

Abstract
Background and purpose:
Previous results have shown that mice lacking in the group 1B phospholipase A2 (Pla2g1b) are resistant to obesity and diabetes induced by feeding a diabetogenic high-fat/high-carbohydrate diet. This study examined the potential of using the Pla2g1b inhibitor methyl indoxam as therapy to suppress diet-induced obesity and diabetes.
Experimental approach:
Male C57BL/6 mice were fed the diabetogenic diet with or without methyl indoxam supplementation. Body weight gain, fasting plasma glucose levels, glucose tolerance and postprandial lysophospholipid absorption were compared.
Key results:
Wild-type C57BL/6 mice fed the diabetogenic diet without Pla2g1b inhibitor showed 31 and 69% body weight gain after 4 and 10 weeks respectively. These animals also showed elevated plasma glucose levels and were glucose intolerant. In contrast, C57BL/6 mice fed the diabetogenic diet with 90 mg·kg−1 of methyl indoxam gained only 5% body weight after 10 weeks. These animals were also euglycaemic and displayed normal glucose excursion rates in glucose tolerance test. Methyl indoxam suppression of diet-induced body weight gain and glucose intolerance was correlated with the inhibition of Pla2g1b-mediated postprandial lysophospholipid absorption.
Conclusions and implications:
These results show that oral supplementation of a diabetogenic diet with the Pla2g1b inhibitor methyl indoxam effectively suppresses diet-induced obesity and diabetes in mice. This suggests that Pla2g1b inhibition may be a potentially effective oral therapeutic option for treatment of obesity and diabetes.
Keywords: phospholipase, diabetes therapy, obesity therapy, intestinal absorption
Introduction
The increasing prevalence of obesity and diabetes due to increasing consumption of meals rich in fat and carbohydrate is a major global health threat that inflicts enormous economic burdens on society. The most common strategies currently used for diabetes therapy include insulin replacement by injection and/or drugs that either increase pancreatic insulin secretion, such as the sulphonylureas; suppress hepatic glucose production, such as metformin; or activate peroxisome proliferator-activated receptor (PPAR) signalling pathways such as rosiglitazone and pioglitazone. Unfortunately, these treatment strategies have undesirable side effects. Metformin is not well tolerated by all patients and is also contraindicated for older patients and those with renal impairment. The use of PPAR-γ activators has been linked to increased risk of cardiovascular disease and may cause or worsen heart failure (Delea et al., 2003). Therefore, there is an increasing demand for effective diabetes drugs with minimal or no adverse effects. The latest diabetes drugs on the market are modulators of the incretin pathway such as glucagon-like peptide-1 (GLP-1) analogues or activators, and inhibitors of dipeptidyl peptidase 4 (Amori et al., 2007; Triplitt et al., 2007). These next-generation drugs are not without disturbing side effects; the Food and Drug Administration recently issued a warning regarding pancreatitis in patients taking the GLP-1 analogue, exenatide (Beyetta) (Bain and Stephens, 2008).
Advances made in transgenic and knock-out technology, and in the characterization of the genetically modified mouse models, have led to the identification of novel targets for drug development in the treatment of a variety of diseases in humans (Zambrowicz and Sands, 2003; Powell, 2006). Our laboratory has recently developed a genetically modified mouse model that targets the pancreatic lipolytic enzyme group 1B phospholipase A2 (Pla2g1b), and showed that Pla2g1b−/− mice are resistant to diet-induced obesity and diabetes (Huggins et al., 2002). This enzyme contributes to obesity and diabetes by digesting dietary and biliary phospholipids in the intestinal lumen, and catalyzing the absorption of the digestive product lysophospholipids, which directly promote postprandial insulin resistance in the liver and other high-energy metabolism tissues (Labontéet al., 2006). Importantly, Pla2g1b is expressed predominantly in the gastrointestinal tract (Richmond and Hui, 2000), suggesting that oral administration of Pla2g1b inhibitors may be sufficient to effectively protect against diet-induced obesity and diabetes. This study was undertaken to determine if pharmacological intervention of phospholipase A2 activity may be a viable option for suppressing diet-induced obesity and diabetes in wild-type mice.
Methods
Inhibitors
The pancreatic phospholipase A2, also known as group 1B phospholipase A2 or PLA2G1B according to the Guide to Receptors and Channels nomenclature (Alexander et al., 2008), can be inhibited by the general PLA2 inhibitor commonly known as methyl indoxam (structure shown in Figure 1). This compound was synthesized according to a published procedure (Singer et al., 2002). The effectiveness of the newly synthesized methyl indoxam was confirmed by in vitro assays based on its ability to inhibit the hydrolysis of phospholipid vesicles with human or mouse Pla2g1b according to the procedure described previously (Singer et al., 2002). The inhibitor was then dissolved in 0.9% carboxyl methylcellulose, 9% polyethylene glycol-400 and 0.05% Tween 80, and then blended into a rodent diabetogenic high-fat/high-carbohydrate diet to achieve final concentrations of the test compound based on an average daily consumption of ∼2.5–3 g each day.
