

Abstract
Background and purpose:
TAK-242, a novel synthetic small-molecule, suppresses production of multiple cytokines by inhibiting Toll-like receptor (TLR) 4 signalling. In this study, we investigated the target molecule of TAK-242 and examined its therapeutic effect in a mouse sepsis model.
Experimental approach:
Binding assay with [3H]-TAK-242 and nuclear factor-κB reporter assay were used to identify the target molecule and binding site of TAK-242. Bacillus calmette guerin (BCG)-primed mouse sepsis model using live Escherichia coli was used to estimate the efficacy of TAK-242 in sepsis.
Key results:
TAK-242 strongly bound to TLR4, but binding to TLR2, 3, 5, 9, TLR-related adaptor molecules and MD-2 was either not observed or marginal. Mutational analysis using TLR4 mutants indicated that TAK-242 inhibits TLR4 signalling by binding to Cys747 in the intracellular domain of TLR4. TAK-242 inhibited MyD88-independent pathway as well as MyD88-dependent pathway and its inhibitory effect was largely unaffected by lipopolysaccharide (LPS) concentration and types of TLR4 ligands. TAK-242 had no effect on the LPS-induced conformational change of TLR4-MD-2 and TLR4 homodimerization. In mouse sepsis model, although TAK-242 alone did not affect bacterial counts in blood, if co-administered with ceftazidime it inhibited the increases in serum cytokine levels and improved survival of mice.
Conclusions and implications:
TAK-242 suppressed TLR4 signalling by binding directly to a specific amino acid Cys747 in the intracellular domain of TLR4. When co-administered with antibiotics, TAK-242 showed potent therapeutic effects in an E. coli-induced sepsis model using BCG-primed mice. Thus, TAK-242 may be a promising therapeutic agent for sepsis.
Keywords: TAK-242, Toll-like receptor 4, intracellular domain, cysteine, sepsis
Introduction
Toll-like receptors (TLRs) play important roles in innate and adaptive immune responses. TLRs are pattern-recognition receptors that recognize microbial pathogens and their components. For example, TLR3, 4, 5, 7 and 9 recognize viral double-stranded RNA (Alexopoulou et al., 2001), lipopolysaccharide (LPS) of gram-negative bacteria (Poltorak et al., 1998; Hoshino et al., 1999), bacterial fragellin (Hayashi et al., 2001), viral single-stranded RNA (Diebold et al., 2004; Heil et al., 2004) and viral and bacterial CpG DNA (Hemmi et al., 2000) respectively. In addition, TLR2 forms heterodimers with TLR1 or TLR6, and recognizes peptidoglycan and lipopeptides of gram-positive bacteria (Ozinsky et al., 2000; Takeuchi et al., 2001; 2002;). MD-2 is associated with the extracellular domain (ECD) of TLR4 and greatly enhances its LPS signalling (Shimazu et al., 1999). The recognition of microbial pathogens and their components by TLRs initiates the activation of intracellular signalling and results in the release of a wide variety of inflammatory mediators, including nitric oxide, prostaglandins, cytokines such as tumour necrosis factor-α (TNF-α), interleukin (IL)-6, and IL-1β, type 1 interferon (IFN), and chemokines (Beutler, 2004). Activation of intracellular signalling requires the interaction between TLRs and adaptor molecules containing Toll/IL-1 receptor (TIR) domain such as MyD88, TIR domain-containing adaptor protein (TIRAP)/MyD88-adaptor-like (Mal), TIR domain-containing adaptor inducing IFN-β (TRIF)/TIR-domain containing adaptor molecule (TICAM)-1 and TRIF-related adaptor molecule (TRAM)/TICAM-2. MyD88 is utilized by all the TLRs except for TLR3; it activates nuclear factor-κB (NF-κB) and induces the expression of inflammatory cytokine genes (Medzhitov et al., 1998). In the case of TLR2 and TLR4, MyD88-dependent signalling requires TIRAP/Mal, which bridges TLR2/TLR4 and MyD88 (Horng et al., 2002; Yamamoto et al., 2002a). TRIF is required in MyD88-independent signalling, and is involved in TLR3 and TLR4 signalling (Yamamoto et al., 2002b; Oshiumi et al., 2003). TRIF leads to activation of NF-κB and IFN regulatory factor 3, and induces the expression of inflammatory cytokines and type 1 IFN genes. TRAM, which bridges TLR4 and TRIF, is a specific adaptor molecule in TLR4 signalling and transmits its signalling to TRIF (Yamamoto et al., 2003). Thus, TLRs activate intracellular signalling via interaction with adaptor molecules containing the TIR domain and induce the release of various inflammatory mediators.
Among TLRs, TLR4 has a dominant role in various inflammatory diseases, including sepsis, asthma and chronic obstructive pulmonary disease (Zuany-Amorim et al., 2002). To treat inflammatory diseases, it may be very important to modulate TLR4 signalling. We have discovered that a novel small-molecule cyclohexene derivative, TAK-242, selectively inhibits TLR4 signalling (Ii et al., 2006), and showed beneficial effects in a mouse endotoxin model (Sha et al., 2007). How TAK-242 inhibits signalling, however, remains unclear. In this study, we tried to identify the target molecule of TAK-242. In addition, to further evaluate the efficacy of TAK-242 in vivo, we examined the efficacy of TAK-242 in a mouse sepsis model using live bacteria. Our data demonstrated that TAK-242 inhibited TLR4 signalling by binding directly to a specific amino acid Cys747 in the intracellular domain (ICD) of TLR4 and had potent therapeutic effects in an E. coli-induced mouse sepsis model.
Methods
Cells
African green monkey kidney fibroblast cell line COS-7 cells and human monocytic cell line P31/FUJ cells were obtained through the Health Science Research Resources Bank (Osaka, Japan). Human embryonic kidney (HEK) 293 cells were purchased from Dainippon Pharma (Osaka, Japan). The mouse macrophage cell line RAW264.7 cells were purchased from American Type Culture Collection (Manassas, VA, USA). COS-7 and HEK293 cells were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal calf serum (FCS). P31/FUJ and RAW264.7 cells were maintained in RPMI 1640 medium supplemented with 10% FCS.
Expression vectors
Expression vectors for TLR4 and TLR2 tagged with FLAG were cloned into pcDNA3.1Zeo (Invitrogen, Grand Island, NY, USA) and pFLAG-CMV-1 (Sigma-Aldrich, St Louis, MO, USA) respectively. pUNO-hTLR3-haemagglutinin (HA), pUNO-hTLR5-HA, pUNO-hTLR6-HA and pUNO-hTLR9-HA were purchased from InvivoGen (San Diego, CA, USA). pNifty-luc encoding a firefly luciferase-linked NF-κB reporter gene and phRL-TK encoding Renilla luciferase as an internal control were purchased from InvivoGen and Promega (Madison, WI, USA) respectively. Expression vectors for TIRAP/Mal and MyD88 tagged with HA were cloned into pALTERMAX (Promega) and pFLAG-CMV-1 respectively. Expression vectors for TRIF and TRAM tagged with FLAG were cloned into pFLAG-CMV-1. Expression vector for MD-2 tagged with FLAG and CD14 were cloned into pEFBos and pSRaNeo respectively. TLR4 chimeras were constructed by PCR-site-directed mutagenesis using FLAG-TLR2 and FLAG-TLR4 expression vectors as templates as described previously (Lee et al., 2004). TLR4-Bla(a) and TLR4-Bla(b), which were fused with β-lactamase enzyme fragments, Bla(a) and Bla(b), respectively, were also constructed as described previously (Lee et al., 2004). Expression vectors for FLAG-TLR4 point mutants were generated by using QuickChange II site-directed mutagenesis kit (Stratagene, La Jolla, CA, USA). All constructions involving PCR were verified by sequencing.
Binding assay with [3H]-TAK-242
COS-7 and HEK293 cells were seeded in 10 cm dishes at 2 to 4 × 105 and 0.6 to 1 × 106 cells per dish, respectively, and incubated for 3 to 4 days. COS-7 cells were transiently transfected with expression vectors encoding FLAG-TLR2, FLAG-TLR4, FLAG-T2N-T4C, FLAG-T4N-T2C or TLR5-HA using Lipofectamine reagent and Plus reagent. HEK293 cells were transiently transfected with expression vectors encoding FLAG-TLR4, TLR3-HA, TLR9-HA, FLAG-TRAM, FLAG-TRIF, HA-MyD88 or HA-TIRAP/Mal using Lipofectamine reagent and Plus reagent. After 2 days of transfection, cells were incubated with 100 nM [3H]-TAK-242 at 37°C for 6 h. For competition assays, the cells were incubated for 2 h with various concentrations of nonradioactive TAK-242 and its enantiomer, then for 4 h with [3H]-TAK-242. After being washed with PBS, cells were lysed with lysis buffer [150 mM NaCl, 0.1% Nonidet P40, 0.05% CHAPS, 30 mM NaF, 1 mM Na3VO4, 50 mM Tris-HCl (pH 7.5) plus a protease inhibitor cocktail]. For lysis of cells transfected with TLR3-HA or TLR9-HA, the lysis buffer contained 100 mM NaCl, 1 mM EDTA, 1 mM NaF, 0.1% SDS, 0.5% sodium deoxycholate, 1% Triton-X100, 1% glycerol, 2 mM Na3VO4, 10 mM Tris-HCl (pH 7.5) plus a protease inhibitor cocktail. Cell lysates were incubated with anti-FLAG M2 antibody (Ab) or anti-HA 12CA5 Ab for 3 h at 4°C. Protein A Sepharose 6MB was then added to the mixtures and further incubated overnight at 4°C. Immunoprecipitates were separated by SDS-PAGE and transferred to polyvinylidene difluoride membranes. The membranes were incubated with anti-FLAG M2 or anti-HA 12CA5 Abs as the first Ab, then ECL anti-mouse IgG, horseradish peroxidase-linked whole Ab (from sheep) as the secondary Ab. Immunoprecipitates were detected using ECL western blotting detection reagents. Radioactive imaging of the immunoprecipitates was analysed with a BAStation.
