

Abstract
The ESCRT system is essential for multivesicular body biogenesis, in which cargo sorting is coupled to the invagination and scission of intralumenal vesicles. The ESCRTs are also needed for budding of enveloped viruses including HIV-1, and for membrane abscission in cytokinesis. In yeast, ESCRT-III consists of Vps20, Snf7, Vps24, and Vps2, which assemble in that order, and require the ATPase Vps4 for their disassembly. The ESCRT-III-dependent budding and scission of intralumenal vesicles into giant unilamellar vesicles was reconstituted and visualized by fluorescence microscopy. Three subunits of ESCRT-III, Vps20, Snf7, and Vps24, were sufficient to detach intralumenal vesicles. Vps2, the ESCRT-III subunit responsible for recruiting Vps4, and the ATPase activity of Vps4 were required for ESCRT-III recycling and supported additional rounds of budding. The minimum set of ESCRT-III and Vps4 proteins capable of multiple cycles of vesicle detachment corresponds to the ancient set of ESCRT proteins conserved from archaea to animals.
The Endosomal Sorting Complex Required for Transport (ESCRT) machinery is essential for several fundamental cellular pathways 1-7. The biogenesis of lysosomes involves the maturation of early endosomes into multivesicular bodies (MVBs) 1, 2, 5. In this pathway, portions of the limiting membrane of the endosome invaginate and then detach into the lumen of the endosome, forming intralumenal vesicles (ILVs). The MVBs then fuse with the lysosome and the ILVs and their contents are degraded. Cell surface receptors destined for downregulation and some lysosomal resident enzymes are sorted into this pathway following their covalent ubiquitination. The ESCRTs are involved both in sorting of ubiquitinated cargo into ILVs and in the morphogenesis of the ILVs themselves 1, 2, 5, 7. The ESCRT complexes are also required for the release of nascent HIV-1 virions from the plasma membrane of human cells 8-10. The ESCRTs are needed for cytokinesis in both animal cells 11, 12 and a subset of the archaea 13, 14.
The detachment of ILVs from the endosomal limiting membrane involves the scission of the narrow membrane neck connecting the nascent vesicle with the limiting membrane (Supplementary Fig. 1). This process is topologically opposite to the mechanism of membrane cleavage by dynamin family proteins 15, which coat the outer surface of membrane tubules and cleave from the outside. Cleavage of the membrane neck connecting a nascent virion to the plasma membrane or connecting two daughter also occurs from the membrane surface contiguous with the inside of the neck (Supplementary Fig. 1). The conserved and ubiquitous involvement of ESCRTs in the inwardly directed cleavage of membrane necks has led to the view that ESCRTs are likely to have membrane scission activity. Thus far the absence of an ESCRT scission assay reconstituted entirely from purified proteins and lipids has prevented a direct test of this hypothesis, and has more broadly impeded progress in the ESCRT field.
The ESCRTs consist of the ESCRT-0, -I, -II, and -III complexes together with Vps4, Alix, and other associated proteins 4, 6, 7. ESCRT-0, -I, and -II are soluble, contain ubiquitin-binding domains, and are primarily involved in cargo sorting and in the recruitment and activation of ESCRT-III. The ESCRT-III subunits Vps20, Snf7, Vps24, and Vps2 are soluble monomers in cytosol 16, and the crystal structure of one of these subunits is known 17. Activated ESCRT-III subunits assemble into a detergent-insoluble complex on the endosomal membrane 16, 18, 19. Overexpression of one of these subunits, human SNF7, leads to plasma membrane evaginations that are coated on the inside and have dimensions comparable to those of membrane necks cleaved by the ESCRTs 20. ESCRT-III disassembly requires hydrolysis of ATP by the AAA ATPase Vps4 21. The N-terminal MIT domain of Vps4 binds to a subset of ESCRT-III proteins via MIM122, 23and MIM224 motifs in their C-termini. ESCRT-III subunits can form filaments 25 and helical tubes in vitro 26 that can be disassembled by Vps4. These observations suggest that the ESCRT-III-Vps4 module is a good candidate for a membrane scission machine. Recently, fluorescence microscopy of bilayer giant unilamellar vesicles (GUVs) has become established as a tool for the reconstitution of reactions in membrane trafficking27-30. We set out to initiate an in vitro assay for the ESCRT machinery and to determine whether the ESCRTs have an intrinsic membrane scission capability using purified ESCRT-III subunits and Vps4 in a GUV-based assay.
