

Abstract
Rigid linkers of variable length were used to connect two high-affinity Nle4-D-Phe7-α-melanocyte stimulating hormone (NDP-α-MSH) or two low-affinity MSH(4) ligands. The linked peptides were synthesized by solid-phase methods. Control experiments indicate there is little or no effect of these linkers on NDP-α-MSH or MSH-(4) binding to the human melanocortin 4 receptor (hMC4R). Tethering two high-affinity ligands gave no binding enhancement, while tethering two low-affinity ligands resulted in binding enhancement that decreased with increased linker length. Furthermore, for the low-affinity ligands, the enhancement of affinity is inversely proportional to the estimated molecular moments of inertia. These results are consistent with a model wherein binding is enhanced when the rate of ligand reattachment to the receptor is fast relative to the rate of ligand diffusion.
INTRODUCTION
Early diagnosis is critical to the successful treatment of many human cancers. Noninvasive methods for early detection depend on the availability of nontoxic reagents that can seek out and selectively bind to cancer cells and report their existence and location by molecular imaging (1, 2). We are engaged in a program to develop reagents that target human cancers that presently are difficult to detect, such as melanoma and pancreatic cancer. Our strategy involves linking together reporter moieties and ligands that bind to cell surface receptors that are expressed in cancer cells (3). The resulting multivalent molecules could display enhanced affinity for the targeted cells (4, 5).
Factors that must be considered in designing linkers for bioactive multimeric ligands include the distance that must be spanned, availability, and ease of use. The distance that must be spanned between targeted receptors on the cell surface will vary depending on the number, kind, and mobility of the receptors, but it is expected to greatly exceed the typical dimensions of small molecules (6). Several small molecule linkers connected in series will therefore be necessary to span the distance between receptors. For peptide ligands, linkers should terminate in amine and/or carboxylic acid functional groups to facilitate incorporation by solid-phase peptide synthesis. Both flexible linkers (7, 8) and rigid linkers (9) are of interest. Each new linker must be validated as noninterfering with the binding of the ligand. We report herein the use of rigid linkers 1–4 (Figure 1), which are of variable length (10) to join two “high-affinity” or two “low-affinity” α-melanocyte stimulating hormone (α-MSH) peptide ligands (11–16), and evaluations of the bioactivities of these linker-spliced peptides.
Figure 1.

Rigid amino acid linkers 1–4 and N-protected derivatives 5–8.
EXPERIMENTAL PROCEDURES
Materials and Methods
The N-9-fluorenylmethyloxycarbonyl (Fmoc) derivative 5 (Figure 1) was purchased from Torviq (Niles, MI). The N-trifluoroacetamide (TFA) derivatives 6–8 (Figure 1) were prepared as previously described (17). Other N-α-Fmoc-protected amino acids and Rink amide resin were purchased from SynPep (Dublin, CA) or from Novabiochem (San Diego, CA). O-[1H-6-Chlorobenzotriazol-1-yl)(dimethyl-amino)methylene]uranium hexafluorophosphate N-oxide (HCTU), 1-hydroxybenzotriazole (HOBt), and 6-chloro-1-hydroxyben-zotriazole (HOCt) were purchased from IRIS Biotech (Mark-tredwitz, Germany). For the N-α-Fmoc-protected amino acids, the following side-chain protecting groups were used: Arg(Nγ-2,2,4,6,7-pentamethyldihydrobenzofuran-5-yl sulfonyl); Glu(Oγ-tBu); His(Nim-triphenylmethyl); Lys(Nε-tert-butyloxycarbonyl); Ser(O-tBu); Trp(Ni-tert-butyloxycarbonyl). Peptide synthesis solvents, reagents, and acetonitrile for HPLC were reagent grade, were acquired from VWR (West Chester, PA) or Sigma-Aldrich (Milwaukee, WI), and were used without further purification unless otherwise noted. Solid-phase synthesis was performed in fritted syringes using a Domino manual synthesizer obtained from Torviq (Niles, MI).
