 50 mM KCl, compared with 26 ± 7% of the KCl response in WT vessels (Figure 4B; n= 6, P < 0.001).
Figure 4.
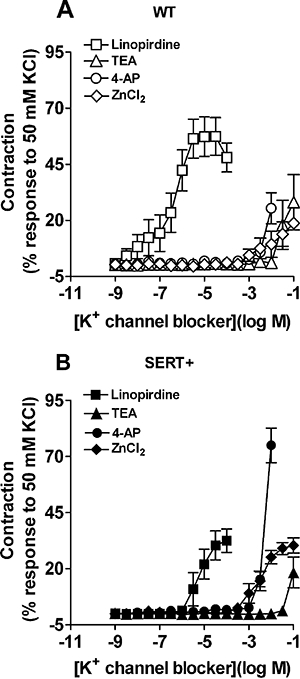
Vasoconstrictor responses to K+ channel blockers in WT and SERT+ mouse pulmonary arteries. Concentration response curves to linopirdine, TEA, 4-AP and ZnCl2 in pulmonary arteries from (A) WT and (B) SERT+ mice. Data are expressed as a percentage of the reference vasoconstrictor response to 50 mM KCl and shown as mean ± SEM from five to eight experiments. 4-AP, 4-aminopyridine; SERT+, mice that over-express the 5-HT transporter; TEA, tetraethylammonium; WT, wild-type mice; ZnCl2, zinc chloride.
Discussion
This is the first report in which Kv7 channels have been directly implicated in PAH. The main finding is that treatment with the selective Kv7 channel activator flupirtine markedly attenuated the development of PAH induced by chronic hypoxia in mice and, moreover, completely reversed established PAH in mice over-expressing the SERT. In chronic hypoxic mice, flupirtine attenuated mRVP, pulmonary vascular remodelling and RVH, suggesting that Kv7 channels are present in mouse pulmonary vasculature and that their activation can prevent the development of hypoxic PAH. We propose that this is because flupirtine hyperpolarizes PASMCs, maintaining a negative membrane potential and preventing voltage-gated Ca2+ influx. This has recently been shown in rat pulmonary artery smooth muscle (Joshi et al., 2009). The beneficial effect of flupirtine on PAH could be ascribed to several possible mechanisms, including its inhibitory effect on pulmonary vasoconstriction or, perhaps, the prevention of Kv channel down regulation, which has been proposed to mediate mitogenic effects on PASMCs (Burg et al., 2008). Recent data from pharmacological studies strongly support the presence of functional Kv7 channels in mouse pulmonary arteries (Joshi et al., 2006).
Several K+ channel subfamilies have been implicated in the regulation of resting membrane potential in PASMCs, including a number of voltage-gated K+ channels (Archer et al., 1998; Remillard et al., 2007) and two-pore domain channels, especially TASK-1 (Gurney et al., 2003; Gardener et al., 2004; Olschewski et al., 2006). It was therefore important to identify which K+ channels are likely to be open constitutively, contributing to PASMC resting membrane potential, and thus regulating resting tone in WT and SERT+ mouse pulmonary arteries. The failure of glibenclamide to significantly affect baseline tone at concentrations known to selectively block KATP channels is consistent with findings on pulmonary arteries from other species (Clapp and Gurney, 1992) and implies that KATP channels contribute little to resting tone in WT or SERT+ mice. Likewise, the lack of a significant contractile response to 4-AP or capsaicin, at concentrations inhibiting Kv2.1, and all Kv1 channels, and TEA at concentrations blocking Kv1.1, Kv1.2, Kv3.1 and BKCa channels (Grissmer et al., 1994; Coetzee et al., 1999), implies that BKCa and most Kv channels also make little contribution to resting tone in WT or SERT+ mice. Selective blockers of TASK channels are not available, so we made use of Zn2+ ions, which at 100 µM depolarized rabbit PASMCs and inhibited a TASK-like K+ current (Gurney et al., 2003). Although ZnCl2 failed to constrict mouse pulmonary arteries until the millimolar range, a role for TASK channels in regulating resting membrane potential in murine PASMCs cannot be ruled out, because Zn2+ can also block voltage-gated Ca2+ channels (Sun et al., 2007), possibly counteracting potential constriction. The selective Kv7 channel blocker, linopirdine, was the only drug that induced potent vasoconstriction in pulmonary arteries from WT and SERT+ mice, at concentrations shown to be selective for Kv7 channels in native tissues (Lamas et al., 1997; Schnee and Brown, 1998) and expression systems (Robbins, 2001). Although interactions of linopirdine with other K+ channels have been reported, these only occurred at higher concentrations (Wladyka and Kunze, 2006). The response to linopirdine is consistent with an earlier study on rat and mouse pulmonary arteries, which showed that it occurs independently of endothelium or nerve-derived mediators (Joshi et al., 2006). Our results therefore strongly suggest that Kv7 channels may be the major K+ channels regulating resting membrane potential and resting tone in mouse PASMCs.