NF-κB reporter assay
HEK293 cells were seeded in 96-well plates at 2 × 104 cells per well and incubated overnight. Cells were transiently transfected with 50 ng TLR expression vectors or empty vector, along with 15 ng pNifty-luc, 15 ng phRL-TK, 10 ng MD-2 expression vector and 10 ng CD14 expression vector per well using Lipofectamine reagent and Plus reagent. After 6 h of transfection, various concentrations of test compounds were added to the wells and further incubated overnight. For LPS stimulation, cells were transiently transfected with 0.5 ng TLR4 expression vector or empty vector per well using Lipofectamine reagent and Plus reagent. On the day after transfection, 100 or 10 ng·mL−1E. coli O111 : B4 LPS and various concentrations of TAK-242 were added to the wells and cells were further incubated for 6 h. The luciferase activity was measured using Dual-Glo luciferase assay system. Transfection efficacy was normalized for cotransfected Renilla luciferase activity.
Detection of TLR4-MD-2 on cell surface
RAW264.7 cells were seeded into 10 cm dishes at 1 × 106 cells per dish and incubated for 2 days at 37°C. Cells were incubated in the presence of 1 µM TAK-242 or 1 µg·mL−1 polymyxin B for 30 min at 37°C, then stimulated with 1 µg·mL−1E. coli O111 : B4 LPS for 30 min at 37°C. Cells (1 × 105 cells) recovered from the dishes were incubated with PE-conjugated anti-mouse TLR4-MD-2 Ab MTS510 for 30 min on ice. After two washes with PBS containing 0.1% BSA and 0.01% NaN3, the cells were analysed with a flow cytometer.
β-Lactamase enzyme fragment complementation assay
HEK293 cells were seeded in 24-well plates at 3 × 105 cells per well and incubated overnight. Cells were transiently transfected with 5 ng TLR4-Bla(a) and 5 ng TLR4-Bla(b), along with 75 ng pNifty-luc, 75 ng phRL-TK, 50 ng MD-2 expression vector and 50 ng CD14 expression vector per well using LipofectAMINE2000. After 24 h of transfection, cells were treated with test compounds for 3 h and then loaded with 1 µM CCF2/AM for 1.5 h at room temperature in the dark. The fluorescence image was analysed by an IN cell analyser 1000. For each well, 15 images were captured with a 360 nm excitation and 460 nm emission filter (for blue fluorescence) and a 360 nm excitation and 535 nm emission filter (for green fluorescence). TLR4 homodimerization was calculated as ratio of the average pixel intensities of blue fluorescence induced by TLR4-Bla(a)-TLR4-Bla(b) interaction and those of green fluorescence (no interaction).
Expression of CD40 molecule and IFN-inducible genes and cytokine production in response to LPS in mouse bone marrow dendritic cells (DCs)
Mouse bone marrow-derived DCs were generated by culturing bone marrow cells in the presence of 10 ng·mL−1 GM-CSF, as described previously (Kaisho et al., 2001). Mouse DCs were cultured in the absence or presence of indicated concentrations of E. coli O55 : B5 LPS for 24 h. TAK-242 was added 1 h before addition of LPS. IL-12 p40 and TNF-α production were measured by ELISA. CD40 expression was analysed by flow cytometry as described previously (Kaisho et al., 2001). DCs were also harvested at indicated times after LPS stimulation and subjected to Northern blot analysis, as described previously (Hoshino et al., 2002).
TNF-α production in response to LPS in PMA-stimulated P31/FUJ cells and RAW264.7 cells
P31/FUJ cells were seeded in 96-well plates at 2 × 105 cells per well and incubated overnight in the presence of phorbol 12-myristate 13-acetate (PMA). PMA-stimulated P31/FUJ cells were cultured with various concentrations of LPS for 4 h in the presence of various concentrations of test compounds. TNF-α production in the culture supernatants was measured by ELISA. RAW264.7 cells were seeded in 96-well plates at 1 × 105 cells per well and incubated overnight. Cells were cultured with 0.5 ng·mL−1E. coli O111 : B4 LPS or 10 µg·mL−1 high mobility group box 1 protein (HMGB-1) for 20 h in the presence of 0.1 ng·mL−1 recombinant mouse IFN-γ and various concentrations of test compounds. TNF-α production in the culture supernatants was measured by ELISA.
E. coli-induced sepsis model in Bacillus calmette guerin (BCG)-primed mice
The animal experiments conducted in this study were approved by animal experiment ethics committee of Takeda Pharmaceutical Company Ltd. Five week-old male C57BL/6 mice (Charles River Japan, Kanagawa, Japan) were injected intravenously with 2 mg live Mycobacterium bovis BCG as described previously (Christ et al., 1995). Twelve or thirteen days later, the BCG-primed mice were inoculated intraperitoneally with approximately 1 × 107 CFU of E. coli O111. Various doses of TAK-242 and 20 mg·kg−1 of ceftazidime were administered intravenously 1 h after the bacterial challenge. Survival was recorded over 7 days. Bacterial counts in blood were determined up to 4 h after the bacterial challenge. Sera were collected up to 4 h after the bacterial challenge, and serum levels of TNF-α, IL-1β, IL-6, IL-10 and MIP-2 were measured by ELISA.
Statistical analysis
All statistical analyses were carried out using sas software (version 6.1, SAS institute, Cary, NC, USA). IC50 values were calculated using sas system procedure NLIN, which produces least squares estimates of the parameters of a nonlinear model (logistic model). Differences in serum cytokine levels between vehicle-treated and TAK-242-treated groups were analysed with one-tailed Williams, one-tailed Shirley-Williams or Wilcoxon's test with Holm's correction. The minimum effective dose in survival data was evaluated by Tarone's test with a simple closed step-down method (Ajit et al., 1996). Effects of TAK-242 and ceftazidime on bacterial counts in blood were evaluated with two-way analysis of variance (anova).
Compounds and reagents
TAK-242, ethyl (6R)-6-[N-(2-chloro-4-fluorophenyl) sulphamoyl]cyclohex-1-ene-1-carboxylate was synthesized by Takeda Pharmaceutical Company Ltd (Osaka, Japan). [3H]-TAK-242 was synthesized by Amersham Pharmacia Biotech UK (1.74 TBq·mM−1; Buckinghamshire, UK).
E. coli O55 : B5 and O111 : B4 LPS was purchased from Sigma-Aldrich. HMGB-1 were purchased from R&D Systems (Minneapolis, MN, USA). Helenalin and resveratrol were purchased from Alexis Biochemicals (Grünberg, Germany) and Wako Pure Chemical Industries (Osaka, Japan), Ltd. respectively. Polymyxin B and Lipid IVa were purchased from Sigma-Aldrich and Peptide Institute (Osaka, Japan) respectively. Lipofectamine reagent and Plus reagent were obtained from Invitrogen; protease inhibitor cocktail, PMA and anti-FLAG M2 Ab, Sigma-Aldrich; anti-HA 12CA5 Ab, Roche (Indianapolis, IN, USA); Protein A Sepharose 6MB, horseradish peroxidase-linked whole Ab and ECL western blotting detection reagents, Amersham Biosciences (Uppsala, Sweden); BAStation, Fuji Film (Tokyo, Japan); Dual-Glo luciferase assay system, Promega; PE-conjugated anti-mouse TLR4-MD-2 Ab MTS510, eBioscience (San Diego, CA, USA); flow cytometer (CytoACE-300), JASCO Corporation (Tokyo, Japan); LipofectAMINE2000 and CCF2/AM, Invitrogen; IN Cell Analyser 1000, GE Healthcare (Buckinghamshire, UK); GM-CSF, PeproTech (London, UK); ELISA for IL-12 p40, TNF-α and MIP-2, Genzyme (Minneapolis, MN, USA); ELISA for TNF-α, BioSource (Camarillo, CA, USA); ELISA for IL-10, R&D Systems; recombinant mouse IFN-γ (Genzyme); BCG, Japan BCG (Tokyo, Japan): ELISA for TNF-α, IL-1β and IL-6, Amersham Pharmacia Biotech.
Results
TAK-242 selectively binds to TLR4, but not to MD-2 or adaptor molecules containing the TIR domain
In a previous study, we showed that TAK-242 acts upstream of TLR4 signalling and that its target molecule might be TLR4, MD-2 or adaptor molecules containing the TIR domain (Ii et al., 2006). We, therefore, examined whether TAK-242 binds to TLR4 and adaptor molecules (MyD88, TIRAP/Mal, TRAM and TRIF) using tritium-labelled TAK-242 ([3H]-TAK-242). Expression levels of adaptor molecules were similar to or greater than that of TLR4 (Figure 1A). Under these conditions, [3H]-TAK-242 strongly bound to TLR4, but did not bind to adaptor molecules (Figure 1A).
Figure 1.