ESCRT-III and Vps4 recruitment in vitro
Purified ESCRT-III proteins aggregate when overexpressed in bacteria or when purified and concentrated, which has posed an obstacle to biochemical analysis. To avoid off-pathway effects due to aggregation, care was taken to preserve all ESCRT-III subunits in their monomeric state. ESCRT-III subunits were expressed as cleavable MBP-fusions26, and maintained at low micromolar concentrations following cleavage. The absence of aggregation was verified for each ESCRT-III protein by size exclusion chromatography (Supplementary Fig. 2).
In the yeast MVB pathway, Vps20 is recruited and activated by ESCRT-II 16, 31. Vps20 and other ESCRT-III proteins are maintained in an inactive, soluble state by autoinhibition by their C-termini 18, 19. C-terminal truncations of the autoinhibitory domains render ESCRT-III subunits constitutively active in vivo. In order to simplify the reconstitution of the system by bypassing the need for ESCRT-II while avoiding spontaneous aggregation, we used a partial C-terminal deletion construct Vps20ΔC. This construct was monomeric as judged by SEC and light scattering (Supplementary Fig. 2).
GUVs with an endosome-like membrane composition 32, 33 were generated by electroformation 34 from POPC:POPS:PI(3)P:cholesterol at a molar ratio of 62:10:3:25. We verified that fluorescently labeled Vps20 and Vps20ΔC bound to these GUVs (Fig. 1a, b). Vps20 did not bind efficiently when the acidic lipid POPS was omitted from the mixture (Fig. 1c), consistent with the expectation that Vps20 binds to negatively charged membranes through a large electropositive surface 17. Yeast ESCRT-III assembles in the following order: Vps20, Snf7, Vps24, Vps2 35. Vps2 is the only one of these subunits to contain the Vps4 binding MIM1 motif 22, 23. The Vps20ΔC construct used in this study lacks the Vps4 MIM2 binding motif 24. Vps4 is not recruited to membranes in the absence of ESCRT-III. Unlabelled ESCRT-IIIs were added sequentially and the recruitment of labeled Vps4 was monitored. Vps4 was recruited to the limiting membrane and internal structures in GUVs when all four ESCRT-III subunits were added beforehand (Fig. 1d). Vps4 was recruited neither to the limiting membrane nor to internal structures if Vps24 and Vps2 were omitted (Fig. 1e). The ESCRT-III subunits, Vps4, and the GUV model membrane thus support ordered ESCRT-III and Vps4 recruitment.
Fig. 1.

ESCRT-III and Vps4 bind to GUV membranes. The GUV membrane is stained red with rhodamine-PE, and selected proteins are labeled green with Alexa-488. a, Vps20 and b, Vps20ΔC are recruited to membranes containing 10% POPS and 3% PI(3)P. c, Vps20ΔC is not recruited to membranes lacking POPS. d, Vps4 colocalizes with GUVs in the presence of Vps20ΔC, Snf7, Vps2, and Vps24. e, Vps4 is not recruited to GUVs if Vps2 and Vps24 are omitted. f, Five rounds of buffer addition to GUVs leads to no ILV formation. g, Vps20ΔC, Snf7, Vps24, Vps2 and Vps4 generate ILVs. h, ILVs were counted in a total 100 GUVs for each condition shown. Error bars were calculated using two independent experiments and calculating the difference between them. “All” refers to Snf7, Vps24, Vps2 and Vps4. Scale bar = 10 μm.