Until further molecular and biophysical information is available, it is only possible to speculate which subtypes of the Kv7 channel are involved in regulating resting membrane potential and resting tone in mouse PASMCs as the β-subunit expression products of the KCNE genes can alter the biophysical and pharmacological properties of the Kv7 channels (Robbins, 2001). From in vitro studies, as flupirtine and retigabine are not active at Kv7.1 channels (Schenzer et al., 2005; Yeung et al., 2008), the effects of these drugs are likely to reflect actions on channels containing Kv7.4 and/or Kv7.5 channel subunits, although the weak effect of TEA on vessel tone argues against a major role for Kv7.4 homomers (Hadley et al., 2000). The very weak effects of Zn+ may rule out an involvement of a Kv7.2 + 7.3 heteromer; however, Zn2+ can potentiate Kv7.5 channels (Jensen et al., 2005), causing vasodilation, which may counteract any vasoconstriction. Pulmonary arteries express KCNQ4 mRNA abundantly, at higher levels than KCNQ1, and KCNQ5 was also present, but not KCNQ2 or KCNQ3 (Joshi et al., 2009). We, therefore, suggest that homomeric Kv7.5, or heteromeric channels containing Kv7.4 subunits, may be involved in the protective effects of flupirtine on the pulmonary circulation.
The lower potency of flupirtine in relaxing SERT+ mouse arteries, compared with WT arteries, mirrors the loss of linopirdine constrictor potency and may reflect reduced KCNQ gene expression or possibly a change in the KCNQ subtype contributing to the channel. Although KCNQ expression has not been investigated in SERT+ mice, the mice have been shown to express reduced levels of Kv1.5 and Kv2.1 mRNA (Guignabert et al., 2006). Reduced KCNQ mRNA expression has also been shown in hypoxic rats (Joshi et al., 2008). 5-HT is thought to modulate (inhibit) the activity of voltage-gated K+ channels via signalling through the 5-HT2A receptor (Cogolludo et al., 2006) and human PASMCs treated with 5-HT show reduced Kv1.5 and Kv2.1 expression (Guignabert et al., 2006). If Kv7 channels are the dominant regulators of resting potential, reduced KCNQ expression would be expected to cause depolarization, thereby promoting the opening of K+ channels with a higher voltage threshold for activation, such as Kv1 or Kv2. An increased contribution of these channels to the resting potential may therefore explain why the loss of linopirdine potency was accompanied by an increase in the potency of 4-AP. A similar increase in 4-AP constrictor potency, due to loss of the background K+ conductance and consequent membrane depolarization, was seen in hypoxic PAH despite concurrent loss of 4-AP sensitive current (Osipenko et al., 1988).
The finding that flupirtine and its more potent analogue, retigabine, relaxed isolated, pre-constricted mouse pulmonary arteries indicates that flupirtine may mediate its beneficial effects on PAH at least in part by acting as a pulmonary arterial vasodilator to improve pulmonary haemodynamics. That the relaxation was completely reversed by linopridine is consistent with flupirtine acting via Kv7 channels. It is interesting that despite the reduced potency in isolated SERT+ arteries, flupirtine in vivo reduced the indices of PAH in SERT+ and chronic hypoxic mice while having no effect on WT mice. As the effect of flupirtine is only apparent in pre-constricted vessels, this probably reflects the minimal pulmonary vasoconstriction present in normoxic WT animals.