Figure 1.
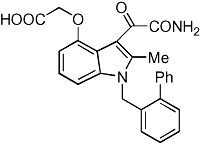
Chemical structure of methyl indoxam. Me: methyl group; Ph: phenyl group.
Human bile samples
Human bile was collected from patients hospitalized at the Gastroenterology Department of La Timone Hospital (Marseille, France) by endoscopic retrograde catheterization of the biliary duct in the framework of standard diagnosis procedures. Samples were pooled without patient identification, and stored frozen at −80°C. These biological samples were accessed for research purposes in accordance with article L.1243-3 of the French Code de la Santé Publique.
In vitro assays
Recombinant human PLA2G1B expressed in Escherichia coli (Singer et al., 2002) and recombinant mouse Pla2g1b produced in transfected insect cells (Rouault et al., 2007) were used. The effectiveness of methyl indoxam at inhibiting hydrolytic activities of the recombinant enzymes was assessed based on the hydrolysis of the fluorescent phospholipid analogue 1-palmitoyl-2-(10-pyrenedecanoyl)-sn-glycero-3-phosphoglycerol at a concentration of 1 µM as described previously (Singer et al., 2002). The influence of methyl indoxam on human PLA2G1B hydrolysis of phospholipids in biliary extracts was assessed as follows. A test meal was prepared by mixing string beans, beef, French fries, butter and sunflower oil in a 8:9:7:1:2 weight ratio. A 2 g aliquot was mixed in the absence or presence of methyl indoxam with 8 mL of human bile containing a 60 mM total bile salt concentration and recombinant human PLA2G1B enzyme solution to a final concentration of 25 µg·mL−1, a concentration that produced a level of phospholipase activity similar to that measured in human duodenal extracts from healthy volunteers. The enzyme-to-inhibitor molar ratio was 1:100. The reaction was performed in a 50 mL temperature-controlled vessel at 37°C. Samples were collected at 0, 10, 20, 30, 40, 60 and 120 min for the analysis of lipolysis products. Lipids were extracted immediately after sampling according to Folch's procedure (Folch et al., 1957), and then separated by high-performance thin-layer chromatography (HPTLC) on silica gel plates (60, 20 × 20 cm). The sample migration was first performed with chloroform/methanol/acetic acid/water (65/35/8/4, v/v) until the solvent front was halfway up the plate. The plate was dried and then placed in a second chamber containing hexane/ether/acetic acid (86/16/1, v/v) until the solvent front reached the top of the plate. The plate was dried again, sprayed with a 10% (w/v) cupric sulphate solution in 8% (w/v) orthophosphoric acid and then heated at 180°C for 10–15 min. The quantitative analysis of phospholipids, lysophospholipids and free fatty acids was performed by directly scanning photodensitometry (CAMAG, Muttenz, Switzerland) of charred lipids on HPTLC.
Animals
Male age-matched (9–11 weeks old) wild-type (Pla2g1b+/+) and Pla2g1b-defective (Pla2g1b−/−) mice, both in C57BL/6 background from the same breeding colony, were fed either standard mouse chow or the diabetogenic high-fat/high-carbohydrate diet with the test compound or vehicle control. Body weights were monitored throughout the experimental period. Blood glucose levels were also monitored after an overnight fast by obtaining blood from the tail vein and analyzing glucose levels in a drop of blood with an Accu-Chek active glucometer. Glucose tolerance tests were performed by injecting (i.p.) a saline solution containing 2 g·glucose·kg−1 body weight into the experimental animal, and then obtaining blood samples at different times for glucose determination. Food consumption was monitored over a 24 h period for 3 consecutive days at weeks 3 and 8 of the experimental period. All animal protocols used in this study were approved by the Institutional Animal Care and Use Committee at the University of Cincinnati.
In vitro absorption
Caco-2 permeability assays were performed according to standard methodology (Artursson et al., 2001). Caco-2 cells were seeded into 24-well transwells at a density of 6 × 104 cells per cm2. Monolayers were grown and differentiated in minimum essential medium supplemented with 20% fetal bovine serum, 100 U·mL−1 penicillin and 100 µg·mL−1 streptomycin at 37°C, 95% humidity, 95% air and 5% CO2. The culture medium was refreshed every 48 h. After 21 days, the cells were washed in transport buffer made up of Hank's buffered salt solution with HEPES, and the monolayer integrity was evaluated by measuring the trans-epithelial electrical resistance (TEER) of each well. Wells with TEER values of 350 Ωcm2 or more were assayed.