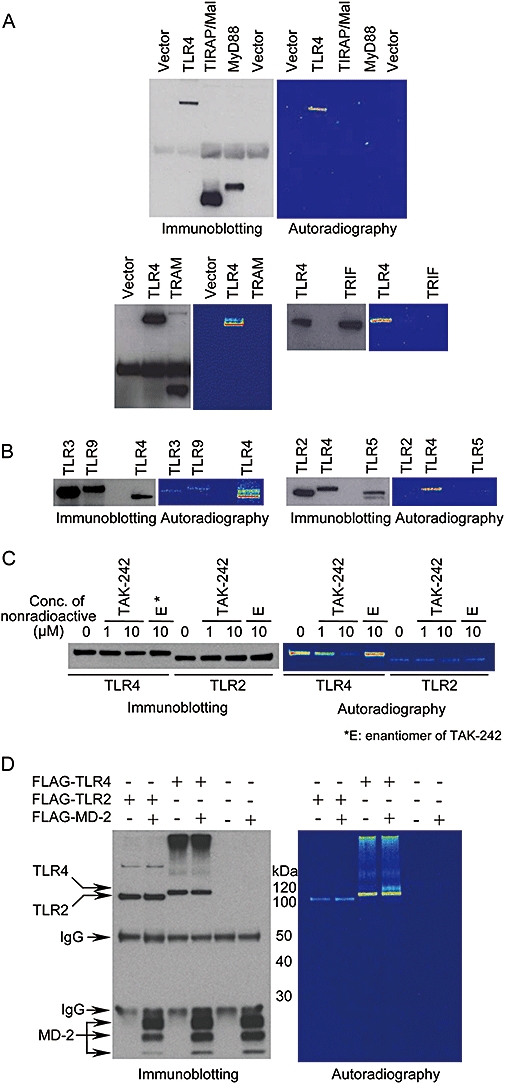
TAK-242 preferentially binds to TLR4, but not to other TLRs, TLR-related adaptor molecules or MD-2. (A) Binding of [3H]-TAK-242 to TLR-related adaptor molecules. HEK293 cells were transiently transfected with expression vector coding FLAG-TRAM, FLAG-TRIF, HA-TIRAP/Mal, HA-MyD88 and FLAG-TLR4. Empty lanes were used for a molecular weight marker. (B) Binding of [3H]-TAK-242 to various TLRs. HEK293 cells were transiently transfected with expression vector coding TLR3-HA, TLR9-HA and FLAG-TLR4. COS-7 cells were transiently transfected with expression vector coding FLAG-TLR2, FLAG-TLR4 and TLR5-HA. Empty lanes were used for a molecular weight marker. (C) Effect of nonradioactive TAK-242 and its enantiomer on the binding of [3H]-TAK-242 to TLR4. COS-7 cells were transiently transfected with expression vector coding FLAG-TLR2 and FLAG-TLR4. (D) Binding of [3H]-TAK-242 to MD-2. COS-7 cells were transiently transfected with FLAG-MD-2, FLAG-TLR2 and FLAG-TLR4. After 2 days of transfection, the cells were incubated with 100 nM of [3H]-TAK-242 for 6 h, then lysed. For competition assay, the cells were incubated with various concentrations of nonradioactive TAK-242 or 10 µM of its enantiomer for 2 h, and with 100 nM [3H]-TAK-242 for 4 h. Cell lysates were immunoprecipitated with anti-FLAG M2 and anti-HA 12CA5 Abs. The immunoprecipitates were subjected to SDS-PAGE and western blotting. Radioactive imaging of the immunoprecipitates was analysed by autoradiography. All data shown are representative of three independent experiments. HEK, human embryonic kidney; Mal, MyD88-adaptor like; TIRAP, TIR domain-containing adaptor protein; TLR, Toll-like receptor; TRAM, TRIF-related adaptor molecule; TRIF, TIR domain-containing adaptor inducing IFN-β.
To determine whether TAK-242 specifically binds to TLR4 among the various TLRs, binding of [3H]-TAK-242 to TLR2, TLR3, TLR4, TLR5 and TLR9 was examined. Expression levels of TLR2, TLR3, TLR5 and TLR9 in COS-7 cells were similar to or greater than that of TLR4 (Figure 1B). Under these conditions, binding of [3H]-TAK-242 to TLR2, TLR3, TLR5 and TLR9 was marginal compared with that to TLR4 (Figure 1B). To confirm the binding of [3H]-TAK-242 to TLR4, a competition assay using nonradioactive TAK-242 or its enantiomer was performed. Nonradioactive TAK-242 inhibited the binding of [3H]-TAK-242 to TLR4 (but not to TLR2) at concentrations of 1 and 10 µM (Figure 1C). On the other hand, an enantiomer of TAK-242, which has an inhibitory effect on cytokine production that is about 350 times less potent than that of TAK-242 (Yamada et al., 2005), did not inhibit the binding of [3H]-TAK-242 to TLR4, even at a concentration of 10 µM (Figure 1C). [3H]-TAK-242 did not bind to MD-2 (Figure 1D). Co-expression of MD-2 and TLR4 did not affect the binding of [3H]-TAK-242 to TLR4 (Figure 1D).
TAK-242 inhibits TLR4 signalling via binding to the ICD of TLR4
To clarify the binding regions of TAK-242 in TLR4, we constructed chimeric molecules of TLR2 and TLR4. T2N-T4C consists of the ECD of TLR2 (amino acids 1-589) and the transmembrane/intracellular domain (TMD/ICD) of TLR4 (amino acids 632-839), whereas T4N-T2C consists of the ECD of TLR4 (amino acids 1-631) and the TMD/ICD of TLR2 (amino acids 590-784) (Lee et al., 2004) (Figure 2A). These chimeric molecules were expressed in COS-7 cells (Figure 2B), and the binding of [3H]-TAK-242 to them was investigated. [3H]-TAK-242 was found to bind to T2N-T4C and TLR4, but did not bind to T4N-T2C and TLR2 (Figure 2B). Next, the inhibitory effect of TAK-242 on constitutive NF-κB activation induced by overexpression of these chimeric proteins was examined. As described previously (Lee et al., 2004), TLR2 and T4N-T2C did not induce constitutive NF-κB activation (Figure 2C). Co-overexpression of TLR2 and TLR6 induced substantial constitutive NF-κB activation with an FL/RL ratio of about 30, although the activation level was much lower than that of TLR4 and T2N-T4C (Figure 2C). TAK-242 inhibited TLR4- and T2N-T4C-mediated constitutive NF-κB activation in a concentration-dependent manner, but TAK-242 did not inhibit TLR2/TLR6-mediated constitutive NF-κB activation (Figure 2C).
Figure 2.
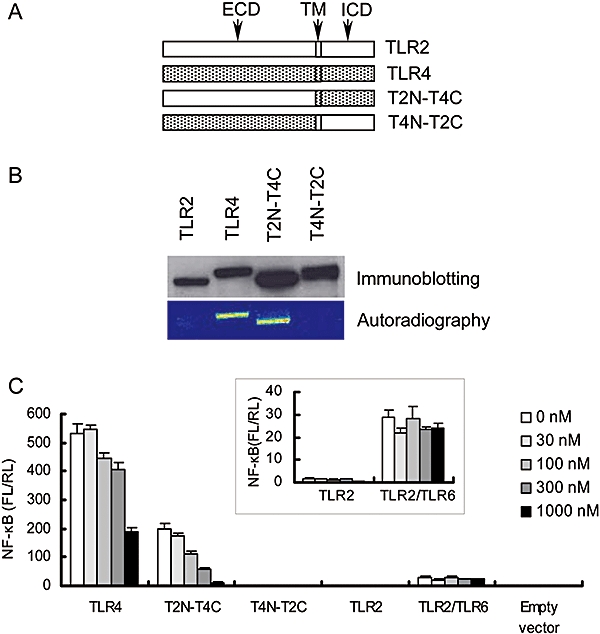
TAK-242 binds to ICD/TMD of TLR4 and inhibits TLR4 signalling. (A) Schematic presentation of TLR4 chimeric constructs. (B) Binding of [3H]-TAK-242 to TLR4 chimeric constructs. COS-7 cells were transiently transfected with expression vectors coding FLAG-TLR2, FLAG-TLR4, FLAG-T2N-T4C and FLAG-T4N-T2C. After 2 days of transfection, the cells were incubated with 100 nM of [3H]-TAK-242 for 6 h. Cell lysates were immunoprecipitated with anti-FLAG M2 Ab, and the immunoprecipitates were subjected to SDS-PAGE and western blotting. Radioactive imaging of the immunoprecipitates was analysed by autoradiography. Data shown are representative from three independent experiments. (C) Effect of TAK-242 on constitutive NF-κB activation in TLR chimeric proteins. HEK293 cells were transiently transfected with expression vectors coding TLR2, TLR4, T2N-T4C, T4N-T2C and TLR2/TLR6 along with pNifty and phRL-TK. Expression vectors coding MD-2 and CD14 were also co-transfected. After 6 h of transfection, cells were incubated with various concentrations of TAK-242 overnight. Luciferase activity was determined using a luciferase assay kit. Data represent the mean ± SEM in triplicate wells. Data shown are representative of three independent experiments. ECD, extracellular domain; ICD, intracellular domain; NF-κB, nuclear factor-κB; TLR, Toll-like receptor; TMD, transmembrane domain.
Considering the chemical structure of TAK-242, we speculated that TAK-242 might work as a Michael acceptor (Dragovich et al., 1998; Matthews et al., 1999; Zhu et al. 2002) and interact with a cysteine or lysine residue. Therefore, alanine substitutions at cysteine (C664A, C706A, C747A and C831A) and arginine substitutions at lysine (K653R, K666R, K694R, K729R, K732R, K773R, K776R, K813R and K819R) were introduced into the TMD/ICD of TLR4 and the binding of [3H]-TAK-242 to these TLR4 mutants was examined. All of these mutants were expressed at similar levels to that of wild-type TLR4 (Figure 3A). [3H]-TAK-242 bound to C664A, C706A, C831A and all the arginine-substituted mutants similar to wild-type TLR4, but the C747A substitution markedly impaired the binding of [3H]-TAK-242 (Figure 3A). Interestingly, the P714H mutant (P714H mutation corresponds to the P712H mutation in C3H/HeJ mice which are hyporesponsive to LPS (Poltorak et al., 1998; Hoshino et al., 1999)) retained the binding of [3H]-TAK-242 (Figure 3A).
Figure 3.