Purification of peptide products was achieved on a Waters 600 HPLC using a reverse-phase column (Vydac C-18, 15–20 m, 22 × 250 mm). Peptide products were eluted with a linear gradient of CH3CN/0.1% CF3CO2H at a flow rate of 5.0 mL/min. Separation was monitored at 230 and 280 nm. The purity of the final peptide products was checked by TLC and by analytical PR-HPLC using a Waters Alliance 2695 separation model with a Waters 2487 dual wavelength detector (220 and 280 nm) on a reverse-phase column (Waters Symmetry 4.6 × 75 mm, 3.5 μm). Peptide products were eluted with a linear gradient of 60% aqueous MeCN/0.1 M NH4OAc or MeCN/0.1% CF3CO2H at a flow rate of 1.0 mL/min. Size exclusion chromatography was performed on a borosilicate glass column (2.6 × 250 mm, Sigma, St. Louis, MO) filled with medium-sized Sephadex G-25 or G-10. The compounds were eluted with an isocratic flow of 1.0 M aqueous AcOH.
Structures were characterized by ESI (Finnigan, Thermoquest LCQ ion trap instrument) or MALDI-TOF (Bruker Rexlex-III, α-cyanocinnamic acid as a matrix). For internal calibration, an appropriate mixture of standard peptides was used with an average resolution of 8000–9000.
Solid-Phase Synthesis (18–19)
Rink resin was washed with DMF, and the N-Fmoc protecting group was removed with 50% piperidine in DMF (1 × 2 min and 1 × 20 min). The resin was washed successively with DMF, 1.0 M HOBt in DMF, and DMF, and the next N-α-Fmoc amino acid was coupled using preactivated 0.3 M HOCt ester in THF (3 equiv of N-α-Fmoc amino acid, 3 equiv of HOCt, and 3 equiv of DIC). The resin slurry was stirred for 2 h or until the bromophenol test became negative. If the test failed, the resin was washed with DMF, and the amino acid was coupled again by the HCTU/2,4,6-lutidine procedure (0.3 M solution of 3 equiv of N-α-Fmoc amino acid, 3 equiv of HCTU, and 6 equiv of 2,4,6-lutidine in DMF) for 3 h or until the Kaiser test was negative. If a positive Kaiser test was observed after the second coupling, the resin was washed with DMF and any reactive amine groups capped by treatment with 50% Ac2O in pyridine for 10 min.
After the first peptide segment had been assembled on the solid support (e.g., see Scheme 1), an N-protected linker (5, 6, 7, or 8) was attached to the free N-terminal amine using the coupling methods previously described. The resin was then split into two portions. The first portion of the resin was treated either with 50% piperidine in DMF to remove the Fmoc protecting group or with 15% anhydrous N2H4 plus 15% MeOH in THF at room temperature for 6 h to remove the trifluoroacetyl protecting group, from the N-terminus of the resin-bound linker. The conditions for TFA cleavage were defined by an empirical study (see Table 1 and the accompanying discussion). The free amine was acetylated using 50% Ac2O in pyridine for 10 min and the [peptide + linker] cleaved off the resin. A cleavage cocktail (10 mL per 1 g of the resin) consisting of CF3CO2H (91%), H2O (3%), HSCH2CH2SH (3%), and PhSMe (3%) was injected into the resin, and the mixture was stirred at room temperature for 3 h. The solution was filtered off, the resin was washed with CF3CO2H (2 × 3 min), the liquid phases were concentrated under a stream of nitrogen, and the product was precipitated using cold Et2O. The peptide pellets were washed three times with cold Et2O, lyophilized, purified, and characterized as described above. For the second portion of the resin, removal of the N-terminal Fmoc or TFA was carried out, and assembly of the second peptide segment was completed using the methods previously described. Following incorporation of the last amino acid, the N-terminal protecting group was removed and the free amine acetylated using 50% Ac2O in pyridine for 10 min. The resin was then thoroughly washed with DMF and CH2Cl2, the [peptide + linker + peptide] was cleaved off the resin using the cleavage cocktail, and the product purified as described above. Mass spectral and HPLC characterization data for the peptide products are given in Table 2 and are discussed later.
Scheme 1. Solid-phase Synthesis of Compounds 9–11 Containing Linked NDP-α-MSH Ligands and Control Compounds 12–14.

