

Abstract
Solar radiation can lead to changes affecting DNA metabolism resulting in loss of DNA integrity. Skin specimens obtained from melanoma patients treated at the Memorial Sloan-Kettering Cancer Center were used to study patterns of DNA fragmentation using the comet assay and levels of deletions in mitochondrial DNA (mtDNA) using real-time PCR. Skin specimens were classified according to the glutathione S-transferase M1 (GSTM1) genotype (either wild type [WT] or null) and patient sunburn history. GSTM1 null individuals with a sunburn history showed increased levels of both DNA fragmentation by comet assays and mtDNA deletions relative to GSTM1 WT patients with little or no sunburn history. Microarray analyses identified a number of genes whose expression was upregulated ≥5-fold in cells from GSTM1-null patients or from those reporting histories of sunburn. These genes encoded small molecule transporters, various growth factor/chemokine receptors, transcription factors and tumor suppressors. Of 17 genes directly involved in DNA repair, three DNA ligases were highly upregulated while the RAD23 UV excision repair gene and the Growth Arrest and DNA Damage gene (GADD45) were downregulated. These findings support the idea that exposure to solar radiation early in life may induce long-term cellular changes that lead to persistent DNA damage and altered patterns of gene expression.
Introduction
The causative association between sun exposure and skin cancer is well established. In in vitro models, irradiation of epidermal cells with sunlight, or its UV components, has been shown to lead to oncogenic mutations that result from DNA strand breaks, pyrimidine dimer formation and other types of DNA lesions (1). Because the neoplastic changes induced in skin by solar radiation are generally not seen for long periods of time after the initial DNA damage, oncogenic processes induced by sun exposure are evidently active in the intervening period that lead up to tumor formation. Despite much research, there is still little consensus about the nature of these oncogenic processes. Estimates of the levels of sun exposure that increase cancer risk are surprisingly low; some sources estimate that even a single instance of severe sunburn in childhood is associated with an increased risk of developing skin cancer in adulthood, particularly melanoma (2). If so, this suggests that irreversible secondary changes in cellular function that result from exposure to solar radiation in childhood may act synergistically with oncogenic processes that ultimately lead to skin cancers in adulthood. In fact, risk of melanoma increases significantly with exposure to ambient UV mostly when the exposure occurs in the first decade of life (3–5).
Inactivating mutations in genes encoding the group of enzymes that comprise the glutathione S-transferase (GSTs) detoxifying system have been associated with the development of various cancers including skin cancer (6,7). In particular, the glutathione S-transferase M1 (GSTM1) null genotype (homozygous null) has been associated with lung cancer in smokers (8–10) and with bladder, colon, stomach and breast cancers (11–14). GSTM1 mutations have been related to chromosomal abnormalities (9,10,15,16) and a lowered DNA repair capacity in individuals exposed to adducting carcinogens (8,17) and radiation (15). The GSTs are also responsible for detoxification of superoxides, peroxides and hydroxyl radicals; reactive oxygen species (ROS) and their secondary products that are produced by a variety of oxidizing stimuli including UV radiation (6). For this reason loss of GST activity potentiates the DNA-damaging effects of UV radiation.
Here, we tested the hypothesis that high levels of solar radiation could induce changes in DNA metabolism that might result in loss of DNA integrity over long periods of time subsequent to sun exposure. For this purpose, we examined levels of DNA fragmentation in chromosomal DNA and deletions in mitochondrial DNA (mtDNA) in normal human skin tissue from melanoma patients.
Materials and Methods
Skin specimens
Skin tissues (100–325 mg) were obtained from melanoma patients who underwent wide skin excisions as part of their standard care at the Memorial Sloan-Kettering Cancer Center (MSKCC). Study participants signed an informed consent form before donating their tissue and the study was approved by the Institutional Review Board. Information on age, sunburn history and phenotypic characteristics was extracted from self-administered epidemiological questionnaires.
The phenotypic index was calculated by combining indices for hair color, eye color and tannability/propensity to burn characteristics as indicators of cutaneous phenotype as previously described (18). The phenotypic indices were further grouped into three sunburn risk categories (low, intermediate and high). A total of 67 specimens of skin tissue were collected of which 22 were from patients rated at low risk, 23 at intermediate risk and 22 from high-risk patients.
Germline DNA
DNA from buccal cells was used to test germline polymorphisms in the GSTM1 gene. Buccal cells were collected with mouthwashes and DNA from buccal cells was extracted using Puregene® kits (Gentra Systems, Inc., Minneapolis, MN) according to the manufacturer's recommendations. DNA concentration was measured by spectrophotometry at 260 nm in a Spectramax Plus 384 (Molecular Devices, Sunnyvale, CA). The DNA quality was determined by the ratio A260/A280.
Analysis of germline GSTM1 variants
Multiplex PCR is used to identify GSTM1 genotypes (7,19,20). The null polymorphism in GSTM1 was detected by multiplex PCR, which consisted of the simultaneous amplification of the GSTM1 fragment and a housekeeping gene fragment (internal control). The PCR products were visualized on agarose gels. The absence of the GSTM1-specific band (273 bp) in the presence of microsomal epoxide hydrolase (EPHX1) used as the internal control band indicated that the sample was indeed homozygous null. The internal control fragments amplified were 322 bp. The PCR reaction included 10–20 ng of DNA which was amplified in a reaction mix containing 200 μm dNTPs, 0.3 μm GSTM1-specific primers (forward 5′-CTGCCCTACTTGATTGATGGG; reverse 5′-CTGGATTGTAGCAGATCATGC); 0.4 μm forward and reverse control EPHX1 primers (forward 5′-TCCCTCTCAACTTGGGGTC; reverse 5′-TTGGGTTCTGAATCTCTCCAA), 2.0 mm MgCl2, 1 m betaine, 0.06 U μL−1 Taq polymerase and 1× PCR buffer (Applied Biosystems, Foster City, CA). For PCR an initial denaturation at 95°C for 2 min was carried out. This was followed by a touchdown PCR protocol with denaturation segments of 95°C for 20 s and extension at 72°C for 40 s. The annealing segments were 65°C for two cycles, 63°C for two cycles, 61°C for two cycles and 60°C for 29 cycles (30 s each). A final extension was carried out at 72°C for 10 min.