ESCRT-III promotes ILV formation
ESCRT subunits were added at submicromolar concentrations in the appropriate order, followed by Vps4. When the electroformed GUVs were shifted to the experimental buffer, a small amount of membrane invagination was observed. Therefore, all results were compared to controls that had undergone identical manipulations and incubations using buffer blanks (Fig. 1f). When the reaction was initiated with Vps20ΔC, a massive increase in the number of ILVs was observed over baseline (Fig. 1g, h). Full-length Vps20 was less active, consistent with its autoinhibition (Fig. 1h). Vps20ΔC was used for all subsequent experiments due to its greater activity. Labeled Vps4 is internalized into ILVs when all ESCRT-III subunits are present (Fig. 1d). Much or all of this pool of Vps4, and the unlabelled ESCRT-III associated with it, may be a passenger on the ILVs. In the absence of cargo, viral Gag, or other material on the membrane, it would not be surprising for a portion of the ESCRT-III and Vps4 to associate non-productively with naked lipids. These data show that ESCRT-III proteins are able to dramatically perturb membrane structure.
ESCRT-III carries out membrane scission
The diffusion of soluble GFP was used to assess permeability between bulk solution and putative ILVs. In each experiment, all of the ESCRT-III subunits and Vps4 were added, while the order of addition of GFP was varied (Fig. 2). When GFP was added with Vps20ΔC, Snf7, or Vps24, all of the ILVs imaged were green (Fig. 2a-c). Therefore, until Vps24 was added, all of the internal structures were still connected to the exterior through the bulk phase. When GFP was added with Vps2 (Fig. 2d), no green structures were observed. Thus at this stage, all of the ILVs were detached from the limiting membrane. No further change in the nature of the ILVs was seen when Vps4 was added in the absence of ATP (Fig. 2e).
Fig. 2.
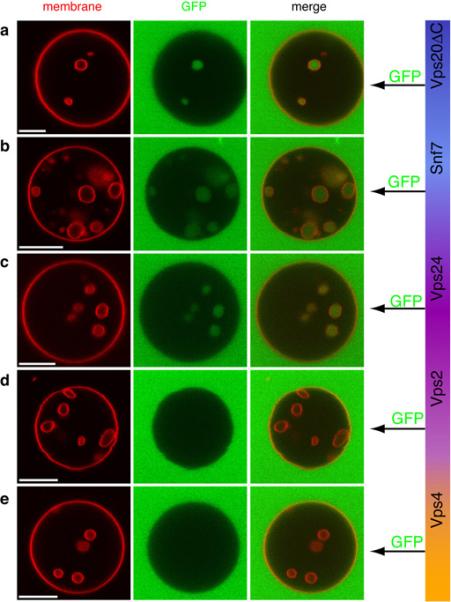
Uptake of the bulk phase and vesicle scission by ESCRT-III. GUVs (red) were incubated with all of Vps20ΔC, Snf7, Vps24, Vps2 and Vps4 in each experiment shown. Each component was added sequentially and incubated 5 min before the addition of the next component. To determine whether ILVs are connected with the bulk phase, the soluble marker GFP (green) was added immediately after either Vps20 ΔC (a), Snf7 (b), Vps24 (c), Vps2 (d), or Vps4 (e). No ATP or other nucleotide was present at any time. These data show that until Vps2 was added, the ILVs were connected with the bulk phase. Scale bar = 10 μm.
ILV detachment was corroborated by a three-dimensional reconstruction of a GUV in which GFP had been added at the Snf7 stage (Fig. 3a, Supplementary Movie 1). The ILVs in this GUV were green (Fig. 3b), indicating that they were formed in the course of the experiment. Another reconstruction was performed for a GUV treated with only Vps20, Snf7, and Vps24 (data not shown), showing complete detachment of ILVs. The ILVs within a GUV were highly mobile (Supplementary Movie 2), consistent with their complete detachment.
Fig. 3.
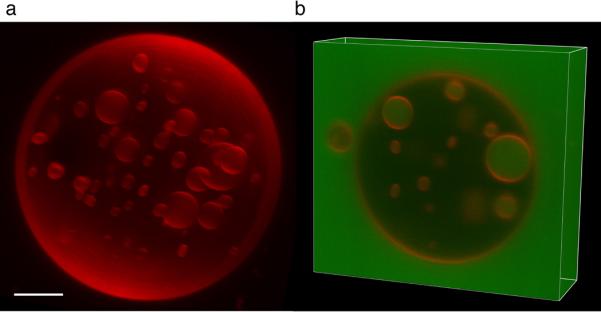
3-D reconstruction of an ESCRT-III treated GUV. a, Three-dimensional reconstruction of a GUV that was incubated with Vps20ΔC, Snf7, Vps24, Vps2, and Vps4, with GFP added together with Snf7. b, A Z stack of the same GUV shows that the ILVs are filled with GFP indicating that they were detached subsequent to the addition of Snf7. Scale bar = 5 μm.
