

Abstract
Background and purpose:
Beside their cholesterol lowering effect, statins exert pleiotropic effects, which include anti-inflammatory, immunosuppressive and anti-proliferative actions. In higher concentrations, statins trigger apoptosis in primary cells and tumour cells. In particular, melanoma cells have been found to be susceptible to statin-induced apoptosis, although only after longer incubation times. The molecular mechanisms behind this delayed drug-induced apoptosis are still unclear.
Experimental approach:
The human melanoma A375 and 518A2 cell lines were exposed to various statins in a time-dependent and dose-dependent manner, and indicators of apoptosis, caspase activity and individual apoptotic pathways were analysed for 3-hydroxy-3-methylglutaryl-coenzyme A reductase dependent and independent effects.
Key results:
Kinetic analysis of statin-induced apoptosis revealed an apoptotic burst for exposure times longer than 24 h. While the extrinsic pathway was not activated within 24 h, longer incubation times corroborated amplification of the mitochondrial pathway with significant activation of caspase 8. Continuous refreshing of the simvastatin-containing medium abrogated the mitochondrial amplification loop via caspase 8. Moreover, conditional medium, supplemented with mevalonic acid in order to nullify a possible contamination by statins, significantly triggered caspase 8 activity. Fas ligand was excluded as a possible candidate to account for the statin-induced autocrine amplification loop.
Conclusions and implications:
Simvastatin and atorvastatin are capable of triggering an ‘autocrine’ suicide factor, which amplifies apoptosis via the extrinsic pathway in human melanoma cells. This pro-apoptotic stimulus implies possible therapeutic potential and may guide feasibility for more potent statins in anti-cancer strategies.
Keywords: melanoma, apoptosis, Bid, caspase 8, cholesterol synthesis, HMG-CoA reductase, statin, mevalonate, Fas ligand
Introduction
The local excision of primary melanoma was first described by William Norris in 1857, and remained the mainstay of therapy for localized melanoma until now (Essner, 2003). In the early stages, excision provides full recovery, while the therapy in late stages remains frustrating. The melanoma is highly resistant to conventional chemotherapy due to the up-regulation of multidrug resistance proteins and anti-apoptotic proteins like Bcl-2 and yet unidentified mechanisms (Soengas and Lowe, 2003). It is therefore important to develop new therapeutic approaches.
The statins, competitive inhibitors of 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase, inhibit the rate-limiting step of the mevalonate pathway, and thereby the synthesis of cholesterol (Goldstein and Brown, 1990). In particular, statins have been shown to synchronize tumour cells by blocking the transition of G1-S phase in the cell cycle and this explains their anti-proliferative effects (Keyomarsi et al., 1991; Saito et al., 2008). In addition, statins are able to trigger apoptosis in various tumour cells in vitro (Dimitroulakos et al., 2001; Werner et al., 2004; Demierre et al., 2005). However, there are cancer types which are less sensitive, like breast cancer cells, prostatic tumour cells or myeloma cells (Dimitroulakos et al., 2001; Wong et al., 2007). The heterologous expression of constitutively active Rho and Ras proteins was not able to convert lovastatin-insensitive myeloma cells into lovastatin-sensitive cells (Wong et al., 2007). Thus, these results suggest that the sensitivity of tumour cells towards statins depends on more than one individual isoprenylated protein and may be indicative of multiple mechanisms, the actions of which depend on the individual cancer cell type (Dimitroulakos et al., 2001; Wong et al., 2007).
In vitro studies on murine and human melanoma cells have shown that statins inhibit proliferation and induce apoptotic stimuli (Shellman et al., 2005). In particular, lovastatin in combination with doxorubicin has potentiated anti-tumour effects compared with the individual compounds (Feleszko et al., 2002). In addition, atorvastatin alone inhibits the Rho signalling pathway, and thereby abrogate the invasion and metastasis of A375 human melanoma cells (Collisson et al., 2002). In accord with these findings in a recent publication, lovastatin and simvastatin have been shown to inhibit the growth, cell migration and invasion of melanoma cells (Glynn et al., 2008). For simvastatin, the induction of apoptosis has been linked to caspase 3 activation and DNA fragmentation in various human and murine melanoma cells (Pan et al., 1990; Saito et al., 2008). Interestingly, the various melanoma cell lines show different susceptibilities towards statin-induced cell cycle arrest and apoptosis. The mechanisms behind these differences are not obvious. In particular, the sequential mechanisms of statin-induced apoptosis are not clear. Most importantly, in a very recent publication, it has been shown that within 24 h of exposure to statin, the viability of melanoma cells is not reduced compared with control cells (Saito et al., 2008). While after 48 h of statin exposure, viability is dramatically reduced. The molecular mechanisms behind this phenomenon have not been identified.
The objectives of the present study were (i) to examine and compare the various apoptosis pathways triggered by statins like simvastatin, lovastatin and cerivastatin in the human melanoma cell lines A375 and 518A2; (ii) to determine a concentration-dependent and lipid-dependent profile for ability of the various statins to initiate apoptosis; (iii) to investigate the kinetics of the onset of statin-induced apoptosis in order to identify the mechanism for the increased susceptibility towards statins upon longer exposure times; and (iv) to evaluate the Fas ligand (FasL)-mediated signalling as a possible mechanism for the activation of the extrinsic pathway of apoptosis.
Methods
Cell culture and protein preparation
All experiments were carried out with human melanoma cell lines A375 or 518A2. Cells were grown in Dulbecco's modified eagle medium (DMEM) high glucose growth medium, supplemented with 10% fetal calf serum, 1% penicillin/streptomycin and kept at 37°C under 5% CO2.
The human melanoma cells were exposed to various concentrations of simvastatin, lovastatin, atorvastatin and cerivastatin as depicted in the figure legends. Thereafter, the cells were washed with phosphate buffer saline (PBS) and subsequently kept in lysis buffer [25 mM HEPES (pH 7.4), 5 mM ethylendiamine tetraacetate (EDTA), 1 mM ethylene glycol tetraacetic acid (EGTA), 5 mM MgCl2, 5 mM dithiothreitol (DTT), 1.4 µg·mL−1 aprotinin, 10 µg·mL−1 leupeptin, 100 µM pefablock]. After a rapid cycle of freezing and thawing, the cells were lysed by sonication for 10 s. The protein slurry was centrifuged at 30 000 g for 20 min at 4°C and separated in a cytosolic and membrane fraction. The protein fractions were stored at −80°C.
Fluorescence-activated cell sorting analysis
Apoptosis in melanoma cells was analysed with rh annexin V/fluorescein isothiocyanate (FITC) apoptosis detection kit. Briefly, melanoma cells were incubated for 48 h with the compounds indicated. Thereafter, the cells were detached with AccuMax (Sigma, St. Louis, MO, USA), washed and resuspended in the binding buffer at a density of 2–5 × 105 cells·mL−1. After the cells had been stained with propidium iodide and annexin V-FITC, samples of 20 000 cells were analysed on a Becton Dickinson FACScan (Franklin Lakes, NY, USA). The data were processed by using WINMDI 2.9 software (http://www.cyto.purdue.edu/flowcyt/software/Winmdi.htm).
Caspase activity
The fluorometric caspase assay relies on the fact that specific caspase substrates change their fluorescence upon cleavage and were carried out as described previously (Werner et al., 2004; Sacher et al., 2005). Briefly, aliquots of the supernatant (10–50 µg) of treated melanoma cells were incubated with 50 µM of the 7-amino-4-trifluoro-methylcoumarin (AFC)-conjugated substrates for caspase 3 (Ac-Asp-Glu-Val-Asp-AFC), caspase 8 (Ac-Leu-Glu-Thr-Asp-AFC) or caspase 9 (Ac-Leu-Glu-His-Asp-AFC) in the reaction buffer [25 mM HEPES (pH 7.4), 20% glycerol and 5 mM DTT]. The probes were incubated for 90 min at 37°C in the dark and thereafter, the change in fluorescence was measured at an excitation wavelength of 405 nm and an emission wavelength of 535 nm using a fluorescence plate reader. In some experiments, the specific caspase activity varies to some extent, which was due to bleaching of the fluorescent substrate. This was, in part, prevented by using fresh aliquots of the fluorescent substrates. The reaction mixture without protein was referred to the background and was subtracted from samples.