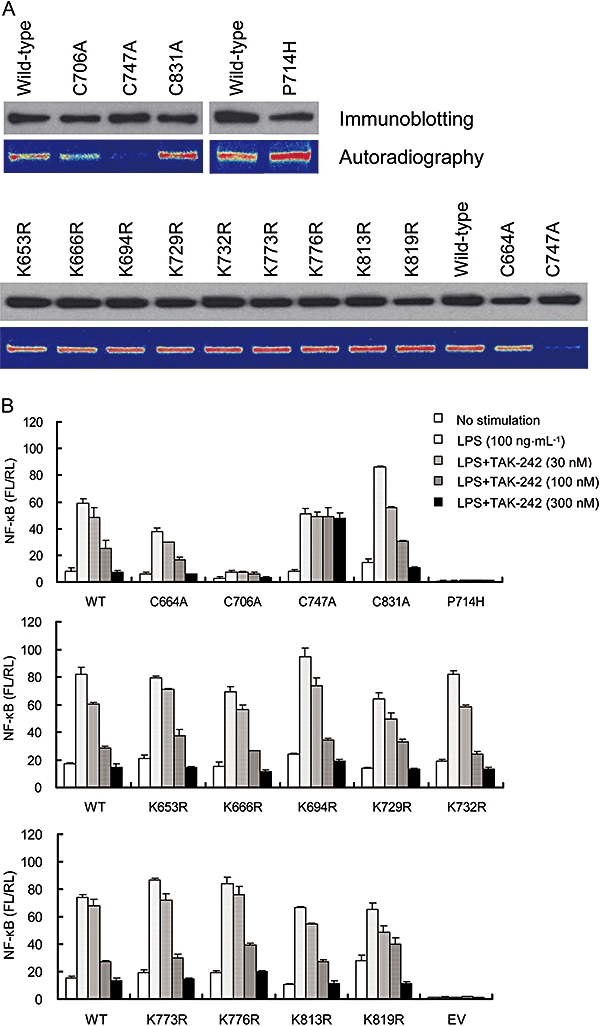
TAK-242 binds to Cys747 in ICD of TLR4 and inhibits TLR4 signalling. (A) Binding of [3H]-TAK-242 to TLR4 mutants with alanine substitutions at cysteine residues and arginine substitutions at lysine residues in the ICD of TLR4. COS-7 cells were transiently transfected with expression vectors coding FLAG-TLR4 and FLAG-TLR4 with single amino acid mutations in TLR4. After 2 days of transfection, the cells were incubated with 100 nM of [3H]-TAK-242 for 6 h. Cell lysates were immunoprecipitated with anti-FLAG M2 Ab, and the immunoprecipitates were subjected to SDS-PAGE and western blotting. Radioactive imaging of the immunoprecipitates was analysed by autoradiography. Data shown are representative of three independent experiments. (B) Effect of TAK-242 on LPS-induced NF-κB activation in TLR4 mutants with alanine substitutions at cysteine residues and arginine substitutions at lysine residues in the ICD of TLR4. HEK293 cells were transiently transfected with expression vectors coding TLR4 and TLR4 with single amino acid mutations along with pNifty and phRL-TK. Expression vectors coding MD-2 and CD14 were also co-transfected. On the day after transfection, cells were incubated with 100 ng·mL−1 LPS and various concentrations of TAK-242 for 6 h. Luciferase activity was determined using luciferase assay kit. Data represent the mean ± SEM in triplicate wells. Data shown are representative of two independent experiments. ICD, intracellular domain; LPS, lipopolysaccharide; NF-κB, nuclear factor-κB; TLR, Toll-like receptor.
Furthermore, we examined the effect of TAK-242 on LPS-induced NF-κB activation through these TLR4 mutants. All mutants except the C706A and P714H mutants showed LPS-induced NF-κB activation similar to wild-type TLR4 (Figure 3B). The LPS-induced NF-κB activation through the P714H mutant was completely attenuated (Figure 3B). On the other hand, the C706A mutant showed substantial LPS-induced NF-κB activation, although its activation level was much lower than that in wild-type TLR4 (Figure 3B). TAK-242 concentration-dependently inhibited LPS-induced NF-κB activation through the C664A, C706A and C831A mutants and all of the mutants with arginine substitutions (Figure 3B). In contrast, the C747A substitution abolished the inhibitory effect of TAK-242 (Figure 3B). To clarify the significance of Cys747 in TLR4 signalling, serine, threonine and valine substitutions at Cys747 were introduced. All of these mutants were expressed at similar levels to that of wild-type TLR4 (Figure 4A). Interestingly, the C747A mutant retained the ability to activate NF-κB, whereas the LPS-induced NF-κB activation through the C747T mutant was much lower than that through wild-type TLR4, and activation through C747S and C747V mutants was almost completely attenuated (Figure 4B). [3H]-TAK-242 did not bind to all of the Cys747 mutants (Figure 4A).
Figure 4.
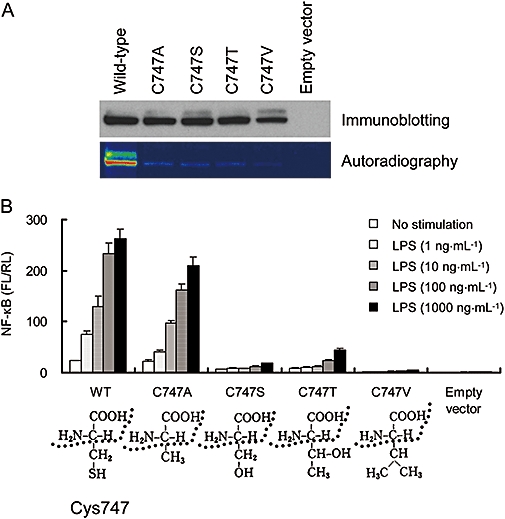
Single mutation at Cys747 in TLR4 affects LPS-induced NF-κB activation. (A) Binding of [3H]-TAK-242 to TLR4 mutants with single mutation at Cys747 in TLR4. COS-7 cells were transiently transfected with expression vectors coding FLAG-TLR4 and FLAG-TLR4 with single amino acid mutations in TLR4. After 2 days of transfection, the cells were incubated with 100 nM of [3H]-TAK-242 for 6 h. Cell lysates were immunoprecipitated with anti-FLAG M2 Ab, and the immunoprecipitates were subjected to SDS-PAGE and western blotting. Radioactive imaging of the immunoprecipitates was analysed by autoradiography. Data shown are representative of two independent experiments. (B) Effect of TAK-242 on LPS-induced NF-κB activation in TLR4 mutants with single mutation at Cys747 in TLR4. HEK293 cells were transiently transfected with expression vectors coding TLR4 and TLR4 with single amino acid mutations along with pNifty and phRL-TK. Expression vectors coding MD-2 and CD14 were also co-transfected. On the day after transfection, cells were incubated with 100 ng·mL−1 LPS and various concentrations of TAK-242 for 6 h. Luciferase activity was determined using a luciferase assay kit. Data represent the mean ± SEM in triplicate wells. Data shown are representative of three independent experiments. HEK, human embryonic kidney; LPS, lipopolysaccharide; NF-κB, nuclear factor-κB; TLR, Toll-like receptor.
The inhibitory effect of TAK-242 is not affected by the concentrations and types of TLR4 ligands
As TAK-242 binds to the ICD of TLR4, the inhibitory effects of TAK-242 on cell activation induced by TLR4 ligands may not be greatly affected by concentration and structure of the ligands. To examine this possibility, the inhibitory effect of TAK-242 in the presence of various concentrations of LPS was examined in PMA-stimulated P31/FUJ cells. When cells were stimulated with 20 ng·mL−1 LPS, TAK-242 and lipid IVa, an LPS antagonist that works extracellularly, inhibited TNF-α production with IC50s of 32 nM and 0.59 ng·mL−1 respectively (Table 1). The inhibitory effect of lipid IVa was diminished about 20-fold when cells were stimulated with 2 µg·mL−1 LPS (Table 1). Under this condition, the IC50 of TAK-242 did not change (Table 1). In addition, a reduction of the inhibitory effect of TAK-242 was less than 10-fold even when cells were stimulated with 20 µg·mL−1 LPS (Table 1).
Table 1.
Effect of LPS concentrations on inhibitory effect of TAK-242 against TNF-α production in PMA-stimulated P31/FUJ cells
| LPS (ng·mL−1) |
IC50 |
|
|---|---|---|
| Lipid IVa (ng·mL−1) | TAK-242 (nM) | |
| 20 | 0.59 (1.00a) | 32 (1.00a) |
| 200 | 2.5 (4.16) | 46 (1.43) |
| 2000 | 11 (18.9) | 37 (1.14) |
| 20000 | 80 (136) | 268 (8.33) |
P31/FUJ cells were stimulated with the indicated concentrations of LPS for 20 h in the presence of various concentrations of test compounds. TNF-α levels in the cuture supernatant were determined by ELISA.
Ratio = IC50 (various concentrations of LPS)/IC50 (20 ng·mL−1 of LPS).
LPS, lipopolysaccharide; TNF-α, tumour necrosis factor-α.
We examined the effects of TAK-242 on cell activation by HMGB-1, another TLR4 ligand structurally different from LPS (Rifkin et al., 2005). When RAW264.7 cells were stimulated with 10 µg·mL−1 HMGB-1 or 0.5 ng·mL−1 LPS in the presence of 0.1 ng·mL−1 IFN-γ, the TNF-α levels in culture supernatants were about 1.4 and 2.4 ng·mL−1 respectively (data not shown). TAK-242 concentration-dependently inhibited HMGB-1-induced TNF-α production as well as LPS-induced TNF-α production (Figure 5). Polymyxin B, which binds with high affinity to lipid A and inactivates its biological activity (Morrison and Jacobs, 1976; Moore et al., 1986), markedly abolished LPS-induced activation but had no effect on HMGB-1-induced TNF-α production, even at a concentration of 1 µg·mL−1 (data not shown).
Figure 5.
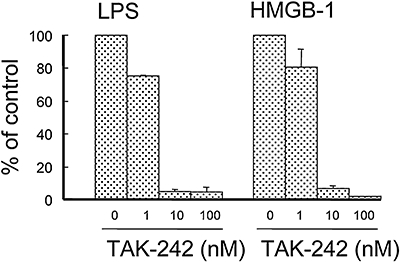
TAK-242 inhibits both LPS-induced and HMGB-1-induced TNF-α production in RAW264.7 cells. Cells were cultured with 0.5 ng·mL−1 LPS or 10 µg·mL−1 HMGB-1 for 20 h in the presence of 0.1 ng·mL−1 IFN-γ and various concentrations of TAK-242. TNF-α levels in the culture supernatants were measured by ELISA. Data represent the mean ± SEM in triplicate wells. HMGB-1, high mobility group box 1 protein; IFN, interferon; LPS, lipopolysaccharide; TNF-α, tumour necrosis factor-α.
TAK-242 has no effect on the LPS-induced conformational change of TLR4-MD-2 and TLR4 homodimerization
The binding of LPS to the TLR4-MD-2 complex causes a conformational change in TLR4-MD-2 (Akashi et al., 2003). Therefore, it is possible that TAK-242 inhibits the LPS-induced conformational change in TLR4-MD-2 and thereby inhibits TLR4 signalling. Anti-TLR4-MD-2 Ab MTS510 recognizes the conformation of TLR4-MD-2 and cannot bind to TLR4-MD-2 after LPS stimulation (Akashi et al., 2003). The effect of TAK-242 on the LPS-induced conformational change was, therefore, examined using MTS510 and RAW264.7 cells. TAK-242 and polymyxin B at a concentration of 1 µM and 1 µg·mL−1, respectively, did not affect the binding of MTS510 to RAW264.7 cells in the absence of LPS stimulation (Figure 6, upper panels). LPS stimulation almost completely inhibited the binding of MTS510 to RAW264.7 cells, whereas polymyxin B inhibited LPS-induced decrease in the binding of MTS510 to RAW264.7 cells (Figure 6, lower panels). In contrast, TAK-242 did not inhibit the LPS-induced decrease in the binding of MTS510 to the cells (Figure 6, lower panels).