Real-time PCR for quantification of mtDNA deletions
QIAamp spin columns (Qiagen, Valencia, CA) were used according to the manufacturer's specifications to extract and purify DNA from about 25 mg of skin adjacent to the excised melanomas. DNA was eluted from the columns and suspended at a final concentration of 100 ng μL−1 in 10 mm Tris, 0.1 mm EDTA pH 7.5. The Roche LightCycler for Real Time PCR was employed to measure total mitochondria and the common deletion (CD). Each PCR capillary contained 1× LightCycler® FastStart DNA Master Mix and 300 nm each of the forward and reverse primers, 200 nm of the FAM-labeled probe and 0.1 U heat-labile uracil-DNA glycosylase (Roche Applied Science) in a total volume of 10 μL.
We quantified two types of mtDNA deletions: (1) the CD comprising 4977 bp is located between bp 8470 and bp 13 447 of the revised Cambridge reference sequence (GenBank AC_000021.2) originally published by Anderson et al. (21) and (2) the recently characterized 5128 bp deletion called Δuv (22). The primers and probes for the CD are described by Pogozelski et al. (23). The forward primer for the CD spans the mtDNA sequence from bp 8416 to bp 8437 and the reverse primer spans the sequence from bp 13 519 to bp 13 498. The probe for the ensuing amplicon is located at bp 13 461–13 480 of the Cambridge sequence and is tagged with the reporter FAM (6-carboxyfluorescein, λmax 518 nm) at the 5′-end and the quencher BHQ1 at the 3′-end. Similarly, the primers for total mtDNA are located at bp 1307–1328 for the forward primer and bp 1433–1414 for the reverse primer. The total amplicon spans a region of the mtDNA that is rarely deleted (23). The total mitochondria probe is located at bp 1340 to 1359 of the Cambridge sequence. Pogozelski et al. (23) also designed two plasmids, one that contains the amplicon for the CD and the other that contains the amplicon for total mitochondria. In each run, as appropriate, the plasmids were run as external controls. The mtDNA copy number per cell (either deleted or intact) was calculated assuming 16 pg of DNA per cell. For the Δuv deletion, the primers used were MT1A and MT2 described by Soong and Arnheim (24) and a 20 bp probe (5′ TTTGAAATATCCACAACCTT 3′) derived from the sequences flanking the deletion cut site. Threshold cycle (Ct) was used to quantify relative levels of deletions in the various samples. These values were then normalized to the total number of mitochondria in each sample to give the final CD and Δuv values.
Explant cultures of cells from skin tissues
Keratinocyte monolayers were grown out from tissue explants and used for the comet assays and microarray analyses. For this purpose, fat was trimmed, tissue washed with 70% ethanol and rinsed twice with serum-free DMEM medium with 0.5 μg hydrocortisone and 5× penicillin/streptomycin. Tissue was minced under sterile conditions into 1–2 mm pieces, transferred to a T75 flask and allowed to dry. After an hour, complete medium was added (DMEM with fetal calf serum, hydrocortisone, 1× penicillin/streptomycin, cholera toxin and EGF). Minced tissue and growing cells were monitored by microscopic examination daily and fibroblasts removed as needed. After 4–6 weeks, keratinocytes were dissociated with 0.25% trypsin and 1.0% collagenase and then stored with complete DMEM media with 10% DMSO in liquid nitrogen for the comet assay.
Alkaline single-cell gel electrophoresis (comet assay)
The comet assay was performed under alkaline conditions as described by Singh et al. (25) with minor modifications. Briefly, superfrosted and commercially precleaned microscope slides (Fisher Scientific, Pittsburgh, PA) were immersed in hot 1% normal-melting-point agarose dissolved in PBS (pH 7.4) and allowed to dry at 37°C on a flat surface for 60 min. Cells were quickly thawed and washed with PBS. Five to ten microliters of cell suspension containing approximately 15 000 cells were mixed with 80 μL of 0.5% low-melting-point agarose in PBS, kept at 37°C in a dry-bath incubator, spread on an agarose precoated slide and covered with a coverslip. The agarose was solidified at 4°C for 10 min. The coverslip was taken off and a third layer of 0.5% low-melting-point agarose was added and covered by a new coverslip. After setting the agarose at 4°C for 10 min and taking the coverslip off, the slides were incubated in complete media for 45 min at 37°C and then lysed for 1 h at 4°C in a freshly prepared lysis buffer (pH 10) containing 2.5 m NaCl, 100 mm EDTA, 100 mm Trizma base, 10% dimethyl sulfoxide and 1% Triton X-100. After lysis, the slides were rinsed three times in 0.4 m Tris-HCl (pH 7.5) for 5 min to remove detergents and salts. The slides were immediately placed side by side on a horizontal electrophoretic unit without power for 30 min in freshly prepared alkaline buffer (300 mm NaOH and 1 mm EDTA, pH > 13) at 4°C. Electrophoresis was carried out in a TECA2222 electrophoresis unit (Ellard Instrumentation Ltd., Monrow, WA) for 30 min at 25 V, adjusting the current to 295–300 mA and with a constant recirculating flow of 100 mL min−1. All these steps were carried out in the dark. Finally, the slides were rinsed in neutralization buffer (0.4 m Tris-HCl, pH 7.5) for 5 min ×3 times, fixed in cold 100% ethanol for 15 min and subsequently air dried. The slides were stored overnight in the dark at room temperature.
Scoring of DNA damage
Immediately before imaging analysis, slides were stained with 60 μL of a 1 μg mL−1 ethidium bromide solution for 10 min and covered with coverslips. Evaluations were made at 400× magnification using a fluorescent microscope (Axioskop, Zeiss, equipped with a 515–560 nm excitation filter, a 590 nm barrier filter and a Anchrophan 20/0.45 Ph2 filter) connected to a CoHu 4912 CCD camera. One hundred consecutive cells (50 from each duplicate slide) were randomly selected with care to avoid borders, and quantified with Optomax Comet Assay III image analysis software (Hollis, NH). The extent of the damage was measured quantitatively by the tail moment (TM), defined as the product of the percentage of DNA in the comet tail and the tail length (26), and the tail intensity (TI), defined as the percentage of DNA in the tail. Results were expressed as the mean and standard deviations of TMs and TIs of 200 cells from four slides from each of two separate cultures and two independent assays.