Methyl indoxam was diluted to 50 µg·mL−1 in transport buffer and added to the apical wells separately. Samples (150 µL) were collected for LC/MS analysis from the basolateral well at 15, 30 and 45 min, and 1, 3 and 6 h time-points; the volume was replaced with pre-warmed transport buffer after each sampling. The apparent permeabilities in cm·s−1 were calculated based on the equation: Papp= (ΔQ/Δt) × (1/C0) × (1/A) where (ΔQ/Δt) is the permeability rate corrected for the sampling volumes over time, C0 is the initial concentration and A is the surface area of the monolayer (0.32 cm2). At the end of the experiment, TEER measurements were taken again, and wells with readings below 350 Ωcm2 indicative of diminished monolayer integrity were not included for analysis. Finally, the wells were washed with transport buffer, and 100 µM of Lucifer yellow was added to the apical wells. Samples were collected at 15, 30 and 45 min time-points for analysis by LC/MS to determine paracellular transport.
Bioavailability measurements
Two groups (n= 18 and 24) of male CD-1 mice were fasted overnight and then treated with methyl indoxam at a dose of 30 mg·kg−1 by oral gavage or at a dose of 3 mg·kg−1 by i.v. injection. The p.o. formula was prepared with 0.9% (w/v) carboxymethyl cellulose, 9% (v/v) PEG-400, 0.05% (v/v) Tween-80 in H2O. The methyl indoxam suspension was mixed and sonicated in a warming, sonicating bath for 30 min, and kept mixed during dosing. The i.v. formula was prepared with 30% (v/v) PEG-400, 5% (v/v) ethanol in H2O, in which methyl indoxam was fully dissolved. Blood samples were collected from the animals (n= 3 at each time-point) at 0.5, 1, 2, 4, 8 and 24 h in the p.o. group, and 5, 15 min and 0.5, 1, 2, 4, 8, 24 h in the i.v. group after dosing. Serum was prepared by centrifugation at 13 000×g for 5 min at 25°C. The concentration of methyl indoxam in serum was determined by LC–MS–MS. Bioavailability (%F) was calculated from the data according to the equation: %F= (AUC0-t, oral/AUC0-t, iv) × (doseiv/doseoral) × 100, where %F= bioavailability, and AUC0-t= area under the concentration–time curve at the last 24 h time-point measured.
Postprandial plasma lysophospholipid measurements
Age-matched male mice fed the diabetogenic diet with or without the methyl indoxam supplement were fasted overnight and then fed a 0.1 mL glucose–lipid mixed meal containing 50% glucose, 2.6 mM egg phosphatidylcholine (PC), 13.33 mM triolein and 2.6 mM cholesterol with or without methyl indoxam. The mice were anaesthetized with ketamine/xylazine after 1 h, and blood was collected by retroorbital bleeding into tubes containing 1 mM EDTA. Plasma was prepared by centrifugation at 2000×g for 10 min at 4°C. The concentration of lysophospholipids in plasma was determined by enzymatic procedures by incubating 8 µL of sample for 30 min at 37°C with 240 µL of reagent containing 100 mM Tris–HCl (pH 8), 0.01% Triton X-100, 1 mM CaCl2, 3 mM N-ethyl-N-(2-hydroxy-3-sulphopropyl)-3-methylaniline sodium dehydrate; 10 kU·L−1 peroxidase, 0.1 kU·L−1 glycerophosphorylcholine phosphodiesterase and 1 kU·L−1 choline oxidase, as described previously (Labontéet al., 2006).
Statistical analysis
Differences between the two genotypes were determined by Student's t-test. Differences in body weights, fasting blood glucose levels or areas under the curve were determined by one-way analysis of variance (anova) followed by the Tukey–Kramer tests. Serum glucose levels during the glucose tolerance test were compared using two-way anova followed by Bonferroni post hoc tests. P < 0.05 was accepted as statistically significant.
Materials
Rodent diabetogenic high-fat/high-carbohydrate diet (D12331) was obtained from Research Diets (New Brunswick, NJ, USA). Recombinant human PLA2G1B expressed in E. coli (Singer et al., 2002) was obtained from Dr Michael Gelb (University of Washington, Seattle, WA, USA), and recombinant mouse Pla2g1b produced in transfected insect cells (Rouault et al., 2007) was a gift from Dr Gérard Lambeau (Institut de Pharmacologie Moléculaire et Cellulaire, CNRS-UMR6097, Valbonne, France). The Accu-Chek active glucometer was from Roche Applied Science (Penzberg, Germany).