Although flupirtine also relaxed pre-constricted mouse mesenteric arteries, this effect may not be due entirely to activation of Kv7 channels, because it was only partly reversed by linopirdine. Our results are consistent with recent reports that flupirtine and retigabine dilated isolated systemic arteries from mice (Yeung et al., 2007) and decreased mean arterial pressure in rats when given intravenously (Mackie et al., 2008). This highlights a potential side effect of flupirtine treatment. It could be argued that the effect of flupirtine on RVP reflects a decrease in cardiac output rather than a change in pulmonary vascular resistance per se, as Kv7 channels are present in the heart (Sanguinetti et al., 1996; Calloe et al., 2007). This is unlikely, because the predominant Kv7 isoform in the heart is Kv7.1 (Sanguinetti et al., 1996), which lacks the transmembrane tryptophan residue thought to be essential for the actions of flupirtine (Schenzer et al., 2005; Wuttke et al., 2005; Bentzen et al., 2006). The lack of effect of flupirtine on HR is consistent with this and minimal cardiac effects are seen in humans undergoing flupirtine treatment (Herrmann et al., 1987). Although a central cardiovascular action of flupirtine cannot be ruled out, intracisternal administration of flupirtine does not alter mean arterial pressure in rabbits (Yoro et al., 2008).
Pulmonary vascular remodelling occurs as a result of PASMC proliferation and attenuated PASMC apoptosis. Reduced K+ channel activity in PAH patients is thought to promote PASMC proliferation through an increase in the concentration of free Ca2+within the cytoplasm (Platoshyn et al., 2000; Burg et al., 2008), allowing for progression through the cell cycle. As flupirtine attenuated hypoxia-induced pulmonary vascular remodelling in WT mice, Kv7 channel dysfunction/down-regulation may be a feature of PAH, and Kv7 channel activation may inhibit PASMC proliferation. Several limitations to this study deserve comment. The study on chronic hypoxic mice concentrated on the preventive effects of flupirtine on the development of hypoxia-induced PAH, which is not easily extrapolated to patients with end-stage disease. Preventative therapy may, however, be useful, for example in carriers within families with a mutation in BMPR-II (Machado et al., 2001). The SERT+ mouse, with elevated pulmonary arterial pressure in the absence of a hypoxic stimulus (MacLean et al., 2004), provided an example of established PAH, which may be a more clinically relevant endpoint. Although no single animal model precisely recapitulates the human disease, several drugs currently in use, including endothelin receptor antagonists (Rubin et al., 2002; Galie et al., 2008) and phosphodiesterase-5 inhibitors (Galie et al., 2005), first demonstrated promising, beneficial results in experimental animal models (Zhao et al., 2001). There is still a continual need to identify potential therapeutic targets and novel pathways that target vascular remodelling/vasoconstriction in order to halt or reverse the progression of this disease. The chronic hypoxic model of pulmonary hypertension still provides useful, as well as potentially relevant, data to the clinical setting, as structural remodelling through proliferation of PASMCs and fibroblasts and vasoconstriction of the pulmonary arteries both contribute to the progression of PAH, irrespective of different underlying causes (Humbert et al., 2004; Simonneau et al., 2004).
In this study we have shown that the Kv7 channel activator flupirtine is a potent pulmonary arterial vasodilator and markedly attenuates elevated RVP and RVH in two independent models of PAH. With the advent of new compounds that are more selective for the Kv7 channel subtypes, further investigation into the effects of these compounds on the pulmonary vasculature is warranted. Drugs that selectively activate Kv7.2–Kv7.5 potassium channels may prove a useful co-strategy to prevent and treat PAH in humans.
Acknowledgments
We are grateful to Astra Zeneca for the gift of retigabine. This work was supported by a PhD studentship from the British Heart Foundation to AM, and project grants from the British Heart Foundation (MRM & AMG).