The bulk phase uptake experiment was repeated while omitting the subunits one at a time. GFP was added after Vps24 in these experiments. Omission of Vps20ΔC led to a sharp decrease in ILV formation (Fig. 4a). Omission of Snf7 almost completely abrogated ILV formation (Fig. 4b). Omission of Vps24, with GFP added in its place, led modestly decreased ILV formation (Fig. 4c). A combination of detached and non-detached vesicles was observed in the absence of Vps24 (Fig. 4c). When Vps2 or Vps4 were omitted (Fig. 4d, e), the GUVs were indistinguishable from the situation with all components (Fig. 1g). We find that efficient ILV formation and detachment require Vps20ΔC, Snf7, and Vps24 (Fig. 4f). When PI(3)P was replaced by another endosomal phosphoinositide, PI(3,5)P2, the reaction proceeded just as with PI(3)P (Supplementary Fig. 3a, c). When phosphoinositides are omitted but the mole fraction of the monoanionic lipid POPS is increased to maintain the same net negative charge, ILV formation is reduced by about half (Supplementary Fig. 3b, c). Thus phosphoinositides promote the reaction but are not strictly required.
Fig. 4.

Contributions of individual ESCRT-III subunits to ILV formation. GUVs were incubated with Vps20ΔC, Snf7, Vps24, Vps2 and Vps4, except that one component at a time was omitted. Omission of Vps20ΔC (a) or Snf7 (b) reduces ILV formation dramatically. Omitting Vps24 (c) leads to delayed detachment of ILVs whereas omission of Vps2 (d) and Vps4 (e) has no significant effect. f, Summary of the omission experiment (n = 100). Each color corresponds to a single experiment where the indicated component was omitted, buffer control - only buffer has been added to GUVs (black). Grey - positive control, reaction initiated in the presence of Vps20 ΔC, Snf7, Vps24, Vps2, and Vps4. Scale bar = 10 μm.
Vps4 recycles ESCRT-III after scission
The bulk phase uptake experiment was modified such that ATP and GFP were added after Vps4 (Fig. 5a). The resulting images showed that the GUVs contained a mixture of GFP-filled and empty vesicles. When ATP and GFP were added, but Vps4 omitted, no GFP-filled vesicles were observed (Fig. 5b), as seen in the presence of Vps4 and absence of ATP (Fig. 2e). Thus the addition of ATP to the ESCRT-III-Vps4 mixture after the termination of one round of ILV formation leads to a second wave of ILV formation. Omission of Vps2 completely inhibited the second wave of ILV formation (Fig. 5c, d), even though Vps2 is not essential for the initial round of formation. This is consistent with expectation, since Vps2 is the only ESCRT-III component in the model system that contains a Vps4-binding MIM motif 22, 23. Specific ESCRT-III-Vps4 interactions are required for recycling, recapitulating the biological function of Vps416. Vps4 is functionally competent in the in vitro system, and the lack of a Vps4 effect on ILV detachment was not due to a lack of Vps4 activity. These observations show that the ATP hydrolysis step is not directly involved in vesicle detachment, but that is indirectly involved by virtue of recycling ESCRT-III for multiple rounds of vesicle detachment.
Fig. 5.

Vps4 and ATP induce a second cycle of ILV formation. a, GUVs (red) were incubated with Vps20ΔC, Snf7, Vps24, Vps2, and Vps4. GFP (green) was added after Vps4. In the absence of Vps4 (b) or Vps2 (c), no GFP-filled ILVs are formed following ATP addition. Scale bar = 10 μm. d, Quantification of the ATP induced second wave of ILV formation. Buffer control: only buffer and ATP were added to GUVs (black bar). Grey bars: GUVs with all protein components (Vps20ΔC, Snf7, Vps24, Vps2, and Vps4) in the presence (dark grey) and absence (light grey) of ATP. Colored bars: the indicated subunit was omitted from the reaction in the presence of all other components and ATP. Two sets of 15 GUVs were analyzed for each reaction and the difference between the two sets was used to calculate error bars.