Reagents and conditions: (a) piperidine. (b) Fmoc/tBu solid-phase synthesis (Refs 18 and 19). (c) N-protected linker (5, 6, or 7), HOCt-DIC, DMF. (d) THF/MeOH/N2H4 (70/15/15). (e) pyridine/Ac2O (50/50). (f) CF3CO2H/HSCH2CH2SH/PhSMe/H2O (91/3/3/3).
Table 1.
Optimization of N-TFA Cleavage Using Resin 34a
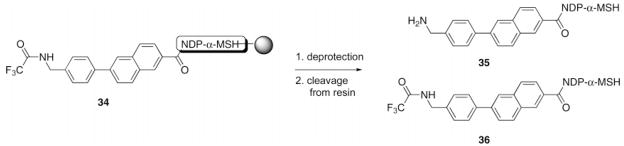 | |||||
|---|---|---|---|---|---|
| yields (%)b |
|||||
| entry | conditions | time (min) | 35 | 36 | impurityc |
| 1 | 10% piperidine, 10% H2O in DMF | 30 | 0 | 100 | 0 |
| 2 | 0.6 M potassium silanolate in THF | 30 | 0 | 100 | 0 |
| 3 | 15% N2H4 in DMF | 75 | 9 | 91 | 0 |
| 4 | 15% N2H4 in DMF | 180 | 34 | 64 | 2 |
| 5 | 15% N2H4, 15% MeOH in THF | 300 | 100 | 0 | 0 |
| 6 | 15% N2H4, 15% MeOH in THF | 180 | 99 | >1 | 0 |
| 7 | 15% N2H4, 15% MeOH in THF | 75 | 78 | 22 | 0 |
| 8 | 15% N2H4, 15% MeOH in THF | 10 | 15 | 85 | 0 |
| 9 | 3% N2H4, 15% MeOH in THF | 75 | 3 | 97 | 0 |
| 10 | 15% 0.5 M LiOH, 15% MeOH in THF | 180 | 49 | 0 | 51 |
| 11 | 15% 0.5 M CsOH, 15% MeOH in THF | 180 | 40 | 0 | 60 |
NDP-α-MSH Rink resin 34 (30 μL of resin slurry in THF) was placed into a small fritted 2 mL syringe and washed with an N-TFA cleavage solvent mixture. After the treatment, the resin was washed with THF and CH2Cl2, and the peptide was cleaved off the resin and side-chain protecting groups removed as described in the Materials and Methods section. The peptide was then dissolved in 30% aqueous MeCN and analyzed by HPLC using a linear MeCN/0.1% CF3CO2H aqueous gradient (10% to 40% in 30 min) at a flow rate of 1.0 mL/min.
Yields were determined as ratios of peak areas at 280 nm on HPLC analyses; product 35 had a retention time of 16.1 min, and product 36 had a retention time of 24.2 min.
Unidentified impurities.
Table 2.
Analytical Data for Synthetic Compounds
| Ac-NDP-α-MSH-linker-NDP-α-MSH-NH2 |
Ac-linker-NDP-α-MSH-NH2 |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| mass spectruma |
HPLCb |
mass spectruma |
HPLCb |
||||||
| compd | ion | calcd | obsd | k′ | compd | ion | calcd | obsd | k′ |
| 9 | (M + 3)3+ | 1122.896 | 1122.90 | 18.13 | 12 | (M + 2)2+ | 890.4446 | 890.40 | 15.11 |
| 10 | (M + 2)2+ | 1721.860 | 1721.81 | 20.24 | 13 | (M + 2)2+ | 995.4462 | 995.40 | 22.69 |
| 11 | (M + 3)3+ | 1173.592 | 1173.50 | 22.43 | 14 | (M + 2)2+ | 993.4618 | 993.91 | 27.11 |
| 32 | (M + 4)4+ | 873.9417 | 873.94 | 21.35 | 33 | (M + 3)3+ | 653.9693 | 653.98 | 25.25 |
| Ac-MSH(4)-linker-MSH(4)-NH2 |
Ac-linker-MSH(4)-NH2 |
||||||||
| mass spectruma |
HPLCb |
mass spectruma |
HPLCb |
||||||
| compd | ion | calcd | obsd | k′ | compd | ion | calcd | obsd | k′ |
|
| |||||||||
| 37 | (M + 1)1+ | 1445.705 | 1445.911 | 9.16 | 41 | (M + 1)1+ | 819.3976 | 819.40 | 7.05 |
| 38 | (M + 1)1+ | 1521.737 | 1522.176 | 10.77 | 42 | (M + 1)1+ | 895.4289 | 895.41 | 9.65 |
| 39 | (M + 1)1+ | 1597.768 | 1597.910 | 13.00 | 43 | (M + 1)1+ | 971.4603 | 971.65 | 13.13 |
| 40 | (M + 1)1+ | 1571.752 | 1571.833 | 12.03 | 44 | (M + 1)1+ | 945.4445 | 945.48 | 11.66 |
MS found molecular peaks (M + 1)1+, (M + 2)2+, (M + 3)3+, and (M + 4)4+. One diagnostic peak is listed here for each compound.