Statistical methods
Data from multiple samples were collected and grouped according to the criteria described in the figures and the standard error of the mean was computed for each group. Statistical significance for paired data points was carried out using Student's t-test.
Microarray analysis
To study gene expression we used a total human 1.2 cDNA microarray (BD Biosciences, Clontech, Inc.; no. 7850-1, 1176 genes). RNA was isolated from cells using a standard guanidine-phenol extraction protocol with reagents supplied as a kit (RNagents; Promega Biotec, Madison, WI). 32P-labeled cDNA probes were created by reverse transcription from the RNA templates using primers, reagents and protocols provided with the cDNA arrays. Hybridizations of the arrays were performed at 68°C at high stringency using protocols and reagents supplied by the manufacturer. For gene expression analysis autoradiographic signals stored on phosphor storage screens were digitized using a Storm 840 phosphoimager (Molecular Dynamics) and analyzed in either TIFF or GEL formats. Relative levels of gene expression from globally normalized hybridization signal intensities for individual gene sequences were then determined using AtlasImage 2.0 software (BD Biosciences-Clontech, Inc.).
Results
DNA damage vs GSTM1 status
The comet assay was used to examine the extent of endogenous DNA breaks in epithelial cell outgrowths from the patient skin specimens taken from wide excisions of melanomas and grown in explant culture. As GSTM1 modulates the extent of damaged DNA that needs to be repaired, we compared TM and TI values in skin specimens from patients grouped according to either the GSTM1 WT or null genotypes (Fig. 1). DNA isolated from individuals with the null genotype exhibited a much higher degree of fragmentation than the WT. Specifically, there was at least a two-fold increase in the DNA damage for the null group compared with WT when the TM measures were compared. Similarly, the average TIs indicated that the percentage of DNA in the tail was considerably higher (∼50%) in the GSTM null cells than in the WT.
Figure 1.
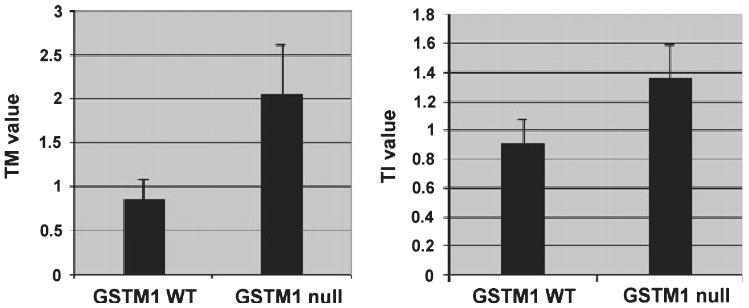
Endogenous DNA damage in cultured keratinocytes derived from patients of WT and null GSTM1 genotype. Comet assays were performed on keratinocyte outgrowths from skin samples from patients who were genotyped as either GSTM1 wild type (WT; n = 11) or GSTM1 null (n = 12) as described in Materials and Methods. Average tail moment (TM; left panel) and tail intensity (TI) values (right panel) are shown (±SEM). The TM values are expressed in arbitrary units. The TI reflects % DNA in the tail. Differences in the mean values were found to be significant at P < 0.05 using Student's t-test.
In parallel experiments, levels of mtDNA deletions in the GSTM1 null and WT groups were quantitated by real-time PCR as separate indicators of DNA damage. We used primers for two types of mtDNA deletions: the well-characterized 4977 bp deletion known as the CD and a larger, recently discovered deletion of 5128 bp induced by UVB radiation of human keratinocytes in vitro (22). The results of these experiments were similar to those of the comet assays in that both types of mtDNA deletions were more prevalent in the GSTM1 null group compared with WT but the differences (more than six-fold) were much more pronounced for the CD (Fig. 2). The CD is known to accumulate in tissues with age (27–29). Although the patient population varied considerably in age ranging from 16 to 85 years, the average and median ages of patients in the GSTM1 null and WT groups was essentially the same (Fig. 3; mean ages of 58.1 vs 56.0 years and median ages of 59.0 vs 55.0 years for null and WT, respectively) indicating that the differences seen were related to the GSTM1 genotype and not to differences in age.
Figure 2.

Mitochondrial DNA deletions in cultured keratinocytes derived from patients of WT and null GSTM1 genotype. Levels of mitochondrial DNA deletions were quantitated by real-time PCR as described in Materials and Methods. The relative levels of the CD (black bars) and Δuv (white bars) normalized to undeleted genomes in skin samples from patients who were genotyped as either GSTM1 WT (n = 14) or GSTM1 null (n = 17) are indicated (±SEM).
Figure 3.
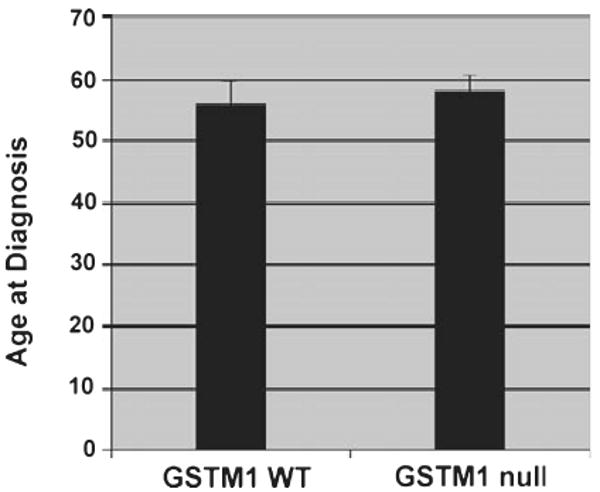
Average age of patients at diagnosis of melanoma grouped according to WT (n = 26) and null (n = 34) GSTM1 genotype (±SEM).