Medium exchange and ‘wash protocol’
In order to control for a secreted component in the medium, melanoma cells were washed every 7 h for an assay duration of 48 h. At each step, the medium was removed, collected and the cells gently washed two times with PBS. Thereafter, the cells were exposed to fresh medium (containing the indicated compounds and drugs) or to the collected conditional medium (containing the indicated drugs). The latter procedure controlled for the mechanical stress during the washing procedure and represents the medium collected before the PBS wash steps. The treated cells were harvested as described above, and the proteins were used for caspase assays or Western blot analysis.
Western blot
Protein samples from cell lysis were used for Western blot analysis. Equal amounts of protein samples (5–15 µg) were dissolved in sample buffer and separated on a sodium dodecyl sulphate polyacrylamide gel. The separated proteins were transferred to nitrocellulose membranes and exposed to the appropriate primary and secondary antibodies. The specific presence of Bid, caspase 8, poly-adenosine diphosphate-ribose polymerase (PARP) and α-tubulin was detected by the electrochemiluminescence (ECL) plus detection system. The intensity of the protein bands was determined using ImageJ software (http://rsbweb.nih.gov/ij) and expressed relative to the intensity of α-tubulin band.
Trypan blue staining
After detachment, cell suspensions were washed with PBS and subsequently resuspended in 1 mL PBS. Aliquots of the cell suspension were incubated with 0.2% trypan blue and incubated for 5 min at room temperature. Thereafter, pictures of the cells were obtained in order to allow offline analysis. The stained cells and total number of cells were counted in at least four visual fields of equal size and magnification. The results are presented as percent of dead cells.
Miscellaneous procedures
The protein concentration was determined according to the Bradford protein assay using bovine serum albumin as a standard (Bradford, 1976). Data are expressed as mean ± standard error mean. The concentration−response curves were subjected to non-linear least squares regression to the Hill equation using the Sigmaplot software (Jandl, Erkrath, Germany). Statistical analysis was carried out with Student's t-test, or for multiple comparisons, with analysis of variance and post hoc Bonferroni correction. A value of P < 0.05 was considered statistically significant.
Materials
FITC apoptosis detection kit, Bender MedSystems (Vienna, Austria); AccuMax, Sigma; Becton Dickinson FACScan; fluorescence plate reader (VICTOR-2), Perkin-Elmer (Wellesley, MA, USA); nitrocellulose membranes, Watman (Dassel, Germany); Bid, FL-195 (Santa Cruz, CA, USA); caspase 8 (1C12), Cell Signalling (Danvers, MA, USA); PARP (C-2-10), Zymed (South San Francisco, CA, USA); α-tubulin (B 5-1-2) and trypan blue, Sigma; ECL plus detection system, GE Healthcare (Bucks, UK); and FasL Ab (14C2 mouse IG1), Bender MedSystems (Vienna, Austria).
Results
Statin-induced apoptosis in A375 and 518A2 melanoma cells
Already, after 24 h, human A375 and 518A2 melanoma cell lines changed their morphology when exposed to simvastatin or lovastatin up to 10 µM (data not shown). Typical for apoptosis, the cells started to shrink, the cytosolic volume decreased compared with the nucleus and finally, the cells detached from the surface. Similar observations have been described by others and confirmed susceptibility of A375 and 518A2 melanoma cells to statin-induced apoptosis. These morphological changes were accompanied by reduced viability of the cells with increasing concentrations of simvastatin. After a 48 h exposure to 10 µM simvastatin, the viability of the cells was diminished to approximately 30–50%, when determined with trypan blue staining (data not shown) or fluorescence-activated cell sorting analysis (Figure 1). The cells were positive for propidium iodide and annexin V staining, indicating a late apoptotic state (Figure 1A,B). The statin-induced accumulation of dead cells was completely prevented by a saturating concentration of mevalonic acid (1 mM), the product of the inhibited enzyme reaction, namely, HMG-CoA reductase activity. A concentration of 100 µM mevalonic acid was not sufficient to fully overcome statin-induced cell death in 518A2 melanoma cells (Figure 1B). Simvastatin-induced apoptosis was further confirmed by cleavage of PARP, another indicator of apoptosis (Figure 1C).
Figure 1.
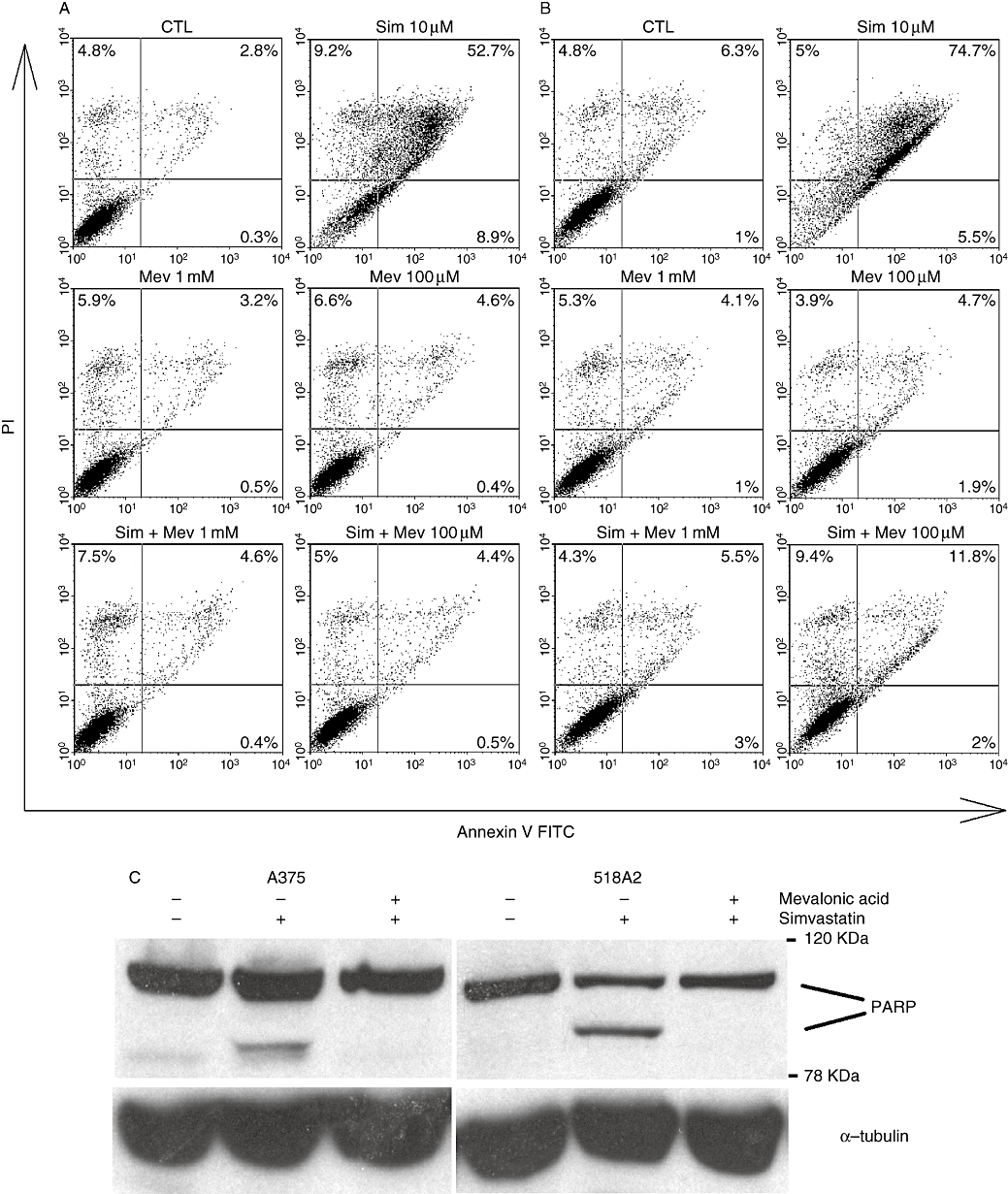
HMG-CoA reductase-dependent apoptosis in human melanoma cells by simvastatin. Human A375 (A) and 518A2 (B) melanoma cells were exposed to 10 µM simvastatin in the absence and presence of 0.1 or 1 mM mevalonic acid for 48 h. Propidium iodide and annexin V-positive cells were quantified by FACS analysis. Apoptosis was also confirmed by PARP cleavage in A375 and 518A2 melanoma cells (C) using Western blot analysis. Tubulin was used as a loading control. Representative experiments are depicted, which were repeated at least twice. CTL, control; FACS, fluorescence-activated cell sorting; FITC, fluorescein isothiocyanate; HMG-CoA, 3-hydroxy-3-methylglutaryl-coenzyme A; Mev, mevalonic acid; PARP, poly-ADP-ribose polymerase; Sim, simvastatin.