Peptides were eluted with a linear MeCN/0.1% CF3CO2H aqueous gradient (10% to 40% in 30 min) at a flow rate of 1.0 mL/min.
Biological Assays
Ligand binding was evaluated using a previously described lanthanide-based binding assay (20). HEK293 cells overexpressing the human melanocortin 4 receptor (hMC4R) were used to assess ligand binding (21). The coding region of the hMC4R gene was expressed in pcDNA3.1 (Invitrogen, Carlsbad, California). HEK293/hMC4R cells were grown in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum (FBS). Cells were plated in Black & White Isoplates (Wallac, 1450-583) at a density of 12 000 cells/well and were allowed to grow for 3 days. On the day of the experiment, media was aspirated from all wells. Ligands of interest were diluted in binding buffer [DMEM, 1 mM 1,10-phenanthroline, 200 mg/L bacitracin, 0.5 mg/L leupeptin, 0.3% bovine serum albumin (BSA)] to result in final dilutions ranging from 10 μM to 4 pM. Eu-labeled NDP-α-MSH was used at a final concentration of 10 nM. 50 μL of the ligand of interest and 50 μL of Eu-NDP-α-MSH were added to each well, and plates were incubated for 40 min at 37 °C. Following the incubation, cells were washed four times with Wash Buffer (50 mM Tris-HCl, 0.2% BSA, 30 mM NaCl) using Molecular Devices SkanWasher. Enhancement solution (Perkin-Elmer, 1244-105) was added (100 μL/well), and the plates were incubated for 30 min at 37 °C prior to reading. The plates were read on a Wallac VICTOR3 instrument using the standard Eu time-resolved fluorescence (TRF) measurement (340 nm excitation, 400 μs delay, and emission collection for 400 μs at 615 nm). Competition curves were analyzed with GraphPad Prism software using the sigmoidal dose–response classical equation for nonlinear regression analysis. Each data point represents the average of at least four samples, with the error bars indicating standard error of the mean.
RESULTS
Solid-Phase Synthesis
The synthesis of compounds 9–11 consisting of two linked “high-affinity” NDP-α-MSH ligands connected in a head-to-tail fashion by linkers 1–3 is depicted in Scheme 1. Also depicted is the synthesis of control compounds 12–14 which are composed of a single “high-affinity” NDP-α-MSH ligand and an N-terminally acylated linker.
The tridecapeptide NDP-α-MSH was constructed on PS–Rink resin (15, initial loading 0.17 mmol/g, 1% DVB). Resin 16 retained all side-chain protecting groups. The N-protected linkers 5–7 were coupled to the N-terminus of 16, giving resins 17–19. The N-Fmoc group of 17 was cleaved by treatment with piperidine, and the N-TFA groups of 18 and 19 were cleaved by treatment with 15% hydrazine and 15% methanol in THF. The resulting resins 20–22 were each split into two portions. For the synthesis of 9–11, the free amine groups of resins 20–22 were coupled with Fmocvaline and solid-phase peptide synthesis continued to completion of the second NDP-α-MSH sequence, giving resins 23–25. The peptide was then capped by N-acetylation, giving resins 26–28. Simultaneous side-chain deprotection and cleavage of the peptides from the Rink resin was effected using a mixture of trifluoroacetic acid, 1,2-ethanedithiol, thioanisole, and water (91/3/3/3) that produced the desired compounds 9–11. For the synthesis of 12–14, resins 20–22 were terminally N-acetylated to give resins 29–31. Treatment of these resins with the CF3CO2H/HSCH2CH2SH/PhSMe/H2O cocktail effected simultaneous side-chain deprotection and cleavage of the peptides from the resin, producing compounds 12–14. Compounds 32 and 33 (Figure 2) that incorporate the phenylnaphthyl linker 4 were similarly prepared (see Scheme S1 in the Supporting Information).