History of sun exposure
As we hypothesized that previous exposure to solar radiation might initiate processes that lead to persistent, long-term DNA damage, we used questionnaire responses regarding history of sunburn to categorize patients into low propensity to burn (0–1 severe sunburns) and high exposure to sun exposure (2–7 severe sunburns) histories. As expected, DNA fragmentation measured in the comet assays was found to be greater in the high sensitivity group according to both TM and TI (Fig. 4). There were also substantial differences in the levels of mtDNA deletions (Fig. 5). The CD was found to be more than 10 times as prevalent in the high exposure group as the low exposure group while the levels of the Δuv deletion were almost 3.5-fold higher in the high exposure group in comparison with the low exposure group. The average age of patients in the two groups (55.3 vs 53.9 years for the low and high sunburn groups respectively) was virtually the same (Fig. 6).
Figure 4.
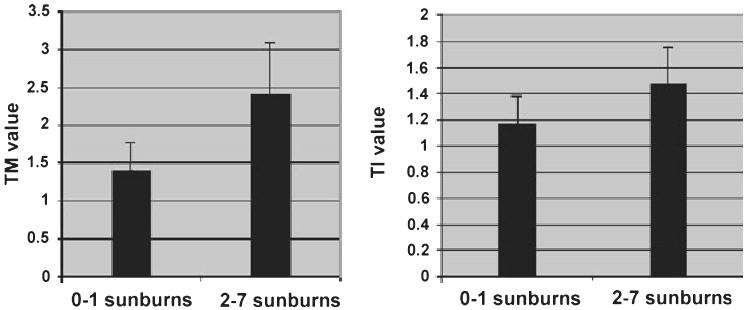
Comet assays of cultured keratinocytes derived from patients grouped according to high (2–7 severe sunburns; n = 11) and low (0–1 severe sunburns; n = 7) sunburn history. Average tail moment (TM; left panel) and tail intensity (TI) values (right panel) are shown (±SEM).
Figure 5.
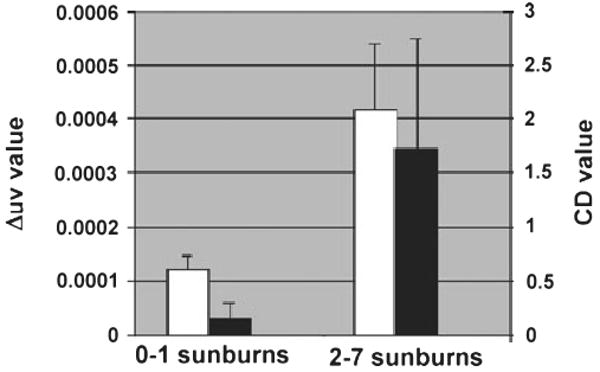
Mitochondrial DNA deletions in cultured keratinocytes derived from patients grouped according to high and low sunburn history. The relative levels of the CD (black bars) and Δuv (white bars) normalized to undeleted genomes in skin samples from patients who reported either 0–1 severe sunburns (n = 7) or 2–7 severe sunburns (n = 19) are indicated (±SEM).
Figure 6.
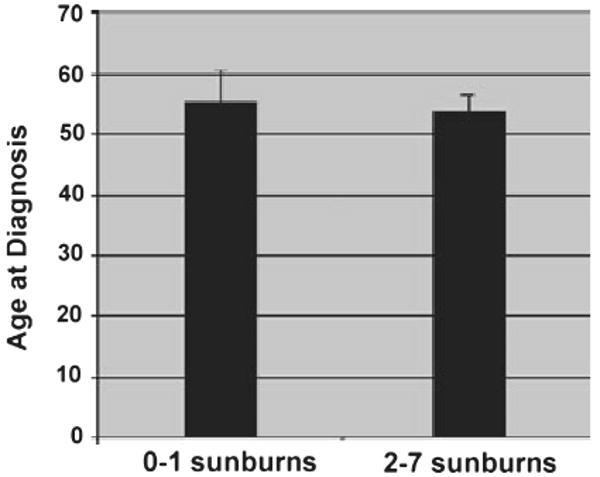
Average age of patients at diagnosis of melanoma grouped according to high (n = 28) or low (n = 12) history of severe sunburn as in Fig. 5 (±SEM).
A relationship between sun exposures and mtDNA deletions was also borne out when the patient population was separated into subgroups according to the body site location of the melanoma (data not shown). Both types of mtDNA deletion were more prevalent in skin specimens taken from the arm or shoulder area than in skin from either the leg⁄hip area or trunk. In fact the levels of the CD were more than 46 times higher in skin specimens taken from the arm⁄shoulder than in skin from the trunk. When compared with the leg⁄hip regions, the frequency of the CD was about 5.2-fold greater in skin from the arm or shoulder. The Δuv deletion also showed similar differences in frequency according to body location, although they were not as striking as for the CD. The Δuv deletions in skin specimens from the arm⁄shoulder were about twice as prevalent compared with deletions in skin taken from the trunk, and when compared with skin taken from leg⁄hip areas the ratio was about 2.4.
Microarray studies
RNAs from keratinocytes growing out from skin explant cultures were used to create cDNA probes for microarray analyses of gene expression. To compare patterns of gene expression related to the GSTM1 genotype, relative levels of gene expression in arrays created from patients of each genotype were determined and the expression ratios (null ⁄WT) were calculated. For gene expression related to sun exposure, the ratio of relative levels of gene expression from the high and low sun exposure groups was compared (high⁄low). Table 1 shows comparison of genes whose expression levels differed by >5-fold according to either GSTM1 genotype or sun exposure history. A number of genes representing a variety of functional classes exhibited wide differences in expression according to GSTM1 genotype or sun exposure. There was a striking concordance in that genes showing the largest differences in expression, when compared according to GSTM1, also showed the largest differences in expression when arrays were grouped according to sun exposure. Of the genes whose expression was modulated five-fold or more, almost all showed higher comparative levels of expression in the GSTM1 null or high sunburn group; only two genes (glucagons and the transcription factor ASH1) showed lower expression in both the GSTM1 null and high sunburn groups.
Table 1.