Figure 6.
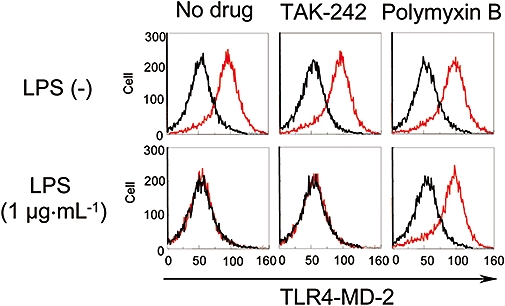
TAK-242 does not inhibit the binding of LPS to TLR4-MD-2. RAW264.7 cells were incubated with 1 µM TAK-242 or 1 µg·mL−1 polymixin B for 30 min, then stimulated with 1 µg·mL−1 LPS for 30 min. The cells were stained with PE-conjugated TLR4-MD-2 Ab MTS510 for 30 min and analysed with a flow cytometer. Representative data are shown. Ab, antibody; LPS, lipopolysaccharide; TLR, Toll-like receptor.
As homodimerization of the ICD of TLR4 is necessary for TLR4 signalling (Lee et al., 2004), its inhibition is also a possible mechanism of action for the inhibory effect of TAK-242 on TLR4 signalling. Therefore, we examined the effect of TAK-242 on constitutive TLR4 homodimerization using the β-lactamase enzyme fragment complementation assay (Lee et al., 2004). TAK-242 inhibited the NF-κB activation, but had no effect on the TLR4 homodimerization even at a concentration of 10 µM (Figure 7A,B). In contrast, helenalin concentration-dependently inhibited both TLR4 homodimerization and NF-κB activation as previously reported (Youn et al., 2006) (Figure 7A,B). Resveratrol had no effect on either TLR4 homodimerization or NF-κB activation (Figure 7A,B).
Figure 7.
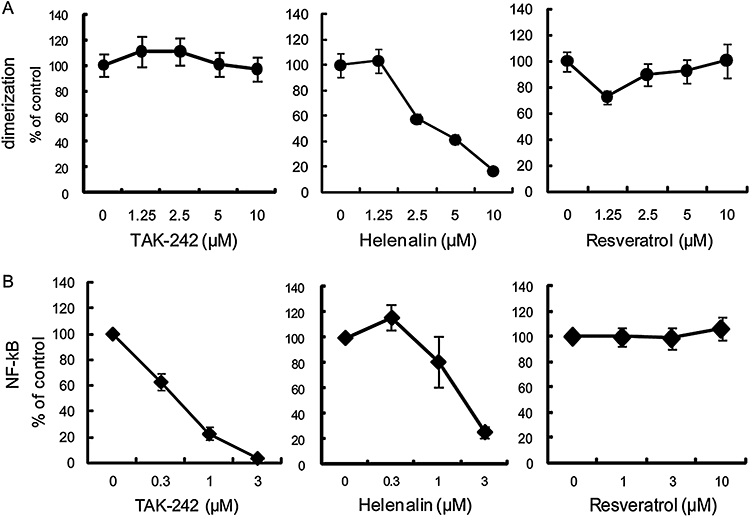
TAK-242 has no effect on TLR4 homodimerization. (A) Effect of TAK-242 on constitutive TLR4 homodimerization induced by overexpresssion of TLR4-Bla(a) and TLR4-Bla(b). HEK293 cells were transiently transfected with TLR-Bla fusion constructs. On the following day, cells were incubated with test compounds for 3 h. Cells were then loaded with CCF2/AM and analysed by an IN cell analyser 1000. Results are shown as the mean ± SEM (n= 30). (B) Effect of TAK-242 on constitutive NF-kB activation induced by overexpression of TLR4-Bla(a) and TLR4-Bla(b). HEK293 cells were transiently transfected with TLR-Bla fusion constructs. After 6 h of transfection, cells were incubated with various concentrations of test compounds overnight. Luciferase activity was determined using a luciferase assay kit. Data represent the mean ± SEM (n= 6). HEK, human embryonic kidney; NF-κB, nuclear factor-κB; TLR, Toll-like receptor.
TAK-242 inhibits the MyD88-independent pathway as well as the MyD88-dependent pathway
There are two signalling pathways under the control of TLR4, MyD88-dependent and -independent pathways (Horng et al., 2002; Yamamoto et al., 2002a,b; Oshiumi et al., 2003). The MyD88-dependent pathway downstream of TLR4 is involved in cytokine production such as TNF-α and IL-12, and the MyD88-independent pathway downstream of TLR4 is involved in the expression of IFN-inducible genes and co-stimulating molecules in DCs (Kaisho et al., 2001; Hoshino et al., 2002). We therefore examined the effect of TAK-242 on these two pathways using DCs. First, IL-12 p40 and TNF-α production induced by LPS was examined to determine the effect of TAK-242 on the MyD88-dependent pathway. TAK-242 concentration-dependently inhibited the LPS-induced IL-12 p40 and TNF-α production from DCs (Figure 8A). This result was consistent with our previous data showing LPS-induced cytokine production from mouse macrophages and human monocytes (Ii et al., 2006). Next, the effect of TAK-242 on LPS-induced up-regulation of the surface expression of CD40 and IFN-inducible genes was also examined in DCs obtained from wild-type and MyD88-deficient mice. As described previously (Kaisho et al., 2001; Hoshino et al., 2002), LPS up-regulated the expression of CD40 and IFN-inducible genes including IFN-inducible protein 10, glucocorticoid-attenuated response gene 16 and immune-responsive gene 1 in DCs obtained from MyD88-deficient mice (Figure 8B,C). TAK-242 concentration-dependently inhibited the increase in surface expression of CD40 and up-regulation of IFN-inducible genes in DCs obtained from MyD88-deficient mice as well as wild-type mice (Figure 8B,C).
Figure 8.
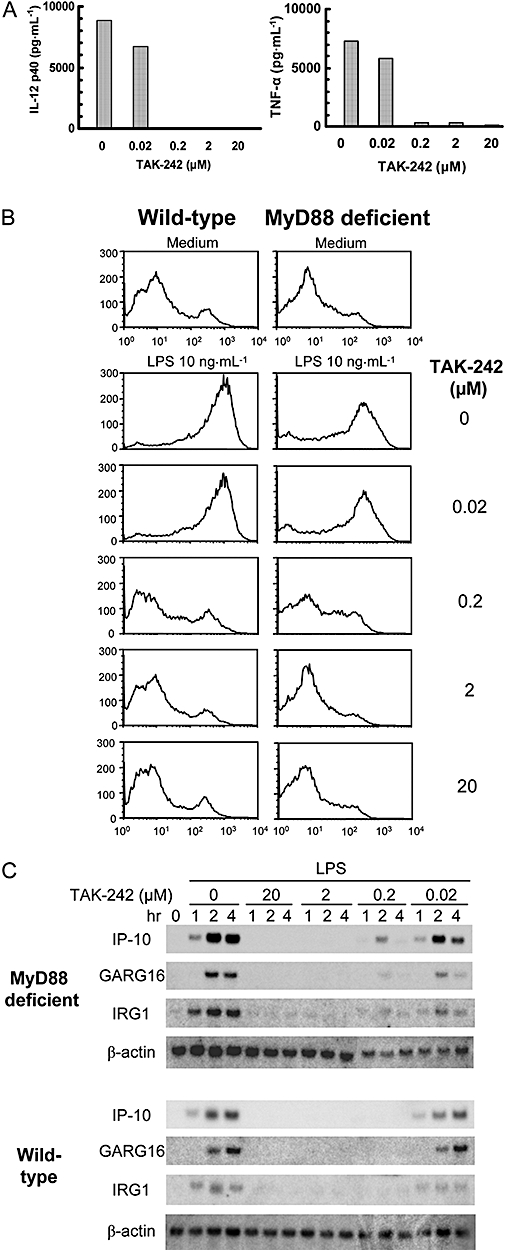
TAK-242 inhibits both MyD88-dependent and -independent signalling. DCs from wild-type and MyD88-knockout mice were incubated with various concentrations of TAK-242 for 1 h, then stimulated with LPS. (A) Effect of TAK-242 on the IL-12 p40 and TNF-α production in LPS-induced DCs. DCs were stimulated for 24 h with 10 ng·mL−1 LPS. IL-12 p40 and TNF-α production were measured by ELISA. Data represent the mean in duplicate wells. (B) Effect of TAK-242 on CD40 expression in LPS-induced DCs. DCs were stimulated for 24 h with 10 ng·mL−1 LPS. Cells were stained with PE-conjugated CD40 Ab and analysed with a flow cytometer. Representative data are shown. (C) Effect of TAK-242 on the expression of IFN-inducible genes in LPS-stimulated DCs. DCs were stimulated with 10 ng·mL−1 LPS for the indicated times. Total RNA was subjected to northern blotting with probes for IP-10, GARG16, IRG1 and β-actin. DCs, dendritic cells; GARG16, glucocorticoid-attenuated response gene 16; IL, interleukin; IP-10, IFN-inducible protein 10; IRG1, immune-responsive gene 1; LPS, lipopolysaccharide; TNF-α, tumour necrosis factor-α.
TAK-242 shows potent therapeutic efficacy in a BCG-primed mouse sepsis model using live E. coli
To evaluate the efficacy of TAK-242 in vivo, the effects of TAK-242 in BCG-primed sepsis model using live E. coli were examined. Although the antibiotic ceftazidime (20 mg·kg−1) alone reduced bacterial counts in blood from 6 log to 3 log CFU 1 h after administration (Figure 9A), it could not protect mice from death (Figure 9B). TAK-242 co-administered with ceftazidime (20 mg·kg−1) dose-dependently improved survival of mice, and TAK-242 at a dose of 3 mg·kg−1 completely protected mice from death (Figure 9B). TAK-242 co-administered with ceftazidime (20 mg·kg−1) significantly inhibited the increases in serum levels of TNF-α, IL-10 and MIP-2 at doses of 0.3 mg·kg−1 or more. Furthermore, TAK-242 at a dose of 1 mg·kg−1 quickly and markedly inhibited the increases in serum cytokine levels of TNF-α, IL-1β, IL-6, IL-10 and MIP-2 even when substantial concentrations of TNF-α, IL-10 and MIP-2 had already been produced by the time of TAK-242 treatment (Figure 9D). Although TAK-242 co-administered with ceftazidime (20 mg·kg−1) significantly inhibited the increases in serum levels of various cytokines, TAK-242 alone even at a dose of 10 mg·kg−1 had no effect on bacterial counts in blood compared with no treatment (vehicle/saline) (Figure 9A).