Glossary
Abbreviations:
- BMP
bone morphogenetic protein
- BMPR-II
bone morphogenetic protein receptor type II
- fPAH
familial pulmonary arterial hypertension
- Kv
voltage-gated potassium
- LV + S
left ventricle plus septum
- mRVP
mean right ventricular pressure
- PAH
pulmonary arterial hypertension
- PASMCs
pulmonary artery smooth muscle cells
- RV
right ventricle
- RVH
right ventricular hypertrophy
- SERT
5-HT (serotonin) transporter
Conflict of interest
None.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Supplemental paper KCNQ modulators reveal a key role for KCNQ potassium channels in regulating the tone of rat pulmonary artery smooth muscle.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Alexander SP, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 3rd edition. Br J Pharmacol. 2008;153(Suppl 2):S1–S209. doi: 10.1038/sj.bjp.0707746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Archer SL, Souil E, Dinh-Xuan AT, Schremmer B, Mercier JC, El Yaagoubi A, et al. Molecular identification of the role of voltage-gated K+ channels, Kv1.5 and Kv2.1, in hypoxic pulmonary vasoconstriction and control of resting membrane potential in rat pulmonary artery myocytes. J Clin Invest. 1998;101:2319–2330. doi: 10.1172/JCI333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentzen BH, Schmitt N, Calloe K, Dalby BW, Grunnet M, Olesen SP. The acrylamide (S)-1 differentially affects Kv7 (KCNQ) potassium channels. Neuropharmacology. 2006;51:1068–1077. doi: 10.1016/j.neuropharm.2006.07.001. [DOI] [PubMed] [Google Scholar]
- Burg ED, Remillard CV, Yuan JXJ. Potassium channels in the regulation of pulmonary artery smooth muscle cell proliferation and apoptosis: pharmacotherapeutic implications. British Journal of Pharmacology. 2008;153:S99–S111. doi: 10.1038/sj.bjp.0707635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calloe K, Nielsen MS, Grunnet M, Schmitt N, Jorgensen NK. KCNQ channels are involved in the regulatory volume decrease response in primary neonatal rat cardiomyocytes. Biochimica et Biophysica Acta-Molecular. Cell Research. 2007;1773:764–773. doi: 10.1016/j.bbamcr.2007.02.008. [DOI] [PubMed] [Google Scholar]
- Chin KM, Rubin LJ. Pulmonary arterial hypertension. J Am Coll Cardiol. 2008;51:1527–1538. doi: 10.1016/j.jacc.2008.01.024. [DOI] [PubMed] [Google Scholar]
- Clapp LH, Gurney AM. ATP-sensitive K+ channels regulate resting potential of pulmonary arterial smooth muscle cells. Am J Physiol. 1992;262:H916–H920. doi: 10.1152/ajpheart.1992.262.3.H916. [DOI] [PubMed] [Google Scholar]
- Coetzee WA, Amarillo Y, Chiu J, Chow A, Lau D, McCormack T, et al. Molecular diversity of K+ channels. Ann N Y Acad Sci. 1999;868:233–285. doi: 10.1111/j.1749-6632.1999.tb11293.x. [DOI] [PubMed] [Google Scholar]
- Cogolludo A, Moreno L, Lodi F, Frazziano G, Cobeno L, Tamargo J, et al. Serotonin inhibits voltage-gated K+ currents in pulmonary artery smooth muscle cells: role of 5-HT2A receptors, caveolin-1, and KV1.5 channel internalization 1. Circ Res. 2006;98:C931–C938. doi: 10.1161/01.RES.0000216858.04599.e1. [DOI] [PubMed] [Google Scholar]
- Daly CJ, Deighan C, Mcgee A, Mennie D, Ali Z, McBride M, et al. A knockout approach indicates a minor vasoconstrictor role for vascular alpha(1B)-adrenoceptors in mouse. Physiol Genomics. 2002;9:85–91. doi: 10.1152/physiolgenomics.00065.2001. [DOI] [PubMed] [Google Scholar]
- Evans AM, Osipenko ON, Gurney AM. Properties of a novel K+ current that is active at resting potential in rabbit pulmonary artery smooth muscle cells. J Physiol (London) 1996;496:407–420. doi: 10.1113/jphysiol.1996.sp021694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedel HA, Fitton A. Flupirtine – a review of its pharmacological properties, and therapeutic efficacy in pain states. Drugs. 1993;45:548–569. doi: 10.2165/00003495-199345040-00007. [DOI] [PubMed] [Google Scholar]