Relative expression levels of genes in patients grouped according to GSTM1 genotype and sunburn history.
| Ratio null⁄WT* | Ratio high/low sunburns* | Protein (gene) | |
|---|---|---|---|
| Transporters | 1360 | 34.9 | Erythrocyte glucose transporter 1 (GLUT1) |
| 218 | 183 | Sodium- and chloride-dependent GABA transporter 3 | |
| 45.4 | 18.9 | Copper-transporting ATPase 2 | |
| 28.8 | 39.0 | Vesicular acetylcholine transporter (VAChT) | |
| 5.46 | 15.5 | High-affinity glutamate transporter | |
| 36.0 | 25.7 | Organic cation transporter 1 | |
| 23.8 | 15.1 | Cardiac muscle sodium channel alpha subunit; HH1 | |
| 10.4 | 11.2 | Zinc transporter 4 | |
| Cell cycle control | 673 | 3820 | wee1Hu CDK tyrosine 15-kinase; wee-1-like protein kinase |
| 107 | 129 | Cyclin-dependent kinase inhibitor 1C (CDKN1C); p57-KIP2 | |
| 6.58 | 5.21 | G1⁄S-specific cyclin D3 (CCND3) | |
| Receptors | 304 | 91.0 | High-affinity nerve growth factor receptor precursor; trk-1 |
| 111 | 97.4 | Transforming growth factor beta receptor III | |
| 69.3 | 59.9 | CC chemokine receptor type 1 (CC CKR1; CCR 1) | |
| 37.0 | 24.3 | Colon carcinoma kinase 4 precursor (CCK4) | |
| 28.6 | 12.2 | Angiopoietin 1 receptor precursor | |
| 23.7 | 17.4 | Macrophage-stimulating protein receptor precursor | |
| 20.4 | 27.1 | Vascular endothelial growth factor receptor 2 precursor (VEGFR2) | |
| 18.2 | 11.1 | N-sam; fibroblast growth factor receptor 1 precursor (FGFR1) | |
| 17.9 | 16.6 | ERBB4 receptor protein-tyrosine kinase; Her4 | |
| 13.1 | 5.84 | Triiodothyronine receptor (THRA1) | |
| 12.3 | 9.79 | ERBB2 receptor protein-tyrosine kinase | |
| 10.9 | 7.47 | Insulin-like growth factor 1 receptor (IGF1R) | |
| 6.03 | 6.78 | Vascular endothelial growth factor receptor 1 (VEGFR1) | |
| 5.10 | 8.49 | Vascular endothelial growth factor receptor 3 precursor (VEGFR3) | |
| Signaling intermediates | 162 | 49.7 | cAMP-dependent 3′,5′-cyclic phosphodiesterase 4D (PDE43); DPDE3 |
| 35.2 | 11.4 | Dual-specificity protein phosphatase 2; PAC-1 | |
| 24.2 | 8.56 | Ribosomal protein kinase B (RSKB) | |
| 23.1 | 6.14 | Myotubularin | |
| 20.7 | 11.0 | Proto-oncogene tyrosine-protein kinase lck; p56-lck | |
| 20.4 | 13.7 | Tyrosine-protein kinase lyn | |
| 9.53 | 5.22 | Anaplastic lymphoma kinase GN (ALK) | |
| 6.74 | 5.73 | T-lymphoma invasion and metastasis inducing TIAM1 | |
| 5.53 | 8.90 | Myosin light chain kinase (MLCK) | |
| Transcription factors | 140 | 68.8 | Breast cancer type 2 susceptibility protein (BRCA2) |
| 43.7 | 46.1 | Signal transducer and activator of transcription 1 alpha⁄beta (STAT1) | |
| 13.3 | 11.4 | Transcription factor DP2 (Humdp2); E2F dimerization partner 2 | |
| 9.37 | 5.36 | Signal transducer and activator of transcription 2 (STAT2); p113 | |
| 9.19 | 8.29 | Human immunodeficiency virus type I enhancer-binding protein 2 (HIV-EP2) | |
| 8.90 | 9.11 | Homeobox protein hLim1; LHX1 | |
| 8.05 | 6.34 | Endothelial transcription factor GATA2 | |
| 7.63 | 11.6 | Homeobox protein HOX-A5; HOX-1C | |
| Growth factors | 28.0 | 46.8 | Brain-derived neurotrophic factor (BDNF)⁄NT-3 |
| 9.66 | 6.11 | Macrophage colony-stimulating factor I receptor precursor (CSF-1-R) | |
| 8.89 | 7.92 | Transforming growth factor beta2 precursor (TGF-beta2; TGFB2) | |
| Intermediary metabolism | 43.1 | 21.0 | Lactotransferrin precursor; lactoferrin |
| 29.5 | 19.4 | Pyruvate dehydrogenase kinase kinase precursor | |
| Tumor suppressors | 58.5 | 24.6 | p53 cellular tumor antigen |
| 34.8 | 21.6 | Retinoblastoma-like protein 2 (RBL2; RB2) | |
| 19.6 | 11.3 | Retinoblastoma-associated protein (RB1); PP110; P105-RB | |
| 9.53 | 8.01 | LUCA15 putative tumor suppressor | |
| Oncogenes | 19.5 | 6.46 | C-fes proto-oncogene |
| 15.9 | 8.65 | H-ras proto-oncogene; transforming G protein | |
| 7.31 | 16.7 | C-mos proto-oncogene serine⁄threonine-protein kinase | |
| Apoptosis | 50.1 | 33.8 | DAXX |
| 14.9 | 13.3 | bcl-2 interacting killer (BIK); NBK apoptotic inducer protein; BP4; BIP1 | |
| Hormones | 5.61 | 10.2 | Leptin precursor; obesity factor; obese protein |
| Translation factors | 26.0 | 42.9 | NIP1 (NIP1) |
Values are the averaged relative expression levels of genes in microarrays for keratinocyte outgrowths from skin specimens from patients grouped by GSTM1 genotype (for GSTM1 WT the values shown represent the average of six microarrays; for the null genotype values represent the average of seven microarrays) or sunburn history as indicated. Only ratios of five-fold or greater in magnitude are shown.
Of the genes screened, 25 represented genes whose products are directly involved in various aspects of DNA repair. As the comet assays and mtDNA deletion experiments indicated that processes involved in DNA repair might be altered in GSTM1-null individuals or those with histories of exposure to solar radiation, we separately examined the levels of expression of DNA repair genes (Table 2). When microarrays were grouped according to GSTM1 genotype, sunburn exposure was evenly represented within the GSTM1 null and WT subgroups (for the null and WT groups the average numbers of severe sunburns were 1.9 and 1.7, respectively). The pattern of expression of DNA repair genes for the GSTM1 null to WT ratios paralleled that for the ratios of high to low sunburn. The greatest differences in gene expression were seen in the DNA ligases which showed average expression ratios of about seven- and 183-fold. The large majority of the DNA repair genes showed either greater expression in GSTM1 null (vs WT) or in high sun exposure (vs low sun exposure) or were not significantly different in expression. Perhaps significantly, however, expression of the UV excision repair protein, RAD23A, showed about 70% reduced expression in the GSTM1-null patients and almost 50% reduced expression in the high sunburn group.