Figure 2.

Dimeric and monomeric NDP-α-MSH compounds 32 and 33.
N-TFA derivatives 6–8 were employed instead of the corresponding Fmoc derivatives of 2–4, since this enhanced the solubility of these more hydrophobic linkers under the conditions employed for solid-phase synthesis.
The intermediate resin 34 was used to establish conditions for the cleavage of the TFA protecting group from resin-bound linkers (Table 1). Published conditions (22) for N-ε-trifluoro-acetyl group deprotection in peptides (aqueous piperidine) gave poor results (Table 1, entry 1). DMF suppressed the cleavage of the N-ε-trifluoroacetyl group (entries 1, 3, and 4). Optimal conditions developed for the removal of the TFA protecting group from the resin-bound linker were treatment with 15% N2H4 and 15% MeOH in THF for 5 h at room temperature (entry 5).
A similar synthetic strategy was employed to synthesize molecules 37–40 containing linked “low-affinity” MSH(4) ligands (His-D-Phe-Arg-Trp; see Schemes S2 and S3 in the Supporting Information) and the corresponding controls 41–44 (Figure 3).
Figure 3.

Dimeric and monomeric MSH(4) compounds 37–44.
All products from solid-phase syntheses were purified by reverse-phase C18 preparative HPLC and were characterized by analytical HPLC, ESI-MS, and/or MALDI-TOF. Characterization data are listed in Table 2.
Biological Assays
Shown in Figure 4 are representative results from a competition binding assay, described in Handl et al. (20) This figure shows the reduction of Eu-NDP-α-MSH binding in the presence of increasing concentrations of com-pounds 10 and 13. From these data, IC50 values were calculated as the concentration of test ligand that reduced Eu-NDP-α-MSH binding by 50%. These values can be quantitatively related to Kd by correcting for the ratio of the Eu-NDP-α-MSH concentration relative to its Kd (11). Similar data sets were obtained multiple times for all compounds. Table 3 lists the IC50 values (averaged over n experiments) for the dimeric and monomeric complexes of NDP-α-MSH and MSH(4). Compounds 9–11 and 32 contained dimeric NDP-α-MSH ligands, compounds 37–40 contained dimeric MSH(4) ligands, and compounds 12–14, 33, and 41–44 were the corresponding monomeric ligand controls. This table also shows the fold increase in avidity as the ratio of IC50 for the dimeric ligands relative to the corresponding monomeric ligands.
Figure 4.

Representative ligand binding curves generated through competitive ligand binding analysis compare the binding affinities of ligands 10 (□) and 13 (■). The calculated IC50 for 10 is 8 nM with an R2 value = 0.91 and for 13 10 nM with an R2 value = 0.93.
Table 3.
Competitive Binding of Linked NDP-α-MSH and MSH(4) Ligands to hMC4R
| compound | monomer/dimer | linkera | IC50 | nb | fold increase |
|---|---|---|---|---|---|
| NDP-α-MSH | none | 5.6 ± 0.2 nM | 3 | ||
| 9 | D | Ph | 3.5 ± 2.2 nM | 4 | 1.4 |
| 12 | M | Ph | 5.0 ± 1.2 nM | 4 | |
| 10 | D | BP | 5.6 ± 3.5 nM | 5 | 0.9 |
| 13 | M | BP | 5.2 ± 5.2 nM | 5 | |
| 11 | D | TP | 8.4 ± 2.9 nM | 5 | 2.0 |
| 14 | M | TP | 16.5 ± 14.7 nM | 5 | |
| 32 | D | PN | 3.7 ± 0.9 nM | 5 | 1.1 |
| 33 | M | PN | 3.9 ± 1.2 nM | 4 | |
| MSH(4) | none | 2.1 ± 0.8 μM | 3 | ||
| 37 | D | Ph | 0.1 ± 0.05 μM | 4 | 22.9 |
| 41 | M | Ph | 1.8 ± 0.2 μM | 4 | |
| 38 | D | BP | 0.6 ± 0.2 μM | 4 | 4.8 |
| 42 | M | BP | 2.8 ± 1.1 μM | 4 | |
| 39 | D | TP | 2.6 ± 1.0 μM | 4 | 1.9 |
| 43 | M | TP | 5.0 ± 3.6 μM | 5 | |
| 40 | D | PN | 0.5 ± 0.2 μM | 4 | 3.2 |
| 44 | M | PN | 1.5 ± 0.7 μM | 4 |
Ph = phenyl linker 1; BP = biphenyl linker 2; TP = terphenyl linker 3; and PN = phenylnaphthyl linker 4.