- Galie N, Ghofrani HA, Torbicki A, Barst RJ, Rubin LJ, Badesch D, et al. Sildenafil citrate therapy for pulmonary arterial hypertension. New England. J Med. 2005;353:2148–2157. doi: 10.1056/NEJMoa050010. [DOI] [PubMed] [Google Scholar]
- Galie N, Olschewski H, Oudiz RJ, Torres F, Frost A, Ghofrani HA, et al. Ambrisentan for the treatment of pulmonary arterial hypertension – results of the Ambrisentan in Pulmonary Arterial Hypertension, Randomized, Double-Blind, Placebo-Controlled, Multicenter, Efficacy (ARIES) Study 1 and 2. Circulation. 2008;117:3010–3019. doi: 10.1161/CIRCULATIONAHA.107.742510. [DOI] [PubMed] [Google Scholar]
- Gardener MJ, Johnson IT, Burnham MP, Edwards G, Heagerty AM, Weston AH. Functional evidence of a role for two-pore domain potassium channels in rat mesenteric and pulmonary arteries. Br J Pharmacol. 2004;142:192–202. doi: 10.1038/sj.bjp.0705691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenwood IA, Yeung SY, Hettiarachi S, Andersson M, Baines DL. KCNQ-encoded channels regulate Na(+) transport across H441 lung epithelial cells. Pflugers Arch. 2009;457:785–794. doi: 10.1007/s00424-008-0557-7. [DOI] [PubMed] [Google Scholar]
- Grissmer S, Nguyen AN, Aiyar J, Hanson DC, Mather RJ, Gutman GA, et al. Pharmacological characterization of five cloned voltage-gated K+ channels, types Kv1.1, 1.2, 1.3, 1.5, and 3.1, stably expressed in mammalian cell lines. Mol Pharmacol. 1994;45:1227–1234. [PubMed] [Google Scholar]
- Guignabert C, Izikki M, Tu LI, Li ZL, Zadigue P, Barlier-Mur AM, et al. Transgenic mice overexpressing the 5-hydroxytryptamine transporter gene in smooth muscle develop pulmonary hypertension. Circ Res. 2006;98:1323–1330. doi: 10.1161/01.RES.0000222546.45372.a0. [DOI] [PubMed] [Google Scholar]
- Gurney AM, Osipenko ON, MacMillan D, McFarlane KM, Tate RJ, Kempsill FEJ. Two-pore domain K channel, TASK-1, in pulmonary artery smooth muscle cells. Circulation Research. 2003;93:957–964. doi: 10.1161/01.RES.0000099883.68414.61. [DOI] [PubMed] [Google Scholar]
- Hadley JK, Noda M, Selyanko AA, Wood IC, Abogadie FC, Brown DA. Differential tetraethylammonium sensitivity of KCNQ1-4 potassium channels. Br J Pharmacol. 2000;129:413–415. doi: 10.1038/sj.bjp.0703086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrmann WM, Kern U, Aigner M. On the adverse reactions and efficacy of long-term treatment with flupirtine – preliminary results of an ongoing 12-month study with 200 patients suffering from chronic pain states in arthrosis or arthritis. Postgrad Med J. 1987;63:87–103. [PubMed] [Google Scholar]
- Humbert M, Morrell NW, Archer SL, Stenmark KR, MacLean MR, Lang IM, et al. Cellular and molecular pathobiology of pulmonary arterial hypertension 11. J Am Coll Cardiol. 2004;43:13S–24S. doi: 10.1016/j.jacc.2004.02.029. [DOI] [PubMed] [Google Scholar]
- Hummel T, Friedmann T, Pauli E, Niebch G, Borbe HO, Kobal G. Dose-related analgesic effects of flupirtine. Br J Clin Pharmacol. 1991;32:69–76. doi: 10.1111/j.1365-2125.1991.tb05615.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen HS, Callo K, Jespersen T, Jensen BS, Olesen SP. The KCNQ5 potassium channel from mouse: a broadly expressed M-current like potassium channel modulated by zinc, pH, and volume changes. Brain Res Mol Brain Res. 2005;139:52–62. doi: 10.1016/j.molbrainres.2005.05.007. [DOI] [PubMed] [Google Scholar]
- Jentsch TJ. Neuronal KCNQ potassium channels: physiology and role in disease. Nat Rev Neurosci. 2000;1:21–30. doi: 10.1038/35036198. [DOI] [PubMed] [Google Scholar]