Table 2.
Averaged expression levels of DNA repair genes in patients grouped according to GSTM1 genotype and sunburn history.
| Protein (gene) | Ratio null⁄WT | Ratio high⁄low sunburns |
|---|---|---|
| DNA ligase IV (LIG4) | 145 | 183 |
| DNA ligase I (LIG1) | 32.0 | 35.4 |
| DNA ligase III (LIG3) | 7.71 | 6.78 |
| Uracil-DNA glycosylase (UNG1) | 6.46 | 6.34 |
| Growth arrest and DNA damage-inducible protein 153 (GADD153) | 3.55 | 4.83 |
| recA-like protein HsRad51 | 3.08 | 1.5 |
| DNA repair protein XRCC1 | 1.48 | 1.02 |
| DNA lyase; AP endonuclease 1 | 1.20 | 2.02 |
| DNA mismatch repair protein PMS1 | 1.20 | 0.62 |
| DNA mismatch repair protein MLH1; COCA2 | 0.89 | 0.97 |
| DNA excision repair protein ERCC2 | 0.84 | 2.08 |
| Repair complementing protein p58⁄HHR23B | 0.82 | 1.22 |
| DNA excision repair protein ERCC5 | 0.64 | 0.9 |
| Growth arrest and DNA damage-inducible protein (GADD45) | 0.59 | 0.89 |
| DNA excision repair protein ERCC3 | 0.58 | 0.93 |
| HHR23A; UV excision repair protein RAD23A | 0.53 | 0.3 |
| Growth arrest and DNA damage-inducible protein 45 beta (GADD45 beta) | 0.17 | 0.58 |
Discussion
The studies described here were designed to test the hypothesis that patterns of DNA damage related to solar radiation may persist in sun-damaged skin over long periods of time. The results of the comet assays and real-time PCR quantitation of mtDNA deletions showed a correlation between sun exposure and increased levels of DNA damage. For mtDNA deletion analyses we used primers specific for two separate mtDNA deletions; the CD and a recently described deletion called Δuv on the basis of the observation that its formation was found to be induced by UV radiation in a line of keratinocytes (22). It was anticipated that Δuv would be a better indicator of UV DNA damage than CD. Although relative levels of Δuv were greater in subjects who experienced more sunburn, the differences were not as great as those seen for the CD. When the occurrence of deletions was compared by body site the CD also showed much greater differences between sun-exposed vs unexposed body site than Δuv (Fig. 5). Eshaghian et al. (30) have previously reported that mtDNA deletions in skin accumulate with age. We also observed an age-related increase in mtDNA deletions, although the difference was more striking for the CD than for Δuv. For example, for patients aged 51–85 years of age vs those diagnosed between 16 and 50 years of age, the CD was approximately six times more prevalent in the older population while for Δuv the age-related increase was only about 71% (data not shown). GSTM1-null subjects also exhibited a higher degree of DNA damage than GSTM1 WT subjects but, again, the differences in levels of the CD were much greater than those seen for the Δuv, although both deletions were more prevalent in the GSTM1 null population. This observation is consistent with the role of GSTs in detoxifying ROS. ROS can be produced by UV radiation and are known to produce deletions in mtDNA as well as cause single stranded DNA breaks as seen in the comet assays (31–33).
Such long-term effects necessarily involve changes in gene expression in that they must be passed on to daughter cells during the process of cell turnover that occurs over periods of years. The microarray analyses were undertaken with a view toward elucidating relative gene expression levels that may identify important target genes. The tumor suppressors p53 and pRb as well as the cell cycle checkpoint regulator, Wee1Hu were relatively upregulated in GSTM1 null and high sun exposure groups and this is consistent with the role of these proteins as negative regulators of the cell cycle in response to DNA damage. The relative differences in expression of many of the other classes of genes, e.g. the transporters and receptors, is much less clear. It is likely that many of these play no causal role in solar-induced oncogenic processes but will turn out to be useful as markers of sun damage. Interestingly, of the array genes directly involved in DNA repair, only the UV excision repair genes RAD23A and GADD45 showed reduced expression in GSTM1 null relative to WT or in high vs low sunburn patients. In contrast, expression levels of DNA ligases, which might be expected to act to reduce the DNA fragmentation seen in the comet assays, were relatively enhanced. However, GST gene expression levels were also relatively lower in the high vs low sunburn patient group (data not shown), particularly GST theta 1 (GSTT1) which was only about 54% (high vs low sunburn). Increased DNA fragmentation might be caused by increased ROS levels when GST activity is lowered. Mitochondrial deletions are thought to reflect increased levels of genotoxic effectors such as ROS rather than loss of DNA repair function (34–37).
Taken together, these findings support the idea that exposure to solar radiation can result in persistent DNA damage and that these long-term effects are paralleled by changes in gene expression.
Acknowledgments
The authors gratefully acknowledge Dr. Marianne Berwick for her intellectual contribution during the developmental and early stages of this study, Ami Patel for data management, the Melanoma Disease Management Team (DMT) for accrual of patients, pathology review and tissue procurement, and Christine Hanlon for managing and maintaining the DMT database at MSKCC. This work was supported by grants GM008168, GM56833, U56 CA096299 and RCMI grant RR3060 from the National Institutes of Health and a PSC-CUNY grant from the State of New York.