The IC50 value given is the average of n independent binding experiments, each done in quadruplicate.
DISCUSSION
The IC50 values for the [linker + NDP-α-MSH] control compounds 12, 13, and 33 were virtually the same as the IC50 for the parental NDP-α-MSH ligand. Similarly, the IC50 values for the [linker + MSH(4)] control compounds 41, 42, and 44 were very close to the IC50 for the parental MSH(4) ligand. These results indicate that attachment of the phenyl, biphenyl, and phenylnaphthyl linkers to the NDP-α-MSH and MSH(4) ligands had little or no effect on the binding of either ligand to hMC4R. In contrast, control compounds 14 and 43 that incorporate the terphenyl linker showed modest (3-fold and 2-fold, respectively) increases in IC50. These results indicate that attachment of the terphenyl linker diminishes slightly NDP-α-MSH and MSH(4) ligand binding affinity to hMC4R.
The expected statistical halving of the IC50 for compounds that contain two ligands was observed only for terphenyl-containing NDP-α-MSH compounds 11 and 14 and for MSH-(4) compounds 39 and 43. This is consistent with the observation that terphenyl-containing compounds bind less well to hMC4R. Tight binding in the cases of 9, 10, and 33, presumably due to slow off-rates for the NDP-α-MSH ligand, effectively removes tethered, unbound ligands from the ligand pool (Figure 5), implying that states B, C, and D are insignificant. Poorer binding of the terphenyl-containing compounds 11 and 14 permits observance of statistical halving of the IC50 for these compounds (Figure 5), implying that states B, C, and D are significant.
Figure 5.

Binding of Linked MSH Ligands to hMC4R. State A: L1 is bound, L2 is unavailable for binding. State B: L1and L2 are unbound, L1 is positioned for binding. State C: L1and L2 are unbound, L2 is positioned for binding. State D: L2 is bound, L1 is unavailable for binding. State E: Compound is dissociated from cell surface (not shown).
A greater than stoichiometric increase in affinity for the hMC4R is observed relative to controls for compounds 37, 38, and 40 that contain two of the more weakly binding MSH(4) ligands joined by noninterfering linkers. This increase in affinity is inversely correlated (Figure 6) with an estimation of the molecular moments of inertia (IM) calculated using eq 1 where M is the compound molecular weight and R is the linker length measured from one benzylic carbon to the other.
Figure 6.

Inverse correlation of compound binding affinity and linker length.
| (1) |
Compound 39 that incorporates the terphenyl linker is displaced from the best-fit line, possibly due to its inherently poorer binding to hMC4R. The existence of this correlation is consistent with a competition between diffusion of the compound away from the receptor (to give state E in Figure 5) and rotation of the compound necessary for presentation of the second ligand to the receptor (equilibration of states B and C). The greater the moment of inertia, the longer this rotation will take, leading to an increased probability of diffusion away from the cell surface before rebinding (to states A and D) can occur.