Results
Methyl indoxam has been reported to be a potent competitive inhibitor of all secretory PLA2 (Singer et al., 2002). The ability of methyl indoxam to inhibit Pla2g1b was confirmed in vitro based on its ability to inhibit the hydrolysis of vesicular PC to lysophosphatidylcholine by either human or mouse Pla2g1b, with an IC50 of 1.12 and 0.59 µM, respectively. Additional experiments were also performed to evaluate the effectiveness of methyl indoxam at inhibiting PLA2G1B hydrolytic activity against physiological substrates such as those present in the intestinal lumen. In these experiments, human biliary extracts were combined with a sample human meal and incubated with 25 µg·mL−1 of recombinant human PLA2G1B in the presence or absence of 40 µM methyl indoxam. Lipids were extracted after various time of incubation and separated by TLC for identification of phospholipids and lysophospholipids. The results, as shown in Figure 2, clearly indicate that methyl indoxam effectively suppressed human Pla2g1b-mediated conversion of phospholipids to lysophospholipids (Figure 2). Similar results were obtained using ex vivo human duodenal extracts recovered after a meal, and incubating the extracts with and without methyl indoxam (data not shown). These results suggest that methyl indoxam may be used in vivoto suppress PLA2G1B activity in the intestinal lumen.
Figure 2.
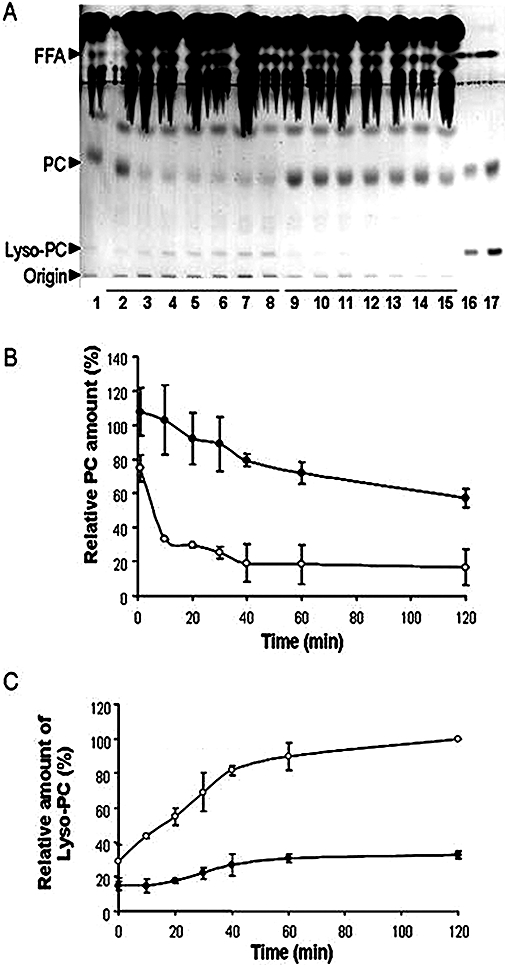
Lipid hydrolysis in a test meal by recombinant human PLA2G1B in the absence or presence of methyl indoxam. (A) The stained lipids after high-performance thin-layer chromatography separation. Line 1, sample without PLA2G1B, lines 2 to 8 correspond to time 0, 10, 20, 30, 40, 60 and 120 min of incubation of Pla2g1b + solvent control in the test meal, respectively; lines 9 to 15 correspond to time 0, 10, 20, 30, 40, 60 and 120 min of incubation of 25 µg·mL−1 PLA2G1B + 40 µM methyl indoxam in the test meal, respectively; lines 16 and 17 correspond to standards [lysophosphatidylcholine (Lyso-PC), phosphatidylcholine (PC) and FFA] 2 and 4 µg each respectively. (B) and (C) The densitometric evaluation of the relative amounts of PC and Lyso-PC during lipid hydrolysis by PLA2G1B in test meal in the presence (closed circles) or absence (open circles) of methyl indoxam respectively. Data represent mean ± SD from three experiments. All data points between samples with and without methyl indoxam are significantly different at P < 0.01.
The ability of methyl indoxam to inhibit mouse Pla2g1b activity in vivoand recapitulate the Pla2g1b gene inactivation phenotype was assessed by feeding C57BL/6 mice with diets with or without the methyl indoxam supplement. The methyl indoxam-treated mice displayed no obvious signs of abnormalities and consumed similar amounts of food to the wild-type C57BL/6 mice (Table 1). Body weights and blood glucose levels after an overnight fast were also similar in the methyl indoxam-treated mice to those in the untreated mice when both groups were maintained on a low-fat diet (data not shown). However, a significant difference in body weights between the two groups was observed when the mice were fed a high-fat/high-carbohydrate diabetogenic diet with or without methyl indoxam. Whereas the wild-type C57BL/6 mice displayed a significant body weight gain after being fed the diabetogenic diet, as expected (Figure 3A), the mice treated with methyl indoxam showed a dosage-dependent suppression of body weight gain. The body weight increase in response to the diabetogenic diet was significantly slower in mice treated with 25 and 50 mg methyl indoxam·kg−1 body weight day−1 (Figure 3A). Importantly, an increase in body weight was not observed in mice treated with 90 mg·kg−1 methyl indoxam each day throughout the 10 week period of feeding the high-fat/high-carbohydrate diet (Figure 3A). The lack of body weight gain observed in diabetogenic diet-fed mice after treatment at the higher dose of methyl indoxam was reminiscent of Pla2g1b−/− mice that were found to display minimal weight gain in response to feeding a high-fat diet (Huggins et al., 2002).