- Jespersen T, Grunnet M, Olesen SP. The KCNQ1 potassium channel: from gene to physiological function. Physiology (Bethesda) 2005;20:408–416. doi: 10.1152/physiol.00031.2005. [DOI] [PubMed] [Google Scholar]
- Jin Z, Liang GH, Cooper EC, Jarlebark L. Expression and Localization of K Channels KCNQ2 and KCNQ3 in the Mammalian Cochlea. Audiol Neurootol. 2008;14:98–105. doi: 10.1159/000158538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi S, Balan P, Gurney AM. Pulmonary vasoconstrictor action of KCNQ potassium channel blockers. Respir Res. 2006;7:31. doi: 10.1186/1465-9921-7-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi S, Sedivy V, Hodyc D, Herget J, Gurney A. A role for KCNQ channels in hypoxia-induced pulmonary vasoconstriction and hypertension. Fund Clin Pharmacol. 2008;22:27. [Google Scholar]
- Joshi S, Sedivy V, Hodyc D, Herget J, Gurney AM. KCNQ modulators reveal a key role for KCNQ potassium channels in regulating the tone of rat pulmonary artery smooth muscle. J Pharmacol Exp Ther. 2009;329:368–376. doi: 10.1124/jpet.108.147785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keegan A, Morecroft I, Smillie D, Hicks MN, MacLean MR. Contribution of the 5-HT1B receptor to hypoxia-induced pulmonary hypertension – converging evidence using 5-HT1B-receptor knockout mice and the 5-HT1B/1D-receptor antagonist GR127935. Circ Res. 2001;89:1231–1239. doi: 10.1161/hh2401.100426. [DOI] [PubMed] [Google Scholar]
- Lamas JA, Selyanko AA, Brown DA. Effects of a cognition-enhancer, linopirdine (DuP 996), on M-type potassium currents (IK(M)) and some other voltage- and ligand-gated membrane currents in rat sympathetic neurons. Eur J Neurosci. 1997;9:605–616. doi: 10.1111/j.1460-9568.1997.tb01637.x. [DOI] [PubMed] [Google Scholar]
- Lane KB, Machado RD, Pauciulo MW, Thomson JR, Phillips JA, III, Loyd JE, et al. Heterozygous germline mutations in BMPR2, encoding a TGF-beta receptor, cause familial primary pulmonary hypertension. The International PPH. Consortium. Nat Genet. 2000;26:81–84. doi: 10.1038/79226. [DOI] [PubMed] [Google Scholar]
- McCallum LA, Greenwood IA, Tribe RM. Expression and function of K(v)7 channels in murine myometrium throughout oestrous cycle. Pflugers Arch. 2009;457:1111–1120. doi: 10.1007/s00424-008-0567-5. [DOI] [PubMed] [Google Scholar]
- Machado RD, Pauciulo MW, Thomson JR, Lane KB, Morgan NV, Wheeler L, et al. BMPR2 haploinsufficiency as the inherited molecular mechanism for primary pulmonary hypertension. Am J Hum Genet. 2001;68:92–102. doi: 10.1086/316947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackie AR, Brueggemann LI, Henderson KK, Shiels AJ, Cribbs LL, Scrogin KE, et al. Vascular KCNQ potassium channels as novel targets for the control of mesenteric artery constriction by vasopressin, based on studies in single cells, pressurized arteries, and in vivo measurements of mesenteric vascular resistance. J Pharmacol Exp Ther. 2008;325:475–483. doi: 10.1124/jpet.107.135764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLean MR, Deuchar GA, Hicks MN, Morecroft I, Shen SB, Sheward J, et al. Overexpression of the 5-hydroxytryptamine transporter gene – effect on pulmonary hemodynamics and hypoxia-induced pulmonary hypertension. Circulation. 2004;109:2150–2155. doi: 10.1161/01.CIR.0000127375.56172.92. [DOI] [PubMed] [Google Scholar]
- Main MJ, Cryan JE, Dupere JRB, Cox B, Clare JJ, Burbidge SA. Modulation of KCNQ2/3 potassium channels by the novel anticonvulsant retigabine. Mol Pharmacol. 2000;58:253–262. doi: 10.1124/mol.58.2.253. [DOI] [PubMed] [Google Scholar]