Figure 9.
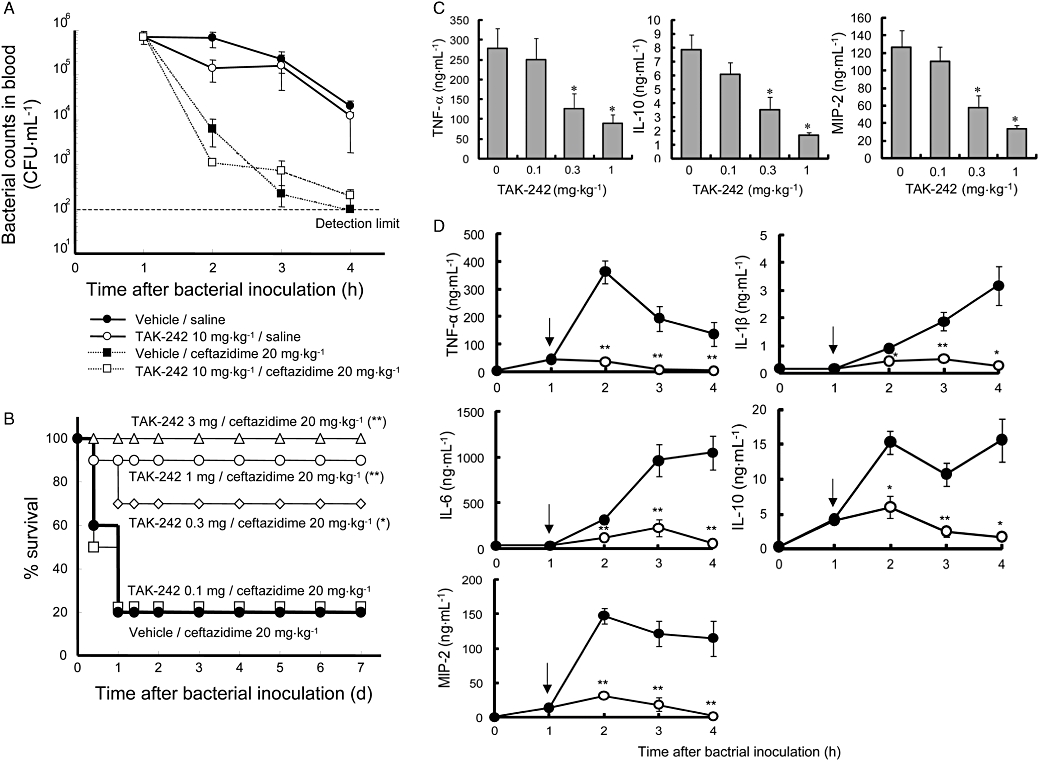
Treatment with TAK-242 protects mice in an E. coli-induced sepsis model using BCG-primed mice. C57BL/6 mice were injected intravenously with 2 mg BCG. Twelve or thirteen days later, the BCG-primed mice were inoculated intraperitoneally with approximately 107 CFU of E. coli O111. Various doses of TAK-242 (or vehicle) were intravenously administered concomitantly with 20 mg·kg−1 ceftazidime (or saline) 1 h after the bacterial challenge. (A) Influence of TAK-242 and ceftazidime on bacterial counts in blood. TAK-242 and ceftazidime were administered 1 h after the bacterial challenge at doses of 10 and 20 mg·kg−1 respectively. Bacterial counts were determined up to 4 h after bacterial challenge. Data represent the mean ± SEM (n= 4). Statistical differences at 4 h after the bacterial challenge were analysed with two-way anova. (B) Effect of TAK-242 co-administered with ceftazidime on survival rate (n= 10). Various doses of TAK-242 were intravenously administered concomitantly with 20 mg·kg−1 ceftazidime 1 h after the bacterial challenge. Survival was recorded over 7 days after the bacterial challenge. Significant differences (compared with vehicle) are denoted by *P≤ 0.05, **P≤ 0.01; Tarone's test with a simple closed step-down method. (C) Effect of TAK-242 co-administered with ceftazidime on serum levels of various cytokines. Various doses of TAK-242 were intravenously administered concomitantly with 20 mg·kg−1 ceftazidime 1 h after the bacterial challenge. The concentrations of TNF-α, IL-10 and MIP-2 were examined 2 h after bacterial challenge. Data represent the mean ± SEM (n= 10). Significant differences (compared with vehicle) are denoted by *P≤ 0.025; one-tailed Williams test (TNF-α) and one-tailed Shirley-Williams test (IL-10 and MIP-2). (D) Effect of TAK-242 co-administered with ceftazidime on time course of serum levels of various cytokines. TAK-242 and ceftazidime were concomitantly administered 1 h after the bacterial challenge at a dose of 1 and 20 mg·kg−1 respectively. The concentrations of TNF-α, IL-1β, IL-6, IL-10 and MIP-2 were examined up to 4 h after bacterial challenge. Data represent the mean ± SEM (n= 8). Significant differences (compared with vehicle) are denoted by *P≤ 0.05 and **P≤ 0.01; Wilcoxon's test with Holm's correction. All data shown are representative of two to three independent experiments. anova, analysis of variance; BCG, Bacillus calmette guerin; IL, interleukin; TNF-α, tumour necrosis factor-α.
Discussion
Our previous study showed that TAK-242 inhibited LPS-induced production of nitric oxide and various cytokines and phosphorylation of MAPKs at similar concentrations (Ii et al., 2006). Furthermore, TAK-242 selectively inhibited the cytokine production induced by ligands for TLR4 (Ii et al., 2006; Kawamoto et al., 2008). Although we could not identify the target molecule of TAK-242 in our previous study, these findings led us to speculate that TAK-242 may act early in the process of TLR4 signalling. In the present study, we found that TAK-242 binds directly to ICD of TLR4 and that the binding site of TAK-242 in ICD of TLR4 is Cys747. These results were consistent with our previous study that TAK-242 selectively inhibits TLR4 signalling mediated by the ICD of TLR4 (Kawamoto et al., 2008). TAK-242 inhibited both MyD88-dependent and -independent pathways subsequent to TLR4 activation in DCs. Furthermore, TAK-242 inhibited phosphorylation of MAPKs and IκB degradation in mouse MyD88-deficient macrophages (data not shown). This is the first study to demonstrate that a small-molecule compound can inhibit TLR4 signalling via direct binding to a specific amino acid in ICD of TLR4.
The activation of immune cells by LPS is initiated by the recognition of LPS through a receptor complex composed of CD14, TLR4 and MD-2 on the cell surface (da Silva Correia et al., 2001). Although LPS antagonists such as E5531 inhibit the binding of LPS to TLR4-MD-2 (Akashi et al., 2003), TAK-242 had no effect on the LPS-induced conformational change of TLR4-MD-2. In addition, TAK-242 does not inhibit the binding of LPS to human PBMCs (Ii et al., 2006). These results suggest that LPS may bind to TLR4-MD-2 even after TAK-242 treatment. Thus, TAK-242 is not an LPS antagonist.
Analysis of the structure-activity relationships of TAK-242 and its derivatives suggests that only TAK-242 is recognized by its target molecule (Yamada et al., 2005). In fact, the enantiomer of TAK-242 has an inhibitory effect on cytokine production but is about 350 times less potent than TAK-242 (Yamada et al., 2005), and did not inhibit the binding of [3H]-TAK-242 to TLR4. Considering the chemical structure of TAK-242, it is possible that this compound functions as a Michael acceptor. Based on this hypothesis, we postulated that TAK-242 might act on the thiol group of cysteine residues or the amino group of lysine residues. Mutational analysis using TLR4 point mutants indicated that TAK-242 did not bind to the C747A mutant and did not inhibit LPS-induced NF-κB signalling through the C747A mutant, suggesting that Cys747 in the ICD of TLR4 is the binding site of TAK-242.
Interestingly, mutational analysis using various Cys747 mutants showed that serine, threonine and valine substitutions at Cys747 impaired TLR4 signalling, although alanine substitution at Cys747 retained the ability to induce signalling. These results suggest that the precise structure of the amino acid side chain at position 747 may be crucial to TLR4 signalling. It seems likely that the binding of TAK-242 to Cys747 changes the microenvironment around Cys747. Conformation of the ICD of TLR4 may be changed to an inactive conformation after binding of TAK-242 to Cys747. Thus, TAK-242 appears to be recognized specifically by amino acid residues of Cys747 within its vicinity, and, of the four cysteine residues in the ICD of TLR4, binds to Cys747, presumably covalently. It should be noted that among the alanine substitutions at cysteine residues tested in the ICD, only the mutation at Cys706 significantly reduced NF-kB activation. Cys706 might be also involved in TLR4 signalling. Mutational analysis using chimeric constructs of TLR2 and TLR4 demonstrated that TAK-242 binds to the TMD/ICD of TLR4, but not the ECD. In addition, TAK-242 suppressed TLR4 signalling via the TMD/ICD of TLR4. These data suggest Cys747 in the ICD is the only amino acid on which TAK-242 works.