Table 1.
Normal food intake in mice with or without methyl indoxam therapy
| Treatment |
Food intake (g·day−1)a |
|
|---|---|---|
| Week 3 | Week 8 | |
| − Methyl indoxam | 2.68 ± 0.45 | 2.95 ± 0.52 |
| + Methyl indoxam | 2.84 ± 0.46 | 2.76 ± 0.60 |
Food consumption was monitored over a 24 h period for 3 consecutive days at weeks 3 and 8 after feeding mice with the diabetogenic diet containing vehicle with or without 90 mg·kg−1·day−1 of methyl indoxam. Each value represents the mean ± SD from eight mice in each group.
Figure 3.
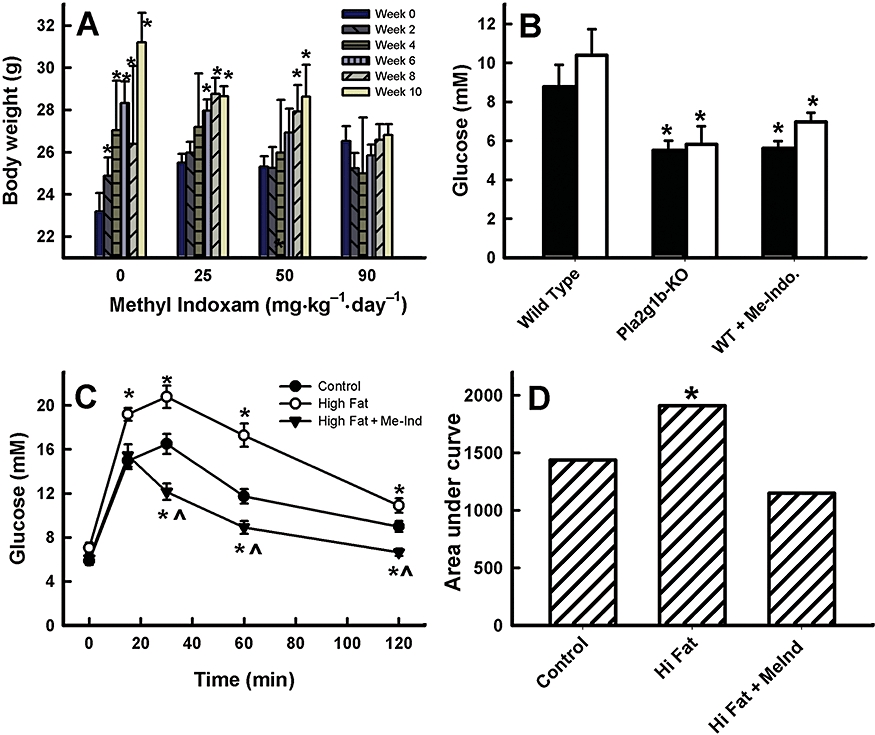
Methyl indoxam suppressed diet-induced obesity and hyperglycaemia in mice. (A) The effect of various doses of methyl indoxam treatment on body weights of mice fed the high-fat/high-carbohydrate diabetogenic diet. Body weights were recorded at 2 week intervals as shown (left to the right bars with each dose of methyl indoxam). *Denotes significant difference from the body weight at week 0 for the same group of mice. (B) Fasting blood glucose levels in wild-type mice fed a diabetogenic diet without inhibitor or with 90 mg·kg−1 methyl indoxam (WT + Me-Indo) and in Pla2g1b−/− (Pla2g1b-ko) mice after 4 (solid columns) and 10 (open columns) weeks. *Denotes significant difference from wild-type control mice at P < 0.05. (C) Glucose tolerance test results after i.p. injection of glucose (2 g·kg−1) into wild-type C57BL/6 mice maintained on control low-fat diet, or high-fat diet without or with 90 mg·kg−1 methyl indoxam treatment for 10 weeks. *Denotes significant difference from wild-type control mice at P < 0.05 and indicates significant difference from the high fat-fed group at P < 0.01. (D) The area under the curve analysis of the glucose tolerance test data presented in (C). *Denotes significant difference from wild-type control mice at P < 0.05. All data are presented as mean ± SD from eight mice in each group.