In order to confirm that statins induce apoptosis, the central executioner caspase 3 was investigated in greater detail (Figure 2). When A375 and 518A2 melanoma cells were exposed to higher simvastatin concentrations (>3 µM), after 24 h, a significant increase in caspase 3 activity was detected. However, a 10-fold increment in caspase 3 activity was observed when the cells were exposed for 48 h to the indicated simvastatin concentrations (Figure 2). Even a concentration of 1 µM simvastatin was sufficient to significantly trigger caspase 3 activity. However, the potency of simvastatin did not change with time of exposure. A rough estimation of the EC50 (at 24 h) ranged from 6.2 µM to 7.8 µM simvastatin, which compares with 9.9–12 µM after 48 h exposure. Thus, the potency of simvastatin at triggering caspase 3 activity was not changed.
Figure 2.
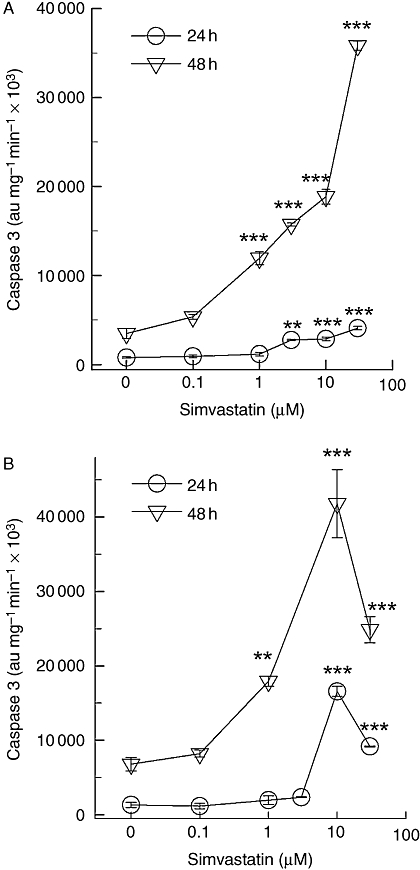
Simvastatin-induced caspase 3 activity. Human A375 (A) and 518A2 (B) melanoma cells were lysed after 24 h or 48 h of incubation with the indicated simvastatin concentrations. The supernatant of the cell lysates (25–40 µg) was incubated with fluorescent caspase 3 substrate (50 µM) for 90 min at 37°C in a reaction buffer described in the Methods section. The estimated EC50 values calculated from the ascendant part of the concentration-response curve gave values between 3.5 µM and 6.5 µM after 24 h, and 1.4–1.7 µM after 48 h of exposure to statin. The symbols represent the mean ± standard error mean (n= 8). Asterisks denote statistical significance versus control (**P < 0.01, ***P < 0.001).
In various tumour cell lines, statins have been shown to trigger the mitochondrial pathway of apoptosis (Chapman-Shimshoni et al., 2003; Werner et al., 2004). We, therefore, investigated caspase 9 activity similar to the results shown in Figure 2. Again, the mitochondrial pathway of apoptosis was activated more than 10-fold, when exposure times were switched from 1 day to 2 days (Figure 3). These effects were also seen with cerivastatin and lovastatin in both melanoma cells lines (data not shown). However, cerivastatin was approximately 100-fold more potent than lovastatin after a 48 h exposure (Figure 4). Thus, the potency of the individual statins at triggering caspase-dependent apoptosis reflects the rank order of potency described for HMG-CoA reductase inhibition. This is an indirect indication that the apoptotic stimulus is highly dependent on lipids.
Figure 3.
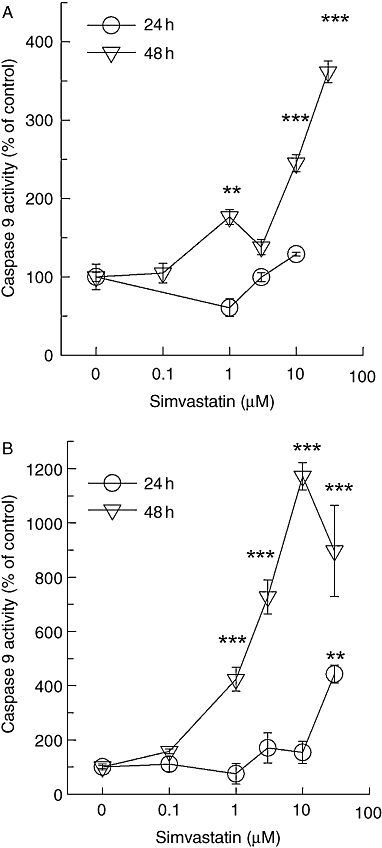
Simvastatin-induced caspase 9 activity. Caspase 9 activity was determined in the supernatant of lysates from A375 (A) and 518A2 (B) melanoma cells treated for 24 h and 48 h with increasing concentrations of simvastatin. Symbols represent mean ± standard error mean (n= 8). Asterisks denote statistical significance versus control (**P < 0.01; ***P < 0.001).
Figure 4.
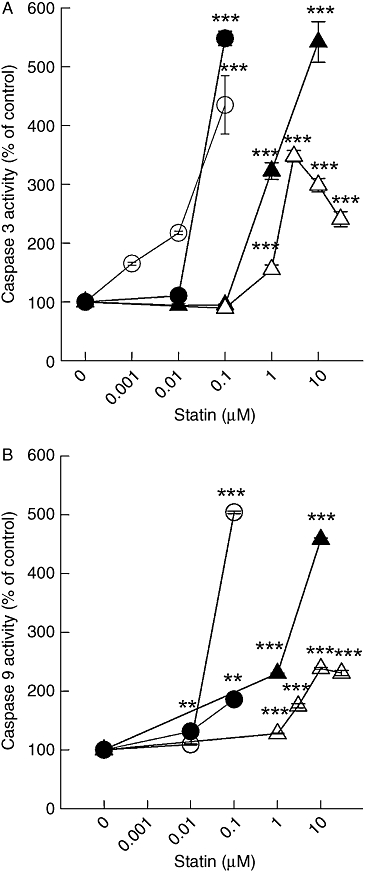
Cerivastatin and lovastatin trigger the intrinsic pathway of apoptosis. Caspases 3 (A) and 9 (B) activity was measured in lysates from A375 (open symbols) and 518A2 (solid symbols) melanoma cells treated for 48 h with lovastatin (triangle) or cerivastatin (circle) as described in Figure 2. The symbols represent the mean ± standard error mean (n= 8). Asterisks denote statistical significance versus control (**P < 0.01; ***P < 0.001).
Interestingly, at higher concentrations (>10 µM), simvastatin and lovastatin demonstrated a biphasic concentration–response relationship (Figures 2B, 3B and 4). Although the mechanism behind this phenomenon is not clear at the moment, carry over of the solvent dimethyl sulfoxide (DMSO) was not responsible for the decline in activity (data not shown).
Caspase 8 activation in 518A2 and A375 melanoma cells
We observed a burst in proteolytic activity for the intrinsic pathway of apoptosis after exposure times longer than 24 h (Figures 2 and 3). This led to the hypothesis that caspase 8 might be activated within this time frame, which then may lead to truncation of Bid and subsequent amplification of the mitochondrial apoptosome formation (Green, 2005; Schafer and Kornbluth, 2006). Similar to caspase 9, up to 10 µM, simvastatin was not able to significantly activate caspase 8 within 24 h. However, after 48 h, a significant stimulation of caspase 8 was evident even at simvastatin concentrations below 10 µM (Figure 5). Similarly, incubation with lovastatin or cerivastatin for 2 days also elicited significant caspase 8 activation in 518A2 and A375 melanoma cells even at 1 µM and 100 nM respectively (data not shown).
Figure 5.