- Mair KM, MacLean MR, Morecroft I, Dempsie Y, Palmer TM. Novel interactions between the 5-HT transporter, 5-HT(1B) receptors and Rho kinase in vivo and in pulmonary fibroblasts. Br J Pharmacol. 2008;155:606–616. doi: 10.1038/bjp.2008.310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrion NV. Control of M-current. Annu Rev Physiol. 1997;59:483–504. doi: 10.1146/annurev.physiol.59.1.483. [DOI] [PubMed] [Google Scholar]
- Morecroft I, Dempsie Y, Bader M, Walther DJ, Kotnik K, Loughlin L, et al. Effect of tryptophan hydroxylase 1 deficiency on the development of hypoxia-induced pulmonary hypertension. Hypertension. 2007;49:232–236. doi: 10.1161/01.HYP.0000252210.58849.78. [DOI] [PubMed] [Google Scholar]
- Nickel B. The antinociceptive activity of flupirtine: a structurally new analgesic. Postgrad Med J. 1987;63(Suppl 3):19–28. [PubMed] [Google Scholar]
- Ohya S, Sergeant GP, Greenwood IA, Horowitz B. Molecular variants of KCNQ channels expressed in murine portal vein myocytes: a role in delayed rectifier current. Circ Res. 2003;92:1016–1023. doi: 10.1161/01.RES.0000070880.20955.F4. [DOI] [PubMed] [Google Scholar]
- Olschewski A, Li Y, Tang B, Hanze J, Eul B, Bohle RM, et al. Impact of TASK-1 in Human Pulmonary Artery Smooth Muscle Cells. Circ Res. 2006;98:1072–1080. doi: 10.1161/01.RES.0000219677.12988.e9. [DOI] [PubMed] [Google Scholar]
- Osipenko ON, Alexander D, MacLean MR, Gurney AM. Influence of chronic hypoxia on the contributions of non-inactivating and delayed rectifier K currents to the resting potential and tone of rat pulmonary artery smooth muscle. Br J Pharmacol. 1988;124:1335–1337. doi: 10.1038/sj.bjp.0702006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platoshyn O, Golovina VA, Bailey CL, Limsuwan A, Krick S, Juhaszova M, et al. Sustained membrane depolarization and pulmonary artery smooth muscle cell proliferation. Am J Physiol. 2000;279:C1540–C1549. doi: 10.1152/ajpcell.2000.279.5.C1540. [DOI] [PubMed] [Google Scholar]
- Platoshyn O, Yu Y, Golovina VA, McDaniel SS, Krick S, Li L, et al. Chronic hypoxia decreases K-V channel expression and function in pulmonary artery myocytes. Am J Physiol. 2001;280:L801–L812. doi: 10.1152/ajplung.2001.280.4.L801. [DOI] [PubMed] [Google Scholar]
- Remillard CV, Tigno DD, Platoshyn O, Burg ED, Brevnova EE, Conger D, et al. Function of Kv1.5 channels and genetic variations of KCNA5 in patients with idiopathic pulmonary arterial hypertension. Am J Physiol. 2007;292:C1837–C1853. doi: 10.1152/ajpcell.00405.2006. [DOI] [PubMed] [Google Scholar]
- Robbins J. KCNQ potassium channels: physiology, pathophysiology, and pharmacology. Pharmacol Ther. 2001;90:1–19. doi: 10.1016/s0163-7258(01)00116-4. [DOI] [PubMed] [Google Scholar]
- Rubin LJ, Badesch DB, Barst RJ, Galie N, Black CM, Keogh A, et al. Bosentan therapy for pulmonary arterial hypertension. N Engl J Med. 2002;346:896–903. doi: 10.1056/NEJMoa012212. [DOI] [PubMed] [Google Scholar]
- Sanguinetti MC, Curran ME, Zou A, Shen J, Spector PS, Atkinson DL, et al. Coassembly of K(v)LQT1 and minK (IsK) proteins to form cardiac I-Ks potassium channel. Nature. 1996;384:80–83. doi: 10.1038/384080a0. [DOI] [PubMed] [Google Scholar]
- Schenzer A, Friedrich T, Pusch M, Saftig P, Jentsch TJ, Grotzinger J, et al. Molecular determinants of KCNQ (K(V)7) K+ channel sensitivity to the anticonvulsant retigabine. J Neurosci. 2005;25:5051–5060. doi: 10.1523/JNEUROSCI.0128-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt WJ, Schuster G, Wacker E, Pergande G. Antiparkinsonian and other motor effects of flupirtine alone and in combination with dopaminergic drugs. Eur J Pharmacol. 1997;327:1–9. doi: 10.1016/s0014-2999(97)89671-9. [DOI] [PubMed] [Google Scholar]