Previous studies have shown that the Pla2g1b−/− mice are also resistant to diet-induced hyperglycaemia (Huggins et al., 2002; Labontéet al., 2006). In the present study, it was found that wild-type mice treated with 90 mg·kg−1·day−1 of methyl indoxam were also resistant to diet-induced hyperglycaemia, and displayed similar fasting blood glucose levels as the Pla2g1b−/− mice after being fed the diabetogenic diet for 4 and 10 weeks (Figure 3B). The reduced fasting blood glucose levels in Pla2g1b−/− mice have been shown previously to be due to improved glucose tolerance (Labontéet al., 2006). In the present study, wild-type mice that had similar blood glucose levels after being fed either the control diet or high-fat diet with or without methyl indoxam for 4 weeks were selected for glucose tolerance measurements. Results showed that whereas the high-fat diet without methyl indoxam treatment displayed impaired glucose tolerance compared to mice fed the control diet, as expected, the methyl indoxam-treated mice showed improved glucose tolerance even when compared to the low fat-fed mice (Figure 3C,D). Taken together, these data indicate that the PLA2 inhibitor methyl indoxam is capable of reproducing the protection against diet-induced obesity and diabetes observed in Pla2g1b−/− mice.
Methyl indoxam is a general PLA2 inhibitor capable of inhibiting the activities of other secretory PLA2 in addition to the inhibition of Pla2g1b. Therefore, we investigated the possibility that orally fed methyl indoxam may be absorbed through the gastrointestinal tract to suppress diet-induced obesity and glucose intolerance through inhibition of systemic PLA2. The first study used well-differentiated Caco-2 cells on Transwell membranes to assess the permeability of methyl indoxam through confluent human intestinal cells. The addition of 50 µg·mL−1 methyl indoxam to the apical side of the confluent Caco-2 cells did not result in any detectable methyl indoxam being absorbed and transported to the basolateral (bottom) chamber of the Transwell apparatus after 6 h. These in vitro findings were supported by the in vivo observation that 12.77% of the orally administered methyl indoxam was absorbed into the blood circulation in a mouse model. Thus, only minimal amounts of methyl indoxam can be absorbed through the gastrointestinal tract, suggesting that methyl indoxam suppresses diet-induced obesity and glucose intolerance by inhibiting Pla2g1b in the digestive tract.
Our previous studies have shown that Pla2g1b inactivation via gene ablation protects against diet-induced obesity and diabetes via inhibition of postprandial lysophospholipid absorption (Labontéet al., 2006). In the present study, the results showed that methyl indoxam treatment also significantly decreased postprandial plasma lysophospholipid levels in mice 60 min after they had been fed a lipid-rich meal (Figure 4). These results are consistent with the conclusion that the predominant mechanism by which methyl indoxam suppresses diet-induced obesity and diabetes is via the inhibition of Pla2g1b activity in the digestive tract.
Figure 4.
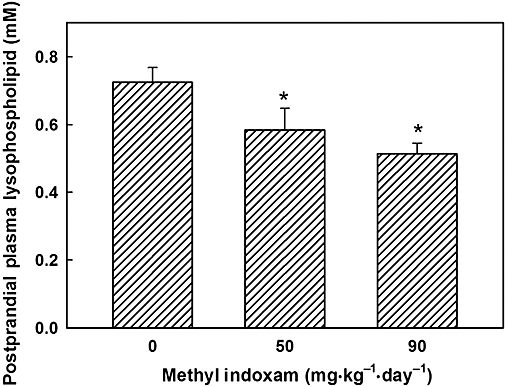
Methyl indoxam treatment decreased postprandial lysophospholipid levels in mice. Age-matched male C57BL/6 mice were fed the high-fat diet with or without the methyl indoxam supplement at the concentration indicated for 10 weeks. The mice were fed a bolus lipid–glucose meal after an overnight fast. Plasma lysophospholipid levels were measured after 1 h by enzymatic methods as described in the Methods section. All data are presented as mean ± SD from eight mice in each group. *Denotes significant difference from wild-type control mice at P < 0.05.
Discussion
The group 1B phospholipase A2 gene locus has recently been identified as an obesity gene locus in humans (Wilson et al., 2006). The gene product Pla2g1b is synthesized primarily by acinar cells of the pancreas and is secreted into intestinal lumen during meal consumption. Previously, the physiological role of this enzyme was thought to be related to phospholipid digestion in the gastrointestinal tract, which is a prerequisite for absorption of lipid nutrients including triglycerides, cholesterol and lipid-soluble vitamins (Mackay et al., 1997; Young and Hui, 1999). However, mice with specific ablation of the Pla2g1b gene displayed normal dietary lipid absorption indicative of a compensatory phospholipid digestion mechanism in the absence of Pla2g1b (Richmond et al., 2001). Surprisingly, the Pla2g1b−/− mice are protected from high-fat/high-carbohydrate diet-induced obesity and insulin resistance (Huggins et al., 2002). Follow-up studies revealed that Pla2g1b promotes diet-induced obesity and diabetes because its digestive product lysophospholipid absorbed postprandially contributes directly to glucose intolerance and hyperglycaemia (Labontéet al., 2006). These studies provided the impetus for the present study that explores the possibility of Pla2g1b inhibition as a potential therapy to suppress obesity and diabetes.