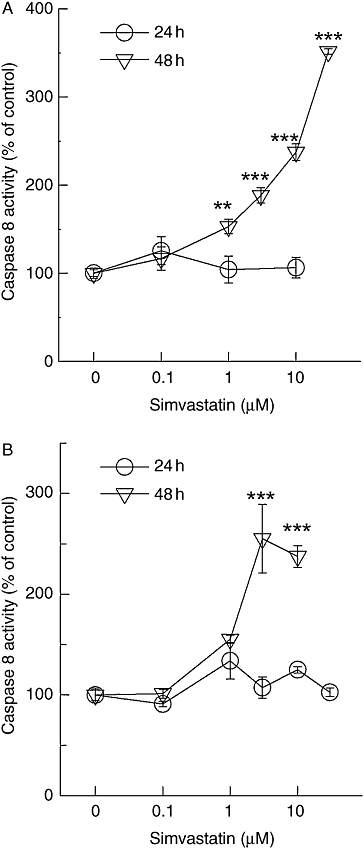
Simvastatin-induced caspase 8 activity. Caspase 8 activity was measured in lysates from A375 (A) and 518A2 (B) melanoma cells treated for 48 h with simvastatin analogous to the description in Figure 2 legend. The symbols represent the mean ± standard error mean (n= 8). Asterisks denote statistical significance versus control (**P < 0.01; ***P < 0.001).
In order to ensure that this caspase 8 activation is not provoked by a component released from dead cells, we treated melanoma cells with vincristine and ionomycin (Table 1). Neither vincristine nor ionomycin activated caspase 8. While vincristine, similar to simvastatin, significantly triggered caspase 3 activity, the ionomycin-induced caspase 3 activity was not significantly elevated. We therefore conclude that simvastatin triggers a specific component in melanoma cells, which is not attributable to a simple release mechanism from dead cells.
Table 1.
Comparison of caspase activation by simvastatin, vincristine and ionomycin
| au.·mg−1·min−1×1000 | Caspase 3 activity | Caspase 8 activity |
|---|---|---|
| Control | 1954.1 ± 135.4 (n = 8) | 479.1 ± 41.2 (n = 8) |
| Simvastatin (10 µM) | 19886.9 ± 1295.5 (n = 8)** | 906.7 ± 67.0 (n = 8)** |
| Vincristine (0.5 µM) | 13861.7 ± 246.1 (n = 4)** | 521.5 ± 48.2 (n = 4) |
| Ionomycin (1 µM) | 2912.1 ± 243.5 (n = 4) | 447.3 ± 62.1 (n = 4) |
Human A375 melanoma cells were exposed to the indicated drugs for 48 h, and the supernatant of the lysed cells was used for determination of caspases 3 and 8 activity. The data represent the mean ± standard error mean. Asterisks denote statistical significance versus control after analysis of variance and post hoc Bonferroni analysis for multiple comparison
P < 0.001).
The activation of the extrinsic pathway of apoptosis was further corroborated by the cleavage of pro-caspase 8 in the presence of increasing concentrations of simvastatin and atorvastatin (Figure 6A). The activated caspase 8 is able to trigger the mitochondrial pathway by truncation of pro-apoptotic Bid. Again, simvastatin and atorvastatin led to a significant reduction of the total amount of intact Bid (Figure 6B). Although we cannot exclude the possibility that statin treatment reduces the expression of Bid, it is more likely that the reduction in the protein band is due to cleavage of the intact protein.
Figure 6.
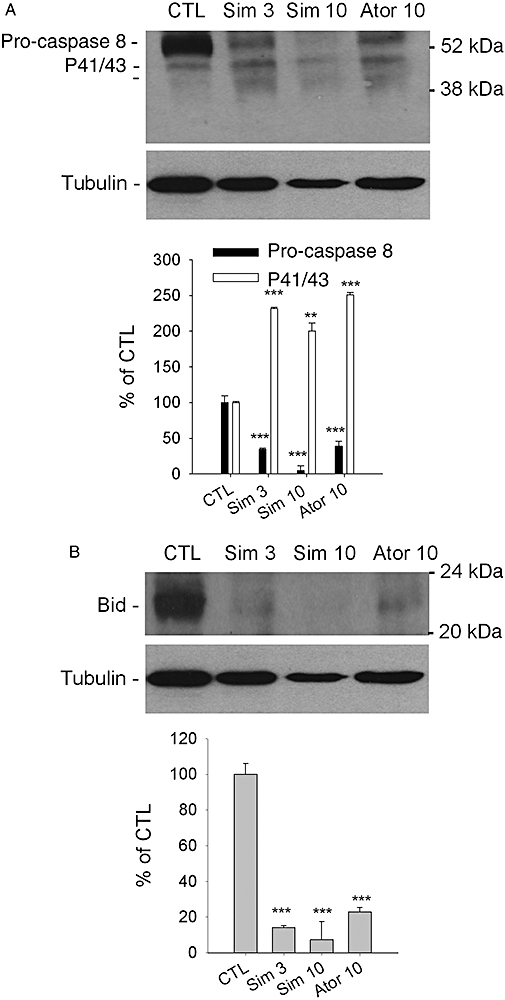
Cleavage of pro-caspase 8 and truncation of Bid by statins. The supernatant of lysed cells was analysed for reduction of pro-caspase 8 (A) and full-length Bid (B). The 518A2 melanoma cells were treated for 48 h with the indicated concentrations (µM) of simvastatin and atorvastatin. The intensity of the protein bands of interest were quantified by the ImageJ software and normalized to the loading control, α-tubulin. Columns and bars represent the mean ± standard error mean (n= 3). Asterisks denote statistical significance versus control (**P < 0.01; ***P < 0.001). Ator, atorvastatin; CTL, control; Sim, simvastatin.
The question remained open as to what extent simvastatin-induced activation of caspases is dependent on inhibition of HMG-CoA reductase. The significant activation of caspases 3, 9 and 8 by 10 µM simvastatin was completely abrogated by the co-application of mevalonate (Figure 7). Thus, in the A375 and 518A2 melanoma cell lines, the simvastatin-induced activation of caspases 3, 8 and 9 revealed strict dependence on the inhibition of HMG-CoA reductase.
Figure 7.
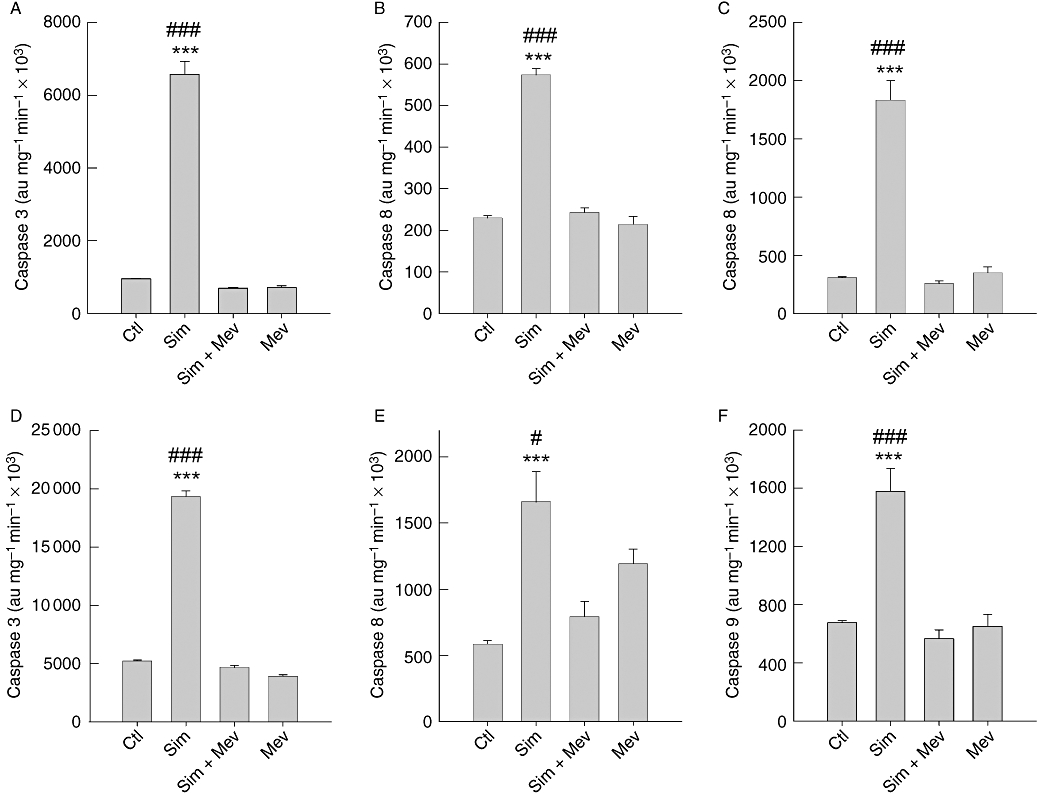
Simvastatin-induced caspase activation is fully dependent on inhibition of HMG-CoA reductase. Simvastatin (10 µM)-induced caspases 3 (A, D), 8 (B, E) and 9 (C, F) activity in A375 (A–C) and 518A2 (D–F) melanoma cells was abrogated by co-application of mevalonic acid (1 mM). Cells were lysed after 48 h and treated for caspase activity according to the legend of Figure 2. The columns and bars represent the mean ± standard error mean (n= 8; ***significance vs. control, P < 0.001; significance vs. simvastatin plus mevalonic acid, #P < 0.05, ###P < 0.001). Ctl, control; HMG-CoA, 3-hydroxy-3-methylglutaryl-coenzyme A; Mev, mevalonic acid; Sim, simvastatin.