Discussion
We have reconstituted ESCRT-dependent membrane scission using a minimal set of protein components and a simple model for the eukaryotic endosomal membrane. The proteins included correspond to the minimal set conserved between eukaryotes and archaea, ESCRT-III and Vps4 13, 14. The model system recapitulates the ordered recruitment of ESCRT-III and Vps4 established in yeast. Snf7 has the highest stoichiometry in vivo relative to other subunits 35, and it is the most important subunit in cargo sequestration 35. The in vitro studies show that Snf7 is the most potent of the subunits in membrane scission. The behavior of the system is consistent with known biological and biochemical data.
The first major insight is that ESCRT-III proteins alone have the intrinsic ability to drive the detachment of ILVs. ESCRT-III is capable of cleaving membrane necks with the appropriate inward topology in this minimal system. It is remarkable that the system does not require any of the upstream ESCRT complexes −0, -I, or -II, cargo, unusual lipids such as lysobisphosphatidic acid 27, or manipulation of membrane tension. Most of these factors are still likely to modulate the reaction, even if they are not essential. For example, the ILVs seen in this study are more heterogeneous and far larger than those seen physiologically36. We previously suggested that ESCRT-I and perhaps other upstream ESCRTs might regulate ILV size37. In a cellular setting the bud neck is scaffolded by upstream ESCRTs and cargo (MVB pathway), viral Gag assembly (HIV-1 budding), or the extension of the central spindle coupled to membrane addition (cytokinesis). In the in vitro assay the ESCRT-III proteins scaffold the formation of their own membrane neck. In vitro scission may be acting at both the intravesicular and extravesicular end of the neck, but the recycling step requires action from the extravesicular side. Substantial amounts of labeled ESCRT-III and Vps4 are on ILVs, suggesting that some of the cleavage is occurring intravesicularly. However, the ability of Vps4 to recycle a portion of the ESCRT-III demonstrates that at least some in vitro scission is driven from the extravesicular end. Under physiological conditions, the presence of cargo, Gag, etc., is likely to inhibit scission from the intravesicular side of the neck. The efficiency of the neck closure and the predominance of extravesicular cleavage would be higher in the physiological setting of a preformed neck. None of this alters the major conclusion that ESCRT-III alone can detach ILVs from the limiting membrane.
The second major insight pertains to the role of ATP hydrolysis by Vps4, the only thermodynamic driving force for the reaction. It has been suggested that ATP hydrolysis by Vps4, whilst anchored tightly to the membrane by ESCRT-III, could power a dynamin-like constriction of the neck leading to cleavage 26. While Vps4 can disassemble ESCRT-III in vitro25, 26, the relationship between disassembly and vesicle scission was unknown. Here we find that Vps4 acts after the membrane scission step to recycle ESCRT-III proteins. This has a precedent in SNARE-dependent vesicle fusion. SNAREs drive vesicle fusion by forming tight complexes that bring opposing membranes into proximity38. SNARE complexes do not dissociate spontaneously, but require the hydrolysis of ATP by the ATPase, NSF after membrane fusion 38. A recycling role for Vps4 is consistent with all available data, and fits well with the SNARE-NSF precedent.
Snf7 is the most critical of the ESCRT-III subunits for membrane scission. Snf7 can form either spirals or extended tubes when overexpressed 20. The tubes suggest a mechanism for the formation of invaginations of the GUV membrane, although we do not propose that the tubes are involved in physiological budding. In MVB biogenesis, the inward bud is filled with cargo, and in HIV-1 budding, with the Gag protein. In the in vitro system, the ability of ESCRT-III components to form inward buds independently of cargo, Gag, or other factors, has fortuitously yielded membrane necks for study as scission substrates. The spirals suggest an attractive mechanism for membrane scission in both in vitro and physiological settings (Fig. 6). In this model, one surface of Snf7 is tightly anchored to the membrane. Other Snf7 surfaces form an inwardly-growing spiral. The growing spiral could progressively squeeze the membrane neck until critical proximity for membrane fusion is reached (Fig. 6, Supplementary Movie 3).
Fig. 6.