References
- 1.Mouret S, Baudouin C, Charveron M, Favier A, Cadet J, Douki T. Cyclobutane pyrimidine dimers are predominant DNA lesions in whole human skin exposed to UVA radiation. Proc Natl Acad Sci USA. 2006;103(37):13765–13770. doi: 10.1073/pnas.0604213103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kennedy C, Bajdik CD, Willemze R, De Gruijl FR, Bouwes Bavinck JN. The influence of painful sunburns and lifetime sun exposure on the risk of actinic keratoses, seborrheic warts, melanocytic nevi, atypical nevi, and skin cancer. J Invest Dermatol. 2003;120:1087–1093. doi: 10.1046/j.1523-1747.2003.12246.x. [DOI] [PubMed] [Google Scholar]
- 3.Holman CD, Armstrong BK. Cutaneous malignant melanoma and indicators of total accumulated exposure to the sun: An analysis separating histogenetic types. J Natl Cancer Inst. 1984;73(1):75–82. [PubMed] [Google Scholar]
- 4.Whiteman DC, Whiteman CA, Green AC. Childhood sun exposure as a risk factor for melanoma: A systematic review of epidemiologic studies. Cancer Causes Control. 2001;12(1):69–82. doi: 10.1023/a:1008980919928. [DOI] [PubMed] [Google Scholar]
- 5.Kricker A, Armstrong BK, Goumas C, Litchfield M, Begg CB, Hummer AJ, Marrett LD, Theis B, Millikan RC, Thomas N, Culver HA, Gallagher RP, Dwyer T, Rebbeck TR, Kanetsky PA, Busam K, From L, Mujumdar U, Zanetti R, Berwick M. Ambient UV, personal sun exposure and risk of multiple primary melanomas. Cancer Causes Control. 2007;18:295–304. doi: 10.1007/s10552-006-0091-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lear JT, Smith AG, Strange RC, Fryer AA. Detoxifying enzyme genotypes and susceptibility to cutaneous malignancy. Br J Dermatol. 2000;142:8–15. doi: 10.1046/j.1365-2133.2000.03339.x. [DOI] [PubMed] [Google Scholar]
- 7.Kanetsky PA, Holmes R, Walker A, Najarian D, Swoyer J, Guerry D, Halpern A, Rebbeck TR. Interaction of glutathione S-transferase M1 and T1 genotypes and malignant melanoma. Cancer Epidemiol Biomarkers Prev. 2001;10:509–513. [PubMed] [Google Scholar]
- 8.Norppa H. Genetic susceptibility, biomarker responses, and cancer. Mutat Res. 2003;544:339–348. doi: 10.1016/j.mrrev.2003.09.006. [DOI] [PubMed] [Google Scholar]
- 9.Norppa H. Cytogenetic biomarkers and genetic polymorphisms. Toxicol Lett. 2004;149:309–334. doi: 10.1016/j.toxlet.2003.12.042. [DOI] [PubMed] [Google Scholar]
- 10.Norppa H. Cytogenetic biomarkers. IARC Sci Publ. 2004;157:179–205. [PubMed] [Google Scholar]
- 11.Parl FF. Glutathione S-transferase genotypes and cancer risk. Cancer Lett. 2005;221:123–129. doi: 10.1016/j.canlet.2004.06.016. [DOI] [PubMed] [Google Scholar]
- 12.Huang K, Sandler RS, Millikan RC, Schroeder JC, North KE, Hu J. GSTM1 and GSTT1 polymorphisms, cigarette smoking, and risk of colon cancer: A population-based case–control study in North Carolina (United States) Cancer Causes Control. 2006;17:385–394. doi: 10.1007/s10552-005-0424-1. [DOI] [PubMed] [Google Scholar]
- 13.Martinez C, Martin F, Fernandez JM, Garcia-Martin E, Sastre J, Diaz-Rubio M, Agundez JA, Ladero JM. Glutathione S-transferases mu 1, theta 1, pi 1, alpha 1 and mu 3 genetic polymorphisms and the risk of colorectal and gastric cancers in humans. Pharmacogenomics. 2006;5:711–718. doi: 10.2217/14622416.7.5.711. [DOI] [PubMed] [Google Scholar]
- 14.Sadat M. Genetic polymorphisms of glutathione S-transferase T1 (GSTT1) and susceptibility to gastric cancer: A meta-analysis. Cancer Sci. 2006;6:505–509. doi: 10.1111/j.1349-7006.2006.00207.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Karahalil B, Sardas S, Kocabas NA, Alhayiroglu E, Karakaya AE, Civelek E. Chromosomal aberrations under basal conditions and after treatment with X-ray in human lymphocytes as related to the GSTM1 genotype. Mutat Res. 2002;515:135–140. doi: 10.1016/s1383-5718(02)00003-7. [DOI] [PubMed] [Google Scholar]
- 16.Au WW. Heritable susceptibility factors for the development of cancer. J Radiat Res (Tokyo) 2006;47(Suppl B):13–17. doi: 10.1269/jrr.47.b13. [DOI] [PubMed] [Google Scholar]
- 17.Pavanello S, Pulliero A, Siwinska E, Mielzynska D, Clonfero E. Reduced nucleotide excision repair and GSTM1-null genotypes influence anti-B[a]PDE-DNA adduct levels in mononuclear white blood cells of highly PAH-exposed coke oven workers. Carcinogenesis. 2005;26:169–175. doi: 10.1093/carcin/bgh303. [DOI] [PubMed] [Google Scholar]
- 18.Kanetsky PA, Rebbeck TR, Hummer AJ, Panossian S, Armstrong BK, Kricker A, Marrett LD, Millikan RC, Gruber SB, Culver HA, Zanetti R, Gallagher RP, Dwyer T, Busam K, From L, Mujumdar U, Wilcox H, Begg CB, Berwick M. Population-based study of natural variation in the melanocortin-1 receptor gene and melanoma. Cancer Res. 2006;66:9330–9337. doi: 10.1158/0008-5472.CAN-06-1634. [DOI] [PubMed] [Google Scholar]
- 19.Kang JH, Oh Y, Chun SM, Seo JY, HeeYoung Shin HY, Kim CW, Ahn HS, Han BD. TotalPlex gene amplification using bulging primers for pharmacogenetic analysis of acute lymphoblastic leukemia. Mol Cell Probes. 2008;22:193–200. doi: 10.1016/j.mcp.2008.02.001. [DOI] [PubMed] [Google Scholar]