The delay in activation of caspase 8 favours the conjecture that an autocrine factor of the human melanoma cells may serve as an amplifier of the statin-induced apoptosis. In order to prove this hypothesis, we performed a wash protocol that would constantly remove such an autocrine factor. The mechanical stress due to the wash procedure and medium change (MW) in the absence of statins did not activate caspases 3, 9 or 8 (Figure 8). Similarly, addition of simvastatin did not activate caspase 8, when the medium was removed and exchanged by fresh medium supplemented with simvastatin (Sim + MW in Figure 8B,E). Of utmost importance, caspases 3, 9 and 8 activity were detected when the wash procedure was followed by readdition of the conditional medium (Sim + W in Figure 8). Whereas activation of caspase 8 was completely reversed, simvastatin-triggered activation of caspases 3 and 9 was only reduced by 50%. These results are compatible with the hypothesis postulating that an autocrine factor is secreted due to simvastatin-induced ‘stress’.
Figure 8.
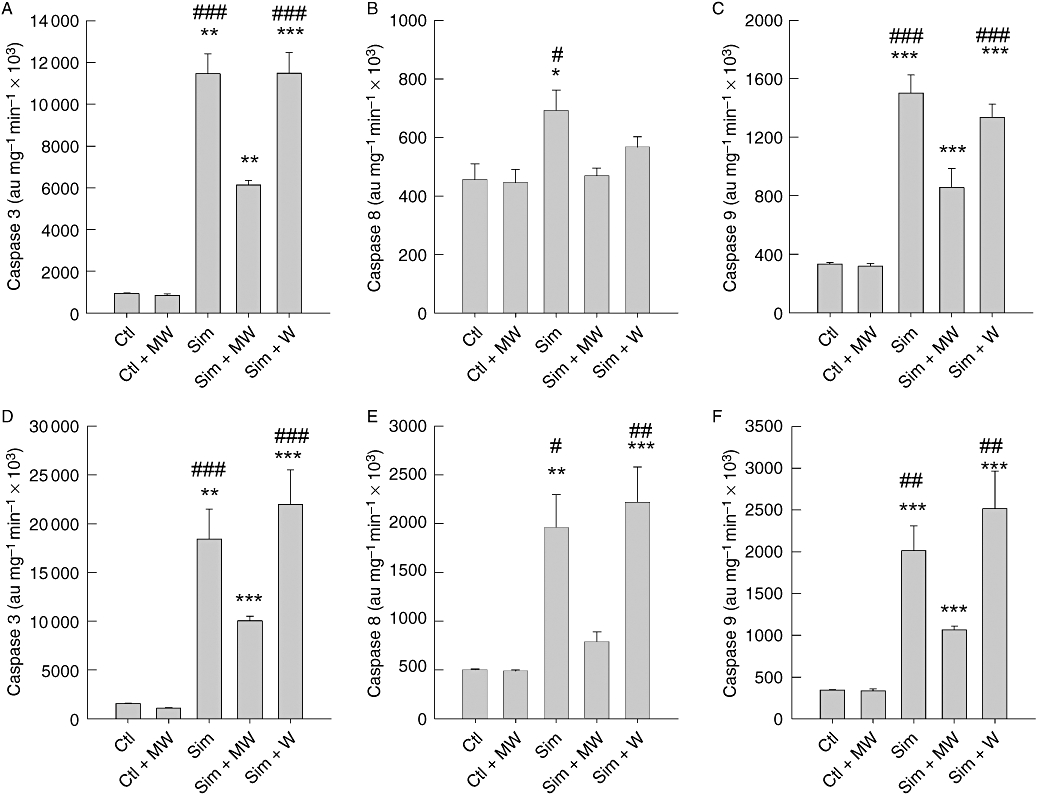
Removal of conditional medium prevents activation of the extrinsic pathway of apoptosis. Caspases 3 (A, C), 8 (B, E) and 9 (C, F) activity was measured in lysates from A375 (A–C) and 518A2 (D–F) melanoma cells, which were washed with PBS every 7 h with (MW) and without medium renewal (W). The cells were treated in the absence (Ctl) and presence of 10 µM simvastatin for 48 h. The columns and bars represent mean ± standard error mean (n= 4). Statistical significance versus control is denoted by asterisks (*P < 0.05; **P < 0.01; ***P < 0.001); significance versus simvastatin plus MW is denoted by pound (#P < 0.05, ##P < 0.01, ###P < 0.001). Ctl, control; PBS, phosphate buffer saline; Sim, simvastatin.
To show that the autocrine factor postulated from the results depicted in Figure 8 is able to trigger the death receptor pathway, samples from the medium exchange experiment were probed for Western blot analysis (Figure 9). Accordingly, renewal of simvastatin-containing medium reversed the cleavage of pro-caspase 8 and the reduction of Bid (Sim + MW in Figure 9A,B). Furthermore, the simple wash protocol and readdition of conditional medium could not prevent cleavage of pro-caspase 8 and Bid. These data are further corroborated by the pro-apoptotic effect of the conditional medium obtained from medium wash experiments.
Figure 9.
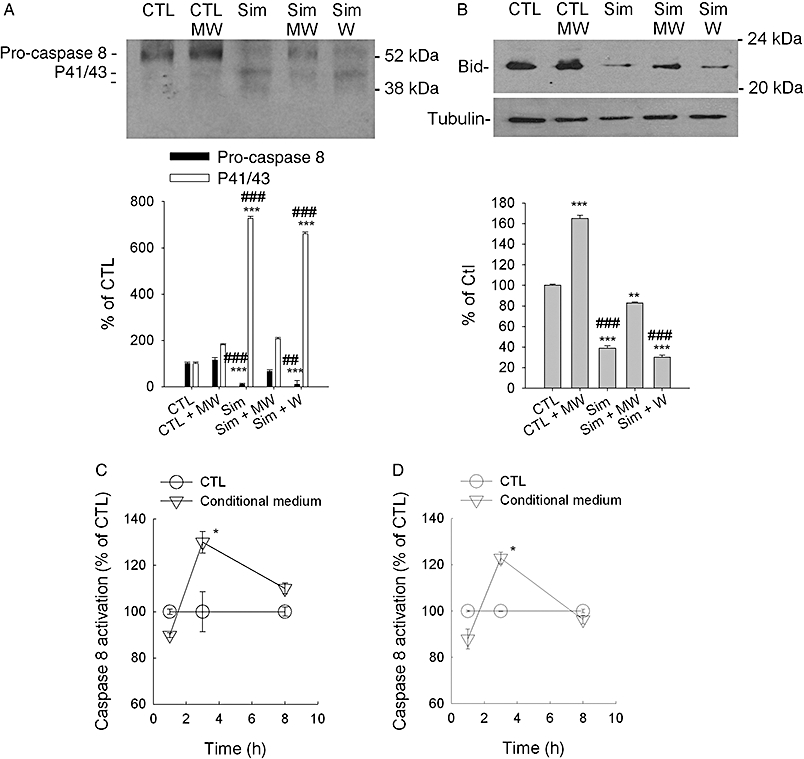
Conditional medium is responsible for caspase 8 pathway activation. Similar to experiments described in Figure 8, the supernatant of 518A2 melanoma cell lysates were analysed for cleavage of caspase 8 (A) and truncation of Bid (B). In the absence (CTL) and presence of 10 µM simvastatin, melanoma cells were washed with PBS every 7 h with (MW) and without medium renewal (W) for 48 h. The intensity of the protein bands of interest were quantified by the ImageJ software and normalized to the loading control, α-tubulin. The Western blots depicted were generated by reblotting of a representative single blot. Columns and bars represent the mean ± standard error mean (n= 3). Statistical significance versus control is denoted by asterisks (**P < 0.01; ***P < 0.001); significance versus simvastatin plus MW is denoted by pound (##P < 0.01; ###P < 0.001). Conditional medium obtained from the above described experiments (518A2 cells) was compared with untreated medium (C, D). The media were supplemented with 1 mM mevalonic acid in order to prevent effects from contaminating simvastatin and were applied to A375 (C) and 518A2 (D) melanoma cells for the indicated time points. Caspase 8 activity was measured from the supernatant of the lysed cells. Symbols and error bars represent the mean ± standard error mean (n= 3). Paired t-test was performed, and asterisks denote statistical significance versus the matched control (*P < 0.05). CTL, control; PBS, phosphate buffer saline; Sim, simvastatin.