Although we found TAK-242 specifically binds to Cys747, it is unclear how TAK-242 inhibits TLR4 signalling after binding to the Cys747. Recently, it has been reported that the myristoylation and phosphorylation of TRAM is essential for TLR4 signalling (McGettrick et al., 2006; Rowe et al., 2006) and that the phosphorylation of TIRAP/Mal by Bruton's tyrosine kinase (BTK) plays a key role in TLR2 and TLR4 signalling (Gray et al., 2006). BTK also interacts with TLR4 (Jefferies et al., 2003). Furthermore, the association of TLR4 and MyD88 recruits serine/threonine kinases, IL-1 receptor-associated kinase (IRAK)-1 and -4. IRAK-1 and -4 are sequentially phosphorylated and dissociated from MyD88, which results in activation of NF-κB (Li et al., 2002; Kollewe et al., 2004). Therefore, TAK-242 may inhibit the myristoylation and/or phosphorylation of these molecules under TLR4 as a consequence of binding of TAK-242 to TLR4. TAK-242, however, does not inhibit signalling induced by overexpression of adaptor molecules such as MyD88, TIRAP, TRIF and TRAM in the NF-κB reporter assay (Kawamoto et al., 2008), and does not bind to them. Thus, TAK-242 does not act directly on these adaptor molecules. It is unclear whether TAK-242 directly inhibits the interaction of TLR4 with other molecules such as the adaptor molecules at the binding site or it indirectly inhibits signalling by inducing conformational changes in TLR4. Although homodimerization of the ICD of TLR4 is necessary for TLR4 signalling (Lee et al., 2004), TAK-242 had no effect on the TLR4 homodimerization. Further analysis is required to clarify the detailed mechanism of action of TAK-242.
As the interaction of TLR4 with endotoxin released from Gram-negative bacteria causes sepsis or septic shock attributable to an excessive acute inflammatory response, it is hypothesized that modulating LPS-induced TLR4 signalling would be useful therapeutically in sepsis. In fact, an LPS antagonist has been shown to have beneficial effects in human experimental endotoxaemia (Lynn et al., 2003; 2004;). TAK-242 is not an LPS antagonist, but inhibits TLR4 signalling. Therefore, we examined the efficacy of TAK-242 in a BCG-primed mouse sepsis model that is a two-hit model of pathogenesis of sepsis and considered to reflect the clinical situation typical of sepsis patients (Ogawa, 1998). Ceftazidime alone reduced bacterial counts in blood, but did not improve survival of mice. This result suggests that an excessive inflammatory response might induce death of mice in this sepsis model. Although substantial amounts of TNF-α, IL-10 and MIP-2 had already been produced at the time of treatment, TAK-242 co-administered with ceftazidime clearly suppressed further increases in serum cytokine levels and protected mice from death. In addition, treatment with TAK-242 was significantly effective in a mouse endotoxin shock model even when administered after LPS challenge (Sha et al., 2007). We need to remember that dozens of candidates of anti-sepsis agents have failed in clinical trials. Anti-sepsis agents are administered after the onset of sepsis in clinical studies, whereas they are often administered before bacterial or LPS challenge in animal studies. Thus, the timing of treatment may contribute to the differences in efficacy observed in the two types of study (Marshall, 2003). TAK-242 was potent in both mouse sepsis and endotoxin shock models even when administered after the onset of the inflammatory response. Therefore, we hope that TAK-242 will have potent clinical efficacy in sepsis patients. One potential clinical concern has been that inhibition of cytokine production, which is a critical constituent of the host immune responses, may have detrimental effects. Therefore, it is possible that patients receiving TAK-242 become more susceptible to subsequent infections. In BCG-primed mice, however, bacterial counts in blood did not increase with TAK-242 treatment. This suggests that TAK-242 may not have a detrimental effect with respect to susceptibility to infection. At present, further investigations are on-going.
Toll-like receptor 4 also functions as a receptor for various endogenous ligands such as fibrinogen, hyaluronic acids, HMGB-1 and heat shock proteins, and endogenous ligand-TLR4 interactions are involved in the development of various human diseases, including sepsis (Rifkin et al., 2005). TAK-242 inhibited TLR4 signalling ligand-independently and the inhibitory effect of TAK-242 on TNF-α production, unlike that of an LPS antagonist lipid IVa, was not greatly affected by the concentrations of LPS. Furthermore, TAK-242 inhibited HMGB-1-induced TNF-α production in a similar concentration-dependent manner to that in LPS-induced TNF-α production in RAW264.7 cells. As Polymyxin B did not suppress HMGB-1-induced TNF-α production at a concentration, 1 µg·mL−1, that markedly diminished LPS-induced TNF-α production (data not shown), the possibility of endotoxin contamination in the HMGB-1 preparation used in this study could be excluded. TAK-242 also suppressed fibrinogen-induced TNF-α production in RAW264.7 cells (data not shown). These data suggest that the effect of TAK-242 is not ultra sensitive to the molecular structure of TLR4 ligands, and seem to be consistent with the finding that TAK-242 works on the ICD of TLR4. Accordingly, TAK-242 may provide potent efficacy in various diseases in which interactions between TLR4 and its endogenous ligands are important.
In conclusion, TAK-242 inhibited TLR4 signalling ligand-independently via binding to Cys747 in the ICD of TLR4. In addition, TAK-242 co-administered with antibiotics showed potent therapeutic effects in an E. coli-induced sepsis model. Thus, TAK-242 may be a promising therapeutic agent for sepsis.
Acknowledgments
We thank Dr K Okonogi and Dr T Kitazaki for critical reviews of the manuscript. We also thank Dr T Kawamoto for the expression vectors for TLR4 tagged with FLAG and CD14.
Glossary
Abbreviations:
- Ab
antibody
- anova
analysis of variance
- BCG
Bacillus calmette guerin
- BTK
Bruton's tyrosine kinase
- DC
dendritic cell
- ECD
extracellular domain
- HA
haemagglutinin
- HEK
human embryonic kidney
- HMGB-1
high mobility group box 1 protein
- ICD
intracellular domain
- IFN
interferon
- IL
interleukin
- LPS
lipopolysaccharide
- Mal
MyD88-adaptor-like
- NF-κB
nuclear factor-κB
- TICAM
TIR-domain containing adaptor molecule
- TIR
Toll/IL-1 receptor
- TIRAP
TIR domain-containing adaptor protein
- TLR
Toll-like receptor
- TMD
transmembrane domain
- TNF-α
tumour necrosis factor-α
- TRAM
TRIF-related adaptor molecule
- TRIF
TIR domain-containing adaptor inducing IFN-β
Conflict of interest
The authors state no conflict of interest.
References
- Ajit CT, Yosef H, Charles WD. Multiple test procedure for dose finding. Biometrics. 1996;52:21–37. [PubMed] [Google Scholar]
- Akashi S, Saitoh S, Wakabayashi Y, Kikuchi T, Takamura N, Nagai Y, et al. Lipopolysaccharide interaction with cell surface Toll-like receptor 4-MD-2: higher affinity than that with MD-2 or CD14. J Exp Med. 2003;198:1035–1042. doi: 10.1084/jem.20031076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature. 2001;413:732–738. doi: 10.1038/35099560. [DOI] [PubMed] [Google Scholar]
- Beutler B. Inferences, questions and possibilities in Toll-like receptor signaling. Nature. 2004;430:257–263. doi: 10.1038/nature02761. [DOI] [PubMed] [Google Scholar]
- Christ WJ, Asano O, Robidoux AL, Perez M, Wang Y, Dubuc GR, et al. E5531, a pure endotoxin antagonist of high potency. Science. 1995;268:80–83. doi: 10.1126/science.7701344. [DOI] [PubMed] [Google Scholar]
- Diebold SS, Kaisho T, Hemmi H, Akira S, Reis e Sousa C. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science. 2004;303:1529–1531. doi: 10.1126/science.1093616. [DOI] [PubMed] [Google Scholar]
- Dragovich PS, Webber SE, Babine RE, Fuhrman SA, Patick AK, Matthews DA, et al. Structure-based design, synthesis, and biological evaluation of irreversible human rhinovirus 3C protease inhibitors. 2. peptide structure-activity studies. J Med Chem. 1998;41:2819–2834. doi: 10.1021/jm9800696. [DOI] [PubMed] [Google Scholar]
- Gray P, Dunne A, Brikos C, Jefferies CA, Doyle SL, O'Neill LA. MyD88 Adapter-like (Mal) is phosphorylated by Bruton's tyrosine kinase during TLR2 and TLR4 signal transduction. J Biol Chem. 2006;281:10489–10495. doi: 10.1074/jbc.M508892200. [DOI] [PubMed] [Google Scholar]
- Hayashi F, Smith KD, Ozinsky A, Hawn TR, Yi EC, Goodlett DR, et al. The innate immune response to bacterial flagellin is mediated by Toll-like receptor 5. Nature. 2001;410:1099–1103. doi: 10.1038/35074106. [DOI] [PubMed] [Google Scholar]
- Heil F, Hemmi H, Hochrein H, Ampenberger F, Kirschning C, Akira S, et al. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science. 2004;303:1526–1529. doi: 10.1126/science.1093620. [DOI] [PubMed] [Google Scholar]
- Hemmi H, Takeuchi O, Kawai T, Kaisho T, Sato S, Sanjo H, et al. A Toll-like receptor recognizes bacterial DNA. Nature. 2000;408:740–745. doi: 10.1038/35047123. [DOI] [PubMed] [Google Scholar]
- Horng T, Barton GM, Flavell RA, Medzhitov R. The adaptor molecule TIRAP provides signaling specificity for Toll-like receptors. Nature. 2002;420:329–333. doi: 10.1038/nature01180. [DOI] [PubMed] [Google Scholar]