The results presented here show that the general Pla2 inhibitor methyl indoxam effectively reproduced the phenotype of Pla2g1b−/− mice that is resistant to obesity and diabetes. Importantly, methyl indoxam was found to be effective when administered orally, suggesting that this compound survived through the acidic environment of the stomach in conferring protection against diet-induced obesity and diabetes. In vitro stability tests show that methyl indoxam is resistant to hydrolysis at low pH (data not shown). Although methyl indoxam is also effective at inhibiting other phospholipase A2 (Singer et al., 2002), our in vitro data with Caco-2 cells were consistent with previously reported observations that methyl indoxam is minimally transported from the apical to the basolateral side of intestinal cells (Mounier et al., 2004). Therefore, it is likely that orally administered methyl indoxam protects against diet-induced obesity and diabetes through direct inhibition of Pla2g1b in the intestinal lumen. However, in vivo measurements revealed that the bioavailability of orally fed methyl indoxam was 12.77%, a low but not insignificant value. Thus, it is possible that methyl indoxam also suppresses other Pla2 enzymes outside of the gastrointestinal lumen. Nevertheless, the observed reduction of postprandial lysophospholipid levels in mice treated with methyl indoxam, along with the previous documentation that Pla2g1b in the digestive tract contributes directly to postprandial lysophospholipids and hyperglycaemia (Labontéet al., 2006), indicates that Pla2g1b inhibition is the predominant mechanism by which methyl indoxam suppresses diet-induced obesity and diabetes. It is also possible, and our data cannot rule out this possibility, that methyl indoxam may also suppress diet-induced obesity and diabetes by inhibiting Pla2g1b binding to M-type receptors (Boilard et al., 2006). Additionally, methyl indoxam may also inhibit other phospholipases in the digestive tract in addition to Pla2g1b, and so suppresses diet-induced obesity and glucose intolerance by reducing lipid absorption (Richmond et al., 2001). Regardless of the precise mechanism by which methyl indoxam treatment reduces diet-induced obesity and diabetes in mice, our results showing that methyl indoxam is also effective at inhibiting human PLA2G1B suggest that methyl indoxam or a similar derivative could be used clinically to reduce obesity and diabetes in patients consuming a high-fat/high-carbohydrate diet. In this regard, a cell-impermeable derivative of methyl indoxam, which would restrict its activity to PLA2G1B inhibition in the intestinal lumen, would be advantageous to eliminate any potential, undesirable systemic effects.
Acknowledgments
The authors thank Drs Frédéric Carrière at the Laboratory of Enzymology at Interfaces and Physiology of Lipolysis (UPR-9025-CNRS, Marseille Cedex, France) for assistance with the assays for human PLA2G1B hydrolysis of test meals, and Ms. Heather Feldhaus for technical assistance with the animal studies. This work was financially supported by a grant (DK69967) from the National Institutes of Health and a research grant from Ilypsa, Inc., a subsidiary of Amgen, Inc. E.D.L. was the recipient of a postdoctoral fellowship (0525340B) from the Great Rivers Affiliate of the American Heart Association.
Glossary
Abbreviations:
- AUC
area under curve
- GLP-1
glucagon-like peptide-1
- Pla2
phospholipase A2
- Pla2g1b
group 1B phospholipase A2
Conflict of interest
D.Y.H. received a research grant and served on the Scientific Advisory Board of Ilypsa, Inc. during the course of this study.
M.J.C., J.S., E.G. and J.B. were employed at Ilypsa, Inc. during the course of this study and are now employees of Relypsa, Inc.
H-T.C. and D.C. were employed at Ilypsa, Inc. during the course of this study and are now employees of Ardelyx, Inc.