In order to exclude an effect induced by the carry over of simvastatin contaminating the medium, we made use of the finding that the activation of caspases 3, 9 and 8 is strictly dependent on the inhibition of HMG-CoA reductase (Figure 7). Conditional medium collected from melanoma cells exposed to simvastatin was therefore supplemented with a saturating concentration of mevalonic acid. Under these conditions, the kinetics of caspase 8 activation revealed a significant activation after only 3 h of exposure to A375 and 518A2 melanoma cells (Figure 9C,D).
A possible candidate for the autocrine suicide factor in melanoma cells is the CD95/CD95 ligand (FasL, CD178) system (Sarrabayrouse et al., 2007). A neutralizing FasL antibody should therefore reduce the simvastatin-induced apoptosis and particularly prevent caspase 8 activation. In both melanoma cells, the simvastatin-induced late apoptosis was not reduced by co-administration of the neutralizing FasL antibody (Figure 10A,B). Similarly, simvastatin-induced increases in caspases 8 and 3 activity were not abrogated by the antibody (Figure 10C,D). Hence, the FasL can therefore be excluded as a possible amplifier of the statin-induced extrinsic apoptosis pathway.
Figure 10.
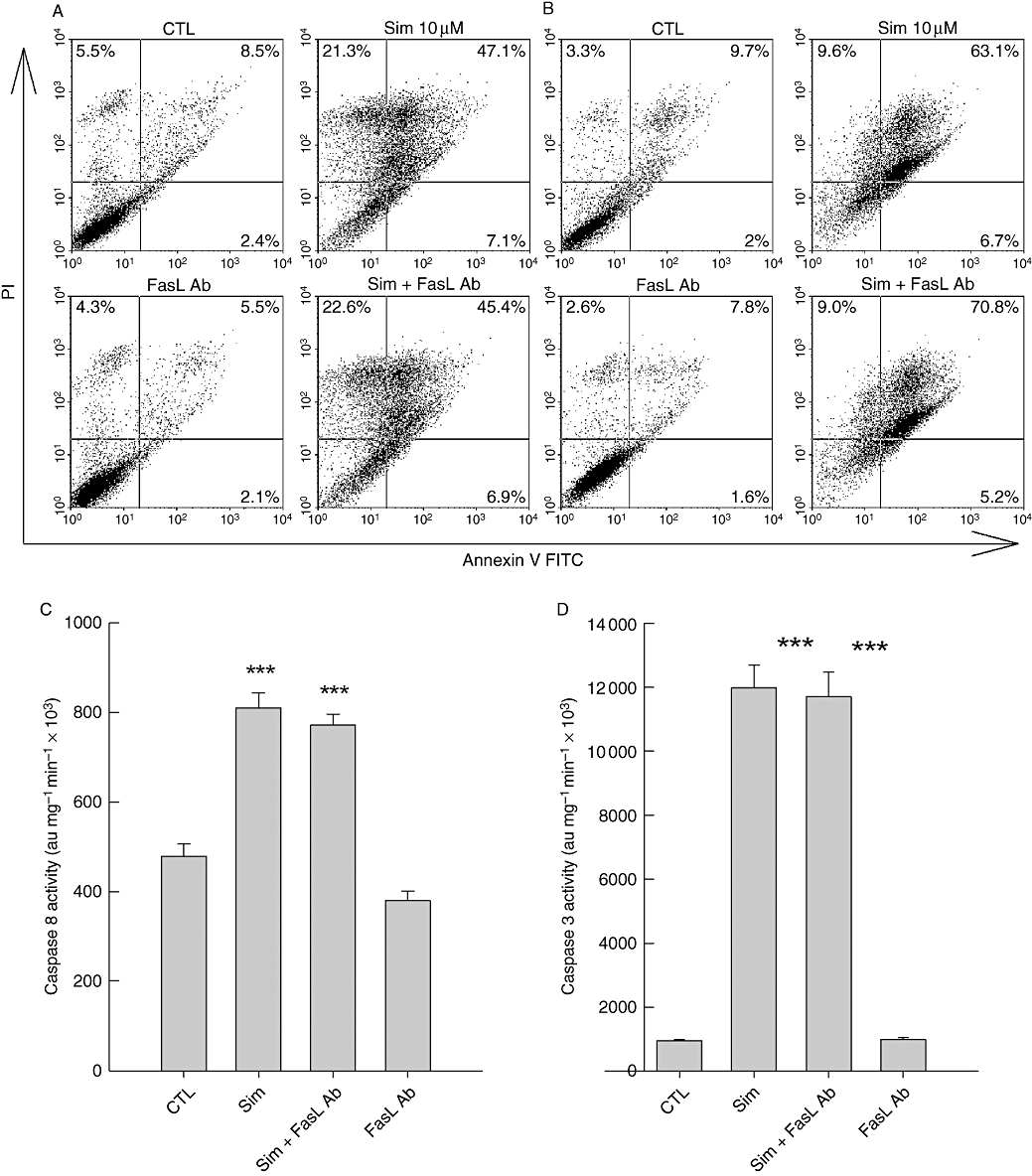
FasL is not responsible for simvastatin-induced activation of caspase 8. Application of a specific neutralizing FasL antibody (1 µg·mL−1) could not prevent simvastatin (10 µM)-induced apoptosis in A375 (A) and 518A2 (B) melanoma cells. FACS analysis was performed as described in the legend of Figure 1. The simvastatin-induced caspases 8 (C) and 3 (D) activity exemplified in A375 melanoma cells was also not altered by the neutralizing FasL antibody (1 µg·mL−1). Simvastatin exposure times in all experiments were 48 h. The bars and errors represent the mean ± standard deviation of a representative experiment carried out in quadruplicate, which was repeated twice. Asterisks represent statistical significance versus control (***P < 0.001). CTL, control; FACS, fluorescence-activated cell sorting; FasL Ab, Fas ligand antibody; FITC, fluorescein isothiocyanate; PI, propidium iodide.
Discussion
Statins-induced apoptosis in melanoma cells
Long-term studies and careful pharmacovigilance have revealed the so-called pleiotropic actions of statins which go far beyond cholesterol lowering (Davignon, 2004; Miida et al., 2004; Demierre et al., 2005; Dale et al., 2006; Farwell et al., 2008; Karp et al., 2008). In particular, statins have been considered as useful in cancer therapy (Demierre et al., 2005). In vitro investigations in human melanoma cells have shown that statins exert anti-proliferative effects, which are mediated by p21-induced cell cycle arrest at the G1/S checkpoint (Saito et al., 2008). Such an up-regulation of p21 is observed in response to DNA damage and under the transcriptional control of p53 (el-Deiry et al., 1994; Saito et al., 2008). However, these effects have been observed with 10 µM simvastatin after exposure times of 48 h.