- Hoshino K, Takeuchi O, Kawai T, Sanjo H, Ogawa T, Takeda Y, et al. Cutting edge: Toll-like receptor 4 (TLR4)-deficient mice are hyporesponsive to lipopolysaccharide: evidence for TLR4 as the Lps gene product. J Immunol. 1999;162:3749–3752. [PubMed] [Google Scholar]
- Hoshino K, Kaisho T, Iwabe T, Takeuchi O, Akira S. Differential involvement of IFN-beta in Toll-like receptor-stimulated dendritic cell activation. Int Immunol. 2002;14:1225–1231. doi: 10.1093/intimm/dxf089. [DOI] [PubMed] [Google Scholar]
- Ii M, Matsunaga N, Hazeki K, Nakamura K, Takashima K, Seya T, et al. A novel cyclohexene derivative, TAK-242, selectively inhibits Toll-like receptor 4-mediated cytokine production through suppression of intracellular signaling. Mol Pharmacol. 2006;69:1288–1295. doi: 10.1124/mol.105.019695. [DOI] [PubMed] [Google Scholar]
- Jefferies CA, Doyle S, Brunner C, Dunne A, Brint E, Wietek C, et al. Bruton's tyrosine kinase is a Toll/interleukin-1 receptor domain-binding protein that participates in nuclear factor kappaB activation by Toll-like receptor 4. J Biol Chem. 2003;278:26258–26264. doi: 10.1074/jbc.M301484200. [DOI] [PubMed] [Google Scholar]
- Kaisho T, Takeuchi O, Kawai T, Hoshino K, Akira S. Endotoxin-induced maturation of MyD88-deficient dendritic cells. J Immunol. 2001;166:5688–5694. doi: 10.4049/jimmunol.166.9.5688. [DOI] [PubMed] [Google Scholar]
- Kawamoto T, Ii M, Kitazaki T, Iizawa Y, Kimura H. TAK-242 selectively suppresses Toll-like receptor 4-signaling mediated by the intracellular domain. Eur J Pharmacol. 2008;584:40–48. doi: 10.1016/j.ejphar.2008.01.026. [DOI] [PubMed] [Google Scholar]
- Kollewe C, Mackensen AC, Neumann D, Knop J, Cao P, Li S, et al. Sequential autophosphorylation steps in the interleukin-1 receptor-associated kinase-1 regulate its availability as an adapter in interleukin-1 signaling. J Biol Chem. 2004;279:5227–5236. doi: 10.1074/jbc.M309251200. [DOI] [PubMed] [Google Scholar]
- Lee HK, Dunzendorfer S, Tobias PS. Cytoplasmic domain-mediated dimerizations of toll-like receptor 4 observed by beta-lactamase enzyme fragment complementation. J Biol Chem. 2004;279:10564–10574. doi: 10.1074/jbc.M311564200. [DOI] [PubMed] [Google Scholar]
- Li S, Strelow A, Fontana EJ, Wesche H. IRAK-4: a novel member of the IRAK family with the properties of an IRAK-kinase. Proc Natl Acad Sci USA. 2002;99:5567–5572. doi: 10.1073/pnas.082100399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynn M, Rossignol DP, Wheeler JL, Kao RJ, Perdomo CA, Noveck R, et al. Blocking of responses to endotoxin by E5564 in healthy volunteers with experimental endotoxemia. J Infect Dis. 2003;187:631–639. doi: 10.1086/367990. [DOI] [PubMed] [Google Scholar]
- Lynn M, Wong YN, Wheeler JL, Kao RJ, Perdomo CA, Noveck R, et al. Extended in vivo pharmacodynamic activity of E5564 in normal volunteers with experimental endotoxemia. J Pharmacol Exp Ther. 2004;308:175–181. doi: 10.1124/jpet.103.056531. [DOI] [PubMed] [Google Scholar]
- McGettrick AF, Brint EK, Palsson-McDermott EM, Rowe DC, Golenbock DT, Gay NJ, et al. Trif-related adapter molecule is phosphorylated by PKCε during Toll-like receptor 4 signaling. Proc Natl Acad Sci USA. 2006;103:9196–9201. doi: 10.1073/pnas.0600462103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall JC. Such stuff as dreams are made on: mediator-directed therapy in sepsis. Nat Rev Drug Discov. 2003;2:391–405. doi: 10.1038/nrd1084. [DOI] [PubMed] [Google Scholar]
- Matthews DA, Dragovich PS, Webber SE, Fuhrman SA, Patick AK, Zalman LS, et al. Structure-assisted design of mechanism-based irreversible inhibitors of human rhinovirus 3C protease with potent antiviral activity against multiple rhinovirus serotypes. Proc Natl Acad Sci USA. 1999;96:11000–11007. doi: 10.1073/pnas.96.20.11000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medzhitov R, Preston-Hurlburt P, Kopp E, Stadlen A, Chen C, Ghosh S, et al. MyD88 is an adaptor protein in the hToll/IL-1 receptor family signaling pathways. Mol Cell. 1998;2:253–258. doi: 10.1016/s1097-2765(00)80136-7. [DOI] [PubMed] [Google Scholar]
- Moore RA, Bates NC, Hancock RE. Interaction of polycationic antibiotics with Pseudomonas aeruginosa lipopolysaccharide and lipid A studied by using dansyl-polymyxin. Antimicrob Agents Chemother. 1986;29:496–500. doi: 10.1128/aac.29.3.496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison DC, Jacobs DM. Binding of polymyxin B to the lipid A portion of bacterial lipopolysaccharides. Immunochemistry. 1976;13:813–818. doi: 10.1016/0019-2791(76)90181-6. [DOI] [PubMed] [Google Scholar]
- Ogawa M. Systemic inflammatory response syndrome – a concept for avoiding organ dysfunction induced by a ‘second attack’. Surg Today. 1998;28:679–681. doi: 10.1007/BF02484611. [DOI] [PubMed] [Google Scholar]
- Oshiumi H, Matsumoto M, Funami K, Akazawa T, Seya T. TICAM-1, an adaptor molecule that participates in Toll-like receptor 3-mediated interferon-beta induction. Nat Immunol. 2003;4:161–167. doi: 10.1038/ni886. [DOI] [PubMed] [Google Scholar]
- Ozinsky A, Underhill DM, Fontenot JD, Hajjar AM, Smith KD, Wilson CB, et al. The repertoire for pattern recognition of pathogens by the innate immune system is defined by cooperation between toll-like receptors. Proc Natl Acad Sci USA. 2000;97:13766–13771. doi: 10.1073/pnas.250476497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poltorak A, He X, Smirnova I, Liu MY, Van Huffel C, Du X, et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 1998;282:2085–2088. doi: 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]
- Rifkin IR, Leadbetter EA, Busconi L, Viglianti G, Marshak-Rothstein A. Toll-like receptors, endogenous ligands and systemic autoimmune disease. Immunol Rev. 2005;204:27–42. doi: 10.1111/j.0105-2896.2005.00239.x. [DOI] [PubMed] [Google Scholar]
- Rowe DC, McGettrick AF, Latz EG, Monks B, Gay NJ, Yamamoto M, et al. The myristoylation of TRIF-related adaptor molecule is essential for Toll-like receptor 4 signal transduction. Proc Natl Acad Sci USA. 2006;103:6299–6304. doi: 10.1073/pnas.0510041103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sha T, Sunamoto M, Kitazaki T, Sato J, Ii M, Iizawa Y. Therapeutic effects of TAK-242, a novel selective Toll-like receptor 4 signal transduction inhibitor, in mouse endotoxin shock model. Eur J Pharmacol. 2007;571:231–239. doi: 10.1016/j.ejphar.2007.06.027. [DOI] [PubMed] [Google Scholar]
- Shimazu R, Akashi S, Ogata H, Nagai Y, Fukudome K, Miyake K, et al. MD-2, a molecule that confers lipopolysaccharide responsiveness on Toll-like receptor 4. J Exp Med. 1999;189:1777–1782. doi: 10.1084/jem.189.11.1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Silva Correia J, Soldau K, Christen U, Tobias PS, Ulevitch RJ. Lipopolysaccharide is in close proximity to each of the proteins in its membrane receptor complex. Transfer from CD14 to TLR4 and MD-2. J Biol Chem. 2001;276:21129–21135. doi: 10.1074/jbc.M009164200. [DOI] [PubMed] [Google Scholar]
- Takeuchi O, Kawai T, Muhlradt PF, Morr M, Radolf JD, Zychlinsky A, et al. Discrimination of bacterial lipoproteins by Toll-like receptor 6. Int Immunol. 2001;13:933–940. doi: 10.1093/intimm/13.7.933. [DOI] [PubMed] [Google Scholar]
- Takeuchi O, Sato S, Horiuchi T, Hoshino K, Takeda K, Dong Z, et al. Cutting edge: role of Toll-like receptor 1 in mediating immune response to microbial lipoproteins. J Immunol. 2002;169:10–14. doi: 10.4049/jimmunol.169.1.10. [DOI] [PubMed] [Google Scholar]
- Yamada M, Ichikawa T, Ii M, Sunamoto M, Itoh K, Tamura K, et al. Discovery of novel and potent small-molecule inhibitors of NO and cytokine production as antisepsis agents: synthesis and biological activity of alkyl 6-(N-substituted sulfamoyl)cyclohex-1-ene-1-carboxylate. J Med Chem. 2005;48:7457–7467. doi: 10.1021/jm050623t. [DOI] [PubMed] [Google Scholar]
- Yamamoto M, Sato S, Hemmi H, Sanjo H, Uematsu S, Kaisho T, et al. Essential role for TIRAP in activation of the signaling cascade shared by TLR2 and TLR4. Nature. 2002a;420:324–329. doi: 10.1038/nature01182. [DOI] [PubMed] [Google Scholar]
- Yamamoto M, Sato S, Mori K, Hoshino K, Takeuchi O, Takeda K, et al. Cutting edge: a novel Toll/IL-1 receptor domain-containing adapter that preferentially activates the IFN-beta promoter in the Toll-like receptor signaling. J Immunol. 2002b;169:6668–6672. doi: 10.4049/jimmunol.169.12.6668. [DOI] [PubMed] [Google Scholar]
- Yamamoto M, Sato S, Hemmi H, Uematsu S, Hoshino K, Kaisho T, et al. TRAM is specifically involved in the Toll-like receptor 4-mediated MyD88-independent signaling pathway. Nat Immunol. 2003;4:1144–1150. doi: 10.1038/ni986. [DOI] [PubMed] [Google Scholar]
- Youn HS, Saitoh SI, Miyake K, Hwang DH. Inhibition of homodimerization of Toll-like receptor 4 by curcumin. Biochem Pharmacol. 2006;72:62–69. doi: 10.1016/j.bcp.2006.03.022. [DOI] [PubMed] [Google Scholar]
- Zhu S, Hudson TH, Kyle DE, Lin AJ. Synthesis and in vitro studies of novel pyrimidinyl peptidomimetics as potential antimalarial therapeutic agents. J Med Chem. 2002;45:3491–3496. doi: 10.1021/jm020104f. [DOI] [PubMed] [Google Scholar]
- Zuany-Amorim C, Hastewell J, Walker C. Toll-like receptors as potential therapeutic targets for multiple diseases. Nat Rev Drug Discov. 2002;1:797–807. doi: 10.1038/nrd914. [DOI] [PubMed] [Google Scholar]