References
- Alexander SPH, Mathie A, Peters JA. Guide to receptors and channels (GRAC) Br J Pharmacol. 2008;153(Suppl 2):S1, S209. doi: 10.1038/sj.bjp.0707746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amori RE, Lau J, Pittas AG. Efficacy and safety of incretin therapy in type 2 diabetes. Systematic review and meta-analysis. JAMA. 2007;298:194–206. doi: 10.1001/jama.298.2.194. [DOI] [PubMed] [Google Scholar]
- Artursson P, Palm K, Luthman K. Caco-2 monolayers in experimental and theoretical predictions of drug transport. Adv Drug Deliv Rev. 2001;46(1–3):27–43. doi: 10.1016/s0169-409x(00)00128-9. [DOI] [PubMed] [Google Scholar]
- Bain SC, Stephens JW. Exenatide and pancreatitis: an update. Expert Opin Drug Saf. 2008;7(6):643–644. doi: 10.1517/14740330802432003. [DOI] [PubMed] [Google Scholar]
- Boilard E, Rouault M, Surrel F, Le Calvez C, Bezzine S, Singer A, et al. Secreted phospholipase A2 inhibitors are also potent blockers of binding to the M-type receptor. Biochemistry. 2006;45:13203–13218. doi: 10.1021/bi061376d. [DOI] [PubMed] [Google Scholar]
- Delea TE, Edelsberg JS, Hagiwara M, Oster G, Phillips LS. Use of thiazolidinediones and risk of heart failure in people with type 2 diabetes: a retrospective cohort study. Diabetes Care. 2003;26:2983–2989. doi: 10.2337/diacare.26.11.2983. [DOI] [PubMed] [Google Scholar]
- Folch J, Lees M, Sloane-Stanley GH. A simple method for the isolation and purification of total lipids from animal tissues. J Biol Chem. 1957;226:497–509. [PubMed] [Google Scholar]
- Huggins KW, Boileau AC, Hui DY. Protection against diet-induced obesity and obesity-related insulin resistance in Group 1B PLA2-deficient mice. Am J Physiol. 2002;283:E994–E1001. doi: 10.1152/ajpendo.00110.2002. [DOI] [PubMed] [Google Scholar]
- Labonté ED, Kirby RJ, Schildmeyer NM, Cannon AM, Huggins KW, Hui DY. Group 1B phospholipase A2-mediated lysophospholipid absorption directly contributes to postprandial hyperglycemia. Diabetes. 2006;55:935–941. doi: 10.2337/diabetes.55.04.06.db05-1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackay K, Starr JR, Lawn RM, Ellsworth JL. Phosphatidylcholine hydrolysis is required for pancreatic cholesterol esterase- and phospholipase A2-facilitated cholesterol uptake into intestinal Caco-2 cells. J Biol Chem. 1997;272:13380–13389. doi: 10.1074/jbc.272.20.13380. [DOI] [PubMed] [Google Scholar]
- Mounier CM, Ghomashchi F, Lindsay MR, James S, Singer AG, Parton RG, et al. Arachidonic acid release from mammalian cells transfected with human group IIA and X secreted phospholipase A2 occurs predominantly during the secretory process and with the involvement of cytosolic phospholipase A2. J Biol Chem. 2004;279:25024–25038. doi: 10.1074/jbc.M313019200. [DOI] [PubMed] [Google Scholar]
- Powell DR. Obesity drugs and their targets: correlation of mouse knockout phenotypes with drug effects in vivo. Obes Rev. 2006;7:89–108. doi: 10.1111/j.1467-789X.2006.00220.x. [DOI] [PubMed] [Google Scholar]
- Richmond BL, Hui DY. Molecular structure and tissue-specific expression of the mouse pancreatic phospholipase A2 gene. Gene. 2000;244:65–72. doi: 10.1016/s0378-1119(00)00006-8. [DOI] [PubMed] [Google Scholar]
- Richmond BL, Boileau AC, Zheng S, Huggins KW, Granholm NA, Tso P, et al. Compensatory phospholipid digestion is required for cholesterol absorption in pancreatic phospholipase A2 deficient mice. Gastroenterology. 2001;120:1193–1202. doi: 10.1053/gast.2001.23254. [DOI] [PubMed] [Google Scholar]
- Rouault M, Le Calvez C, Boilard E, Surrel F, Singer A, Ghomashchi F, et al. Recombinant production and properties of binding of the full set of mouse secreted phospholipases A2 to the mouse M-type receptor. Biochemistry. 2007;46(6):1647–1662. doi: 10.1021/bi062119b. [DOI] [PubMed] [Google Scholar]
- Singer AG, Ghomashchi F, Le Calvez C, Bollinger J, Bezzine S, Rouault M, et al. Interfacial kinetic and binding properties of the complete set of human and mouse groups I, II, V, X, and XII secreted phospholipases A2. J Biol Chem. 2002;277(50):48535–48549. doi: 10.1074/jbc.M205855200. [DOI] [PubMed] [Google Scholar]
- Triplitt C, McGill JB, Porte D, Conner CS. The changing landscape of type 2 diabetes: the role of incretin-based therapies in managed care outcomes. J Manag Care Pharm. 2007;13(Suppl S):S2, S16. doi: 10.18553/jmcp.2007.13.9-c.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson SG, Adam G, Langdown M, Reneland R, Braun A, Andrew T, et al. Linkage and potential association of obesity-related phenotypes with two genes on chromosome 12q24 in a female dizygous twin cohort. Eur J Hum Genet. 2006;14:340–348. doi: 10.1038/sj.ejhg.5201551. [DOI] [PubMed] [Google Scholar]
- Young SC, Hui DY. Pancreatic lipase–colipase mediated triglyceride hydrolysis is required for cholesterol transport from lipid emulsions to intestinal cells. Biochem J. 1999;339:615–620. [PMC free article] [PubMed] [Google Scholar]
- Zambrowicz BP, Sands AT. Knockouts model the 100 best-selling drugs – will they model the next 100? Nat Rev Drug Discov. 2003;2:38–51. doi: 10.1038/nrd987. [DOI] [PubMed] [Google Scholar]