We have therefore investigated the signalling steps that precede this terminal cell cycle arrest, which is, in part, a consequence of apoptosis. Unequivocally, statins as a group induce caspase-dependent apoptosis (Figures 2–4). Interestingly, cerivastatin, which represents the most potent inhibitor of HMG-CoA reductase, even at nano-molar concentrations (10–100 nM), showed significant stimulation of caspases 3 and 9 (Figure 4). But also, lovastatin, simvastatin (Figures 2–4) as well as atorvastatin (data not shown) are able to trigger these proteolytic enzymes, although in concentrations about 100-fold above that of cerivastatin. Thus, the dependence on HMG-CoA reductase is retained at the level of induction of apoptosis.
In addition to being concentration dependent, the statin-induced apoptosis is also strictly time dependent. We investigated the typical statin simvastatin in greater detail and found that concentrations below 10 µM were virtually ineffective at triggering caspases 3 and 9 activity within 24 h. This is in agreement with observations in other melanoma cell lines, where reduction in cell viability and activation of caspase 3 was observed after exposure to simvastatin or lovastatin only at incubation times longer than 24 h (Shellman et al., 2005; Saito et al., 2008). The 5–10-fold increase in caspases 3 and 9 activation, which we observed after 48 h, was accompanied by caspase 8 activation (Figure 5). This represents a switch from the intrinsic to the extrinsic pathway of apoptosis, which may indicate the synthesis of a pro-apoptotic signalling molecule (Green, 2005; Schafer and Kornbluth, 2006).
This conjecture is supported by several findings: (i) renewal of the medium in the presence of simvastatin was sufficient to abrogate the activation of caspase 8 and thereby prevent the apoptotic burst; (ii) the apoptotic burst of statin-induced caspases 3, 8 and 9 activation was strictly dependent on inhibition of the HMG-CoA reductase and was prevented by mevalonic acid (Figure 7); and (iii) application of conditional medium was sufficient to induce caspase 8 activity.
Simvastatin has been shown to trigger an increase in Ca2+ in human skeletal muscle cells, which represents the initial signal in the myotoxic effects of statins (Sacher et al., 2005; Sirvent et al., 2008). Therefore, ionomycin was used to elevate intracellular Ca2+ concentrations, but it failed to activate caspase 8. Similar observations have been obtained with vincristine (Table 1). Thus, simple induction of cell death or activation of caspase 3 is not sufficient to release the suicide factor which activates caspase 8, as this is observed with statins.
The nature of the autocrine factor is currently unknown. However, we can exclude FasL, which has been shown to be released upon the application of lovastatin to murine melanoma cells (Sarrabayrouse et al., 2007). FasL may play a crucial role in tumour counterattack, a defence mechanism against tumour infiltrating lymphocytes (Igney and Krammer, 2005). However, in our melanoma cell lines, a neutralizing FasL antibody was not able to prevent simvastatin-induced apoptosis, or caspase activation.
The effective statin concentrations used in our investigation to trigger an autocrine factor are in the range of 1–10 µM. The peak plasma concentrations of statins during a conventional therapy of hypercholesterinaemia are in the range of 10–100 nM (Bellosta et al., 2004). Thus, this treatment is unlikely to induce plasma concentrations of statins that are sufficient to result in the apoptosis we observed in vitro. This conjecture may explain the controversial effects seen in clinical trials on the incidence of cancer in general and in melanoma in particular. Alternatively, clinical trials exist, where simvastatin and lovastatin were administered at concentrations ranging from 15 mg·kg−1·day−1 to 45 mg·kg−1·day−1 (Thibault et al., 1996; van der Spek et al., 2006). Such high dose regimes result in peak plasma concentrations of approximately 4 µM, which is clearly within the range that will trigger apoptosis in human melanoma cells in vitro. It should therefore be feasible to facilitate statin-induced apoptosis via autocrine stimulation of the extrinsic pathway of apoptosis. In human differentiated primary skeletal muscle cells, we have shown that activation of caspase 8 is not triggered within 36 h (Sacher et al., 2005). Thus, at least for this cellular compartment, possible side effects due to statin-induced activation of caspase 8 are unlikely.
The fact that statins are so well tolerated when taken over long periods of times made it feasible now to analyse the possible anti-cancer action of statins in melanoma. In particular, the Breslow thickness has been found to be reduced in the statin-treated group (Koomen et al., 2007). A retrospective cohort study in a veteran population has concluded that, compared with the general population, statin users had a significantly lower risk of developing cancer, especially lung and colorectal cancer (Farwell et al., 2008). In addition, another retrospective study that included a total of 30 076 patients with acute myocardial infarction has suggested that the administration of lipophilic statins at high doses is associated with a reduction in cancer incidence (Karp et al., 2008).
Conversely, in a recent meta-analysis, no protective effect of statins on melanoma has been found (Bonovas et al., 2006; Dale et al., 2006; Freeman et al., 2006). However, this finding is not conclusive, as most of these trials were not designed to analyse the anti-cancer properties of statins. Moreover, the hydrophilic statin pravastatin, unlike simvastatin, lovastatin and atorvastatin, shows limited apoptotic potency in vitro (Shellman et al., 2005). Also, in other types of tumour cells, hydrophilic statins like pravastatin have shown impaired cytostatic and anti-neoplastic effects in vitro (Campbell et al., 2006; Sivaprasad et al., 2006). Because cancer and, in particular, melanoma has not been the primary outcome of these studies, it is currently difficult to evaluate the clinical potential of statins in melanoma treatment from these trials.
Given the fact that melanoma is highly resistant to standard chemotherapeutic schemes highlight the importance of finding new therapeutic approaches. In the present study, we showed that the addition of different statins to melanoma cells can induce apoptotic cell death, which is the goal of an effective cancer treatment. On the basis of these findings, we confirm for the first time that statins like simvastatin but also atorvastatin are able to trigger a pro-apoptotic autocrine stimulus. At the moment, the molecular identity of the autocrine factor is unknown; however, it will be the task of future investigations to understand these novel molecular pathways triggered by statins and evaluate their therapeutic potential.
Acknowledgments
The authors are grateful to Dr Edgar Selzer for providing us with the human melanoma cell lines A375 and 518A2. This work was supported by Herzfelder'sche Familienstiftung and the Gen-AU Dragon Project of the Austrian Federal Ministry of Science and Research to M.H.
Glossary
Abbreviations:
- AFC
7-amino-4-trifluoro-methylcoumarin
- ECL
electrochemi-luminescence
- FACS
fluorescence-activated cell sorting
- FasL
Fas ligand
- FITC
fluorescein isothiocyanate
- HMG-CoA reductase
3-hydroxy-3-methylglutaryl-coenzyme A reductase
- PARP
poly-adenosine diphosphate-ribose polymerase
Conflict of interest
None.