CONCLUSION
Low- and high-affinity forms of melanocortin ligands were attached to rigid linkers in either monomeric or dimeric forms. Binding affinities of these ligands were tested in whole cells expressing the hMC4 receptor using a sensitive competition assay based on detection of lanthanide (europium) with time-resolved fluorescence. Consistent with previous results, addition of linkers resulted in a reduction in binding affinity that was dependent on the size of the linker. Notably, the short phenyl and biphenyl linkers did not adversely affect binding affinities of monomeric ligands. Also consistent with previous results, there was an increase in binding affinity in dimers compared to the corresponding monomers, and the increase was inversely related to the calculated moment of inertia for the molecules. We interpret these results to indicate that the increased affinity of dimers was due to increased local concentrations of ligand, also known as the statistical effect of Kiessling (5). Somewhat surprisingly, this enhancement of dimers was only observed for the low-affinity MSH(4) ligands and not for the high-affinity NDP-α-MSH ligands. We interpret this to suggest that the statistical effect enhancement is critically dependent on a low-affinity ligand, with potentially faster on-rates relative to the rate of diffusion.
Acknowledgments
This work was supported by NIH grants R33 CA 95944 and RO1 CA 97360.
Footnotes
Supporting Information Available: Additional synthetic schemes and details of the solid-phase synthesis and compound characterization. This material is available free of charge via the Internet at http://pubs.acs.org/BC.
LITERATURE CITED
- 1.Gillies RJ, Hoffman JM, Lam KS, Menkens AE, Piwnica-Worms DR, Sullivan DC, Weissleder R. Meeting Report: High-Throughput Technologies for In Vivo Imaging Agents . Molecular Imaging. 2005;4:98–103. doi: 10.1162/15353500200505115. [DOI] [PubMed] [Google Scholar]
- 2.Gillies RJ, Hruby VJ. Expression-driven Reverse Engineering of Targeted Imaging and Therapeutic Agents. Expert Opin Ther Targets. 2003;7:137–139. doi: 10.1517/14728222.7.2.137. [DOI] [PubMed] [Google Scholar]
- 3.Handl HL, Vagner J, Han H, Mash E, Hruby VJ, Gillies RJ. Hitting Multiple Targets with Multimeric Ligands. Expert Opin Ther Targets. 2004;8:565–586. doi: 10.1517/14728222.8.6.565. [DOI] [PubMed] [Google Scholar]
- 4.Mammen M, Chio SK, Whitesides GM. Polyvalent Interactions in Biological Systems: Implications for Design and Use of Multivalent Ligands and Inhibitors. Angew Chem Int Ed. 1998;37:2754–2794. doi: 10.1002/(SICI)1521-3773(19981102)37:20<2754::AID-ANIE2754>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 5.Kiessling LL, Gestwicki JE, Strong LE. Synthetic Multivalent Ligands in the Exploration of Cell-surface Interactions. Curr Opin Chem Biol. 2000;4:696–703. doi: 10.1016/s1367-5931(00)00153-8. [DOI] [PubMed] [Google Scholar]
- 6.Small molecule dimensions are on the order of ten Angstroms while the distance between receptors is on the order of tens to hundreds of Angstroms; see Tron L, Szollosi J, Szabo G, Jr, Matyus L, Damjanovich S. Cell Surface Dynamics and Distance Relationship of Integral Membrane Proteins. Symposia Biologica Hungarica. 1984;26:307–328.
- 7.Vagner J, Handl HL, Gillies RJ, Hruby VJ. Novel Targeting Strategy Based on Multimeric Ligands for Drug Delivery and Molecular Imaging: Homooligomers of α-MSH. Bioorg Med Chem Lett. 2004;14:211–215. doi: 10.1016/j.bmcl.2003.09.079. [DOI] [PubMed] [Google Scholar]
- 8.Raju N, Ranganathan RS, Tweedle MF, Swenson RE. A New Dendrimer Scaffold for Preparing Dimers or Tetramers of Biologically Active Molecules. Tetrahedron Lett. 2005;46:1463–1465. [Google Scholar]
- 9.Maison W, Frangioni JV, Pannier N. Synthesis of Rigid Multivalent Scaffolds Based on Adamantane. Org Lett. 2004;6:4567–4569. doi: 10.1021/ol048055j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Distances between the benzylic carbon atoms: 1, 5.8 Å; 2, 10.0 Å; 3, 14.3 Å; and 4, 12.2 Å; determined using AM1 in Spartan ‘02 v1.0.4e.