- 20.Kadouri L, Kote-Jarai Z, Hubert A, Baras M, Abeliovich D, Hamburger T, Peretz T, Eeles RA. Glutathione S-transferase M1, T1 and P1 polymorphisms, and breast cancer risk, in BRCA1⁄2 mutation carriers. Br J Cancer. 2008;98:2006–2010. doi: 10.1038/sj.bjc.6604394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Anderson S, Bankier AT, Barrell BG, de Bruijn MH, Coulson AR, Drouin J, Eperon IC, Nierlich DP, Roe BA, Sanger F, Schreier PH, Smith AJ, Staden R, Young IG. Sequence and organization of the human mitochondrial genome. Nature. 1981;290:457–465. doi: 10.1038/290457a0. [DOI] [PubMed] [Google Scholar]
- 22.Fang J, Pierre Z, Liu S, Hwang BJ, Hill HZ, Hubbard K, Steinberg M. Novel mitochondrial deletions in human epithelial cells irradiated with an FS20 ultraviolet light source in vitro. J Photochem Photobiol. 2006;184:340–346. [Google Scholar]
- 23.Pogozelski WK, Hamel CJ, Woeller CF, Jackson WE, Zullo SJ, Fischel-Ghodsian N, Blakely WF. Quantification of total mitochondrial DNA and the 4977-bp common deletion in Pearson's syndrome lymphoblasts using a fluorogenic 5′-nuclease (TaqMan) real-time polymerase chain reaction assay and plasmid external calibration standards. Mitochondrion. 2003;2:415–427. doi: 10.1016/S1567-7249(03)00033-3. [DOI] [PubMed] [Google Scholar]
- 24.Soong NW, Arnheim N. Detection and quantification of mitochondrial DNA deletions. Methods Enzymol. 1996;264:421–431. doi: 10.1016/s0076-6879(96)64038-5. [DOI] [PubMed] [Google Scholar]
- 25.Singh NP, McCoy MT, Tice RR, Schneider EL. A simple technique for quantitation of low levels of DNA damage in individuals cells. Exp Cell Res. 1988;175(1):184–191. doi: 10.1016/0014-4827(88)90265-0. [DOI] [PubMed] [Google Scholar]
- 26.Orlow I, Park BJ, Mujumdar U, Patel H, Siu-Lau P, Clas BA, Downey R, Flores R, Bains M, Rizk N, Dominguez G, Jani J, Berwick M, Begg CB, Kris MG, Rusch V. DNA damage and repair capacity in patients with lung cancer: Prediction of multiple primary tumors. J Clin Oncol. 2008;26:3560–3566. doi: 10.1200/JCO.2007.13.2654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Simonetti S, Chen X, DiMauro S, Schon EA. Accumulation of deletions in human mitochondrial DNA during normal aging: Analysis by quantitative PCR. Biochim Biophys Acta. 1992;1180:113–122. doi: 10.1016/0925-4439(92)90059-v. [DOI] [PubMed] [Google Scholar]
- 28.Wei YH, Lee HC. Oxidative stress, mitochondrial DNA mutation, and impairment of antioxidant enzymes in aging. Exp Biol Med. 2002;227:671–682. doi: 10.1177/153537020222700901. [DOI] [PubMed] [Google Scholar]
- 29.Pak JW, Herbst A, Bua E, Gokey N, McKenzie D, Aiken JM. Mitochondrial DNA mutations as a fundamental mechanism in physiological declines associated with aging. Aging Cell. 2003;2:1–7. doi: 10.1046/j.1474-9728.2003.00034.x. [DOI] [PubMed] [Google Scholar]
- 30.Eshaghian A, Vleuge RA, Canter JA, McDonald MA, Stasko T, Sligh JE. Mitochondrial DNA deletions serve as biomarkers of aging in the skin, but are typically absent in nonmelanoma skin cancers. J Invest Dermatol. 2006;126:336–344. doi: 10.1038/sj.jid.5700088. [DOI] [PubMed] [Google Scholar]
- 31.Cortopassi GA, Wong A. Mitochondria in organismal aging and degeneration. Biochim Biophys Acta. 1999;1410:183–193. doi: 10.1016/s0005-2728(98)00166-2. [DOI] [PubMed] [Google Scholar]
- 32.Donnelly ET, Liu Y, Paul TK, Rockwell S. Effects of motexafin gadolinium on DNA damage and X-ray-induced DNA damage repair, as assessed by the comet assay. Int J Radiat Oncol Biol Phys. 2005;62:1176–1186. doi: 10.1016/j.ijrobp.2005.04.014. [DOI] [PubMed] [Google Scholar]
- 33.Blaszczyk A, Skolimowski J, Materac A. Genotoxic and antioxidant activities of ethoxyquin salts evaluated by the comet assay. Chem Biol Interact. 2006;162:268–273. doi: 10.1016/j.cbi.2006.07.009. [DOI] [PubMed] [Google Scholar]
- 34.Shoffner JM, Lott MT, Voljavec AS, Soueidan SA, Costigan DA, Wallace DC. Spontaneous Kearns-Sayre⁄chronic external ophthalmoplegia plus syndrome associated with a mitochondrial DNA deletion: A slip-replication model and metabolic therapy. Proc Natl Acad Sci USA. 1989;86:7952–7956. doi: 10.1073/pnas.86.20.7952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Marlin DJ, Johnson L, Kingston DA, Smith NC, Deaton CM, Mann S, Heaton P, Vugt FVan, Saunders K, Kydd J, Harris PA. Application of the comet assay for investigation of oxidative DNA damage in equine peripheral blood mononuclear cells. J Nutr. 2004;134:2133S–2140S. doi: 10.1093/jn/134.8.2133S. [DOI] [PubMed] [Google Scholar]
- 36.Prithivirajsingh S, Story MD, Bergh SA, Geara FB, Ang KK, Ismail SM, Stevens CW, Buchholz TA, Brock WA. Accumulation of the common mitochondrial DNA deletion induced by ionizing radiation. FEBS Lett. 2004;571:227–232. doi: 10.1016/j.febslet.2004.06.078. [DOI] [PubMed] [Google Scholar]
- 37.de Grey AD. Reactive oxygen species production in the mitochondrial matrix: Implications for the mechanism of mitochondrial mutation accumulation. Rejuvenation Res. 2005;8:13–17. doi: 10.1089/rej.2005.8.13. [DOI] [PubMed] [Google Scholar]