References
- Bellosta S, Paoletti R, Corsini A. Safety of statins: focus on clinical pharmacokinetics and drug interactions. Circulation. 2004;109:III50–III57. doi: 10.1161/01.CIR.0000131519.15067.1f. [DOI] [PubMed] [Google Scholar]
- Bonovas S, Filioussi K, Tsavaris N, Sitaras NM. Statins cancer risk: a literature-based meta-analysis and meta-regression analysis of 35 randomized controlled trials. J Clin Oncol. 2006;24:4808–4817. doi: 10.1200/JCO.2006.06.3560. [DOI] [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantization of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Campbell MJ, Esserman LJ, Zhou Y. Breast cancer growth prevention by statins. Cancer Res. 2006;66:8707–8714. doi: 10.1158/0008-5472.CAN-05-4061. [DOI] [PubMed] [Google Scholar]
- Chapman-Shimshoni D, Yuklea M, Radnay J, Shapiro H, Lishner M. Simvastatin induces apoptosis of B-CLL cells by activation of mitochondrial caspase 9. Exp Hematol. 2003;31:779–783. doi: 10.1016/s0301-472x(03)00192-9. [DOI] [PubMed] [Google Scholar]
- Collisson EA, Carranza DC, Chen IY, Kolodney MS. Isoprenylation is necessary for the full invasive potential of rho A overexpression in human melanoma cells. J Inv Dermatology. 2002;119:1172–1176. doi: 10.1046/j.1523-1747.2002.19519.x. [DOI] [PubMed] [Google Scholar]
- Dale KM, Coleman CI, Henyan NN, Kluger J, White CM. Statins and cancer risk: meta-analysis. JAMA. 2006;295:74–80. doi: 10.1001/jama.295.1.74. [DOI] [PubMed] [Google Scholar]
- Davignon J. Beneficial cardiovascular pleiotropic effects of statins. Circulation. 2004;109:III39–III43. doi: 10.1161/01.CIR.0000131517.20177.5a. [DOI] [PubMed] [Google Scholar]
- Demierre MF, Higgins PD, Gruber SB, Hawk E, Lippman SM. Statins and cancer prevention. Nat Rev Cancer. 2005;5:930–942. doi: 10.1038/nrc1751. [DOI] [PubMed] [Google Scholar]
- Dimitroulakos J, Ye LY, Benzaquen M, Moore MJ, Kamel-Reid S, Freedman MH, et al. Differential sensitivity of various paediatric cancers and squamous cell carcinomas to lovastatin-induced apoptosis: therapeutic implications. Clin Cancer Res. 2001;7:158–167. [PubMed] [Google Scholar]
- el-Deiry WS, Harper JW, O'Connor PM, Velculescu VE, Canman CE, Jackman J, et al. WAF1/CIP1 is induced in p53-mediated G1 arrest and apoptosis. Cancer Res. 1994;54:1169–1174. [PubMed] [Google Scholar]
- Essner R. Surgical treatment of malignant melanoma. Surg Clin North Am. 2003;83:109–156. doi: 10.1016/S0039-6109(02)00205-0. [DOI] [PubMed] [Google Scholar]
- Farwell WR, Scranton RE, Lawler EV, Lew RA, Brophy MT, Fiore LD, et al. The association between statins and cancer incidence in a veterans population. J Natl Cancer Inst. 2008;100:134–139. doi: 10.1093/jnci/djm286. [DOI] [PubMed] [Google Scholar]
- Feleszko W, Mlynarczuk I, Olszewska D, Jalili A, Grzela T, Lasek W, et al. Lovastatin potentiates antitumour activity of doxorubicin in murine melanoma via an apoptosis dependent mechanism. Int J Cancer. 2002;100:111–118. doi: 10.1002/ijc.10440. [DOI] [PubMed] [Google Scholar]
- Freeman SR, Drake AL, Heilig LF, Graber M, McNealy K, Schilling LM, et al. Statins, fibrates, and melanoma risk: a systematic review and meta-analysis. J Natl Cancer Inst. 2006;98:1538–1546. doi: 10.1093/jnci/djj412. [DOI] [PubMed] [Google Scholar]
- Glynn SA, O'Sullivan D, Eustace AJ, Clynes M, O'Donovan N. The 3-hydroxy-3-methylglutaryl-coenzyme A reductase inhibitors, simvastatin, lovastatin and mevastatin inhibit proliferation and invasion of melanoma cells. BMC Cancer. 2008;8:9. doi: 10.1186/1471-2407-8-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein JL, Brown MS. Regulation of the mevalonate pathway. Nature. 1990;343:425–430. doi: 10.1038/343425a0. [DOI] [PubMed] [Google Scholar]
- Green DR. Apoptotic pathways: ten minutes to dead. Cell. 2005;121:671–674. doi: 10.1016/j.cell.2005.05.019. [DOI] [PubMed] [Google Scholar]
- Igney FH, Krammer PH. Tumour counterattack: fact or fiction? Cancer Immunol Immunother. 2005;54:1127–1136. doi: 10.1007/s00262-005-0680-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karp I, Behlouli H, LeLorier J, Pilote L. Statins and cancer risk. The Am J Med. 2008;121:302–309. doi: 10.1016/j.amjmed.2007.12.011. [DOI] [PubMed] [Google Scholar]
- Keyomarsi K, Sandoval L, Band V, Pardee AB. Synchronization of tumour and normal cells from G1 to multiple cell cycles by lovastatin. Cancer Res. 1991;51:3602–3609. [PubMed] [Google Scholar]
- Koomen ER, Joosse A, Herings RM, Casparie MK, Bergman W, Nijsten T, et al. Is statin use associated with a reduced incidence, a reduced Breslow thickness or delayed metastasis of melanoma of the skin? Eur J Cancer. 2007;43:2580–2589. doi: 10.1016/j.ejca.2007.09.004. [DOI] [PubMed] [Google Scholar]
- Miida T, Hirayama S, Nakamura Y. Cholesterol-independent effects of statins and new therapeutic targets: ischemic stroke and dementia. J Atheroscler Thromb. 2004;11:253–264. doi: 10.5551/jat.11.253. [DOI] [PubMed] [Google Scholar]
- Pan HY, DeVault AR, Wang-Iverson D, Ivashkiv E, Swanson BN, Sugerman AA. Comparative pharmacokinetics and pharmacodynamics of pravastatin and lovastatin. J Clin Pharmacol. 1990;30:1128–1135. doi: 10.1002/j.1552-4604.1990.tb01856.x. [DOI] [PubMed] [Google Scholar]
- Sacher J, Weigl L, Werner M, Szegedi C, Hohenegger M. Delineation of myotoxicity induced by 3-hydroxy-3-methylglutaryl CoA reductase inhibitors in human skeletal muscle cells. J Pharmacol Exp Ther. 2005;314:1032–1041. doi: 10.1124/jpet.105.086462. [DOI] [PubMed] [Google Scholar]
- Saito A, Saito N, Mol W, Furukawa H, Tsutsumida A, Oyama A, et al. Simvastatin inhibits growth via apoptosis and the induction of cell cycle arrest in human melanoma cells. Melanoma Res. 2008;18:85–94. doi: 10.1097/CMR.0b013e3282f60097. [DOI] [PubMed] [Google Scholar]
- Sarrabayrouse G, Synaeve C, Leveque K, Favre G, Tilkin-Mariamé AF. Statins stimulate in vitro membrane FasL expression and lymphocyte apoptosis through RhoA/ROCK pathway in murine melanoma cells. Neoplasia. 2007;9:1078–1090. doi: 10.1593/neo.07727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer ZT, Kornbluth S. The apoptosome: physiological, developmental, and pathological modes of regulation. Dev Cell. 2006;10:549–561. doi: 10.1016/j.devcel.2006.04.008. [DOI] [PubMed] [Google Scholar]
- Shellman YG, Ribble D, Mille L, Gendall J, Vanbuskirk K, Kelly D, et al. Lovastatin-induced apoptosis in human melanoma cell lines. Melanoma Res. 2005;15:83–89. doi: 10.1097/00008390-200504000-00001. [DOI] [PubMed] [Google Scholar]
- Sirvent P, Mercier J, Lacampagne A. New insights into mechanisms of statin-associated myotoxicity. Curr Opin Pharmacol. 2008;8:333–338. doi: 10.1016/j.coph.2007.12.010. [DOI] [PubMed] [Google Scholar]
- Sivaprasad U, Abbas T, Dutta A. Differential efficacy of 3-hydroxy-3-methylglutaryl CoA reductase inhibitors on the cell cycle of prostate cancer cells. Mol Cancer Ther. 2006;5:2310–2316. doi: 10.1158/1535-7163.MCT-06-0175. [DOI] [PubMed] [Google Scholar]
- Soengas MS, Lowe SW. Apoptosis and melanoma chemoresistance. Oncogene. 2003;22:3138–3151. doi: 10.1038/sj.onc.1206454. [DOI] [PubMed] [Google Scholar]
- Thibault A, Samid D, Tompkins AC, Figg WD, Cooper MR, Hohl RJ, et al. Phase I study of lovastatin, an inhibitor of the mevalonate pathway, in patients with cancer. Clin Cancer Res. 1996;2:483–491. [PubMed] [Google Scholar]
- van der Spek E, Bloem AC, van de Donk NW, Bogers LH, van der Griend R, Kramer MH, et al. Dose-finding study of high-dosesimvastatin combined with standard chemotherapy in patients with relapsed or refractory myeloma or lymphoma. Haematologica. 2006;91:542–545. [PubMed] [Google Scholar]
- Werner M, Sacher J, Hohenegger M. Mutual amplification of apoptosis by statin induced mitochondrial stress and doxorubicin toxicity in human rhabdomyosarcoma cells. Brit J Pharm. 2004;143:715–724. doi: 10.1038/sj.bjp.0705928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong WW, Clendening JW, Martirosyan A, Boutros PC, Bros C, Khosravi F, et al. Determinants of sensitivity to lovastatin-induced apoptosis in multiple myeloma. Mol Cancer Ther. 2007;6:1886–1897. doi: 10.1158/1535-7163.MCT-06-0745. [DOI] [PubMed] [Google Scholar]