- Schnee ME, Brown BS. Selectivity of linopirdine (DuP 996), a neurotransmitter release enhancer, in blocking voltage-dependent and calcium-activated potassium currents in hippocampal neurons. J Pharmacol Exp Ther. 1998;286:709–717. [PubMed] [Google Scholar]
- Simonneau G, Galie N, Rubin LJ, Langleben D, Seeger W, Domenighetti G, et al. Clinical classification of pulmonary hypertension. J Am Coll Cardiol. 2004;43:5S–12S. doi: 10.1016/j.jacc.2004.02.037. [DOI] [PubMed] [Google Scholar]
- Smirnov SV, Robertson TP, Ward JPT, Aaronson PI. Chronic hypoxia is associated with reduced delayed rectifier K+ current in rat pulmonary-artery muscle-cells. Am J Physiol. 1994;266:H365–H370. doi: 10.1152/ajpheart.1994.266.1.H365. [DOI] [PubMed] [Google Scholar]
- Sun HS, Hui K, Lee DW, Feng ZP. Zn2+ sensitivity of high- and low-voltage activated calcium channels. Biophys J. 2007;93:1175–1183. doi: 10.1529/biophysj.106.103333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szelenyi I, Nickel B, Borbe HO, Brune K. Mode of antinociceptive action of flupirtine in the rat. Br J Pharmacol. 1989;97:835–842. doi: 10.1111/j.1476-5381.1989.tb12023.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatulian L, Delmas P, Abogadie FC, Brown DA. Activation of expressed KCNQ potassium currents and native neuronal M-type potassium currents by the anti-convulsant drug retigabine. J Neurosci. 2001;21:5535–5545. doi: 10.1523/JNEUROSCI.21-15-05535.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wladyka CL, Kunze DL. KCNQ/M-currents contribute to the resting membrane potential in rat visceral sensory neurons. J Physiol. 2006;575:175–189. doi: 10.1113/jphysiol.2006.113308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wuttke TV, Seebohm G, Bail S, Maljevic S, Lerche H. The new anticonvulsant retigabine favors voltage-dependent opening of the K(v)7.2 (KCNQ2) channel by binding to its activation gate. Mol Pharmacol. 2005;67:1009–1017. doi: 10.1124/mol.104.010793. [DOI] [PubMed] [Google Scholar]
- Yeung S, Schwake M, Pucovsky V, Greenwood I. Bimodal effects of the Kv7 channel activator retigabine on vascular K+ currents. Br J Pharmacol. 2008;155:62–72. doi: 10.1038/bjp.2008.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeung SY, Pucovsky V, Moffatt JD, Saldanha L, Schwake M, Ohya S, et al. Molecular expression and pharmacological identification of a role for K(v)7 channels in murine vascular reactivity. Br J Pharmacol. 2007;151:758–770. doi: 10.1038/sj.bjp.0707284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoro SG, Urosevic D, Fellmann L, Greney H, Bousquet P, Feldman J. G-protein inwardly rectifying potassium channels are involved in the hypotensive effect of I1-imidazoline receptor selective ligands. J Hypertens. 2008;26:1025–1032. doi: 10.1097/HJH.0b013e3282f5ed44. [DOI] [PubMed] [Google Scholar]
- Young KA, Ivester C, West J, Carr M, Rodman DM. BMP signaling controls PASMC KV channel expression in vitro and in vivo. Am J Physiol Lung Cell Mol Physiol. 2006;290:L841–L848. doi: 10.1152/ajplung.00158.2005. [DOI] [PubMed] [Google Scholar]
- Yuan JXJ, Aldinger AM, Juhaszova M, Wang J, Conte JV, Gaine SP, et al. Dysfunctional voltage-gated K+ channels in pulmonary artery smooth muscle cells of patients with primary pulmonary hypertension. Circulation. 1998;98:1400–1406. doi: 10.1161/01.cir.98.14.1400. [DOI] [PubMed] [Google Scholar]
- Zhao L, Mason NA, Morrell NW, Kojonazarov B, Sadykov A, Maripov A, et al. Sildenafil inhibits hypoxia-induced pulmonary hypertension. Circulation. 2001;104:424–428. doi: 10.1161/hc2901.093117. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.