- 11.The “high-affinity” ligand employed here was based on NDP-α-MSH (Ser-Tyr-Ser-Nle-Glu-His-D-Phe-Arg-Trp-Gly-Lys-Pro-Val); see References 12 and 13. The “low-affinity” ligand employed was based on the minimal active sequence for full agonist activity of α-MSH (His-D-Phe-Arg-Trp); see References 14–16.
- 12.Sawyer TK, Sanfilippo PJ, Hruby VJ, Engel MH, Heward CB, Burnett JB, Hadley ME. 4-Norleu-cine, 7-D-phenylalanine-α-melanocyte-stimulating Hormone: A Highly Potent α-Melanotropin with Ultralong Biological Activity. Proc Natl Acad Sci USA. 1980;77:5754–5758. doi: 10.1073/pnas.77.10.5754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hadley ME, Anderson B, Heward CB, Sawyer TK, Hruby VJ. Calcium-dependent Prolonged Effects on Melanophores of [4-Norleucine, 7-D-phenylalanine]-α-melanotropin. Science. 1981;213:1025–1027. doi: 10.1126/science.6973820. [DOI] [PubMed] [Google Scholar]
- 14.Hruby VJ, Wilkes BC, Hadley ME, Al-Obeidi F, Sawyer TK, Staples DJ, DeVaux AE, Dym O, Castrucci AM, Hintz MF, Riehm JP, Rao KR. α-Melanotropin: the Minimal Active Sequence in the Frog Skin Bioassay. J Med Chem. 1987;30:2126–2130. doi: 10.1021/jm00394a033. [DOI] [PubMed] [Google Scholar]
- 15.de Lauro Castrucci AM, Hadley ME, Sawyer TK, Wilkes BC, Al-Obiedi F, Staples DJ, DeVaux AE, Dym O, Hintz MF, Riehm JP, Rao KR, Hruby VJ. Alpha-melanotropin: the Minimal Active Sequence in the Lizard Skin Bioassay. Gen Comp Endocrinol. 1989;73:157–163. doi: 10.1016/0016-6480(89)90066-x. [DOI] [PubMed] [Google Scholar]
- 16.Haskell-Luevano C, Hendrata S, North C, Sawyer TK, Hadley ME, Hruby VJ, Dickinson C, Gantz I. Discovery of Prototype Peptidomimetic Agonists at the Human Melanocortin Receptors MC1R and MC4R. J Med Chem. 1997;40:2133–2139. doi: 10.1021/jm960840h. [DOI] [PubMed] [Google Scholar]
- 17.Monguchi Y, Vagner J, Handl HL, Jana U, Begay LJ, Hruby VJ, Gillies RJ, Mash EA. Design, Synthesis, and Validation of Rigid Linkers for Bioactive Peptides. Tetrahedron Lett. 2005;46:7589–7592. [Google Scholar]
- 18.Merrifield RB. Solid Phase Peptide Synthesis. I The Synthesis of a Tetrapeptide. J Am Chem Soc. 1963;85:2149–2154. [Google Scholar]
- 19.Hruby VJ, Meyer JP. Chemical Synthesis of Peptides. In: Hecht SM, editor. Bioorg Chem: Peptides and Proteins. Oxford Univ. Press: New York; 1998. pp. 27–64. [Google Scholar]
- 20.Handl HL, Vagner J, Yamamura HI, Hruby VJ, Gillies RJ. Lanthanide-based time-resolved fluorescence of in cyto ligand-receptor interactions. Anal Biochem. 2004;330:242–250. doi: 10.1016/j.ab.2004.04.012. [DOI] [PubMed] [Google Scholar]
- 21.The hMC4R vector was originally received from Dr. Ira Gantz; see Gantz I, Miwa H, Konda Y, Shimoto Y, Tashiro T, Watson SJ, DelValle J, Yamada T. Molecular Cloning, Expression, and Gene Localization of a Fourth Melanocortin Receptor. J Biol Chem. 1993;268:15174–15179.
- 22.Goldberger RF, Anfinsen CB. The Reversible Masking of Amino Groups in Ribonuclease and Its Possible Usefulness in the Synthesis of the Protein. Biochemistry. 1962;1:401–405. [Google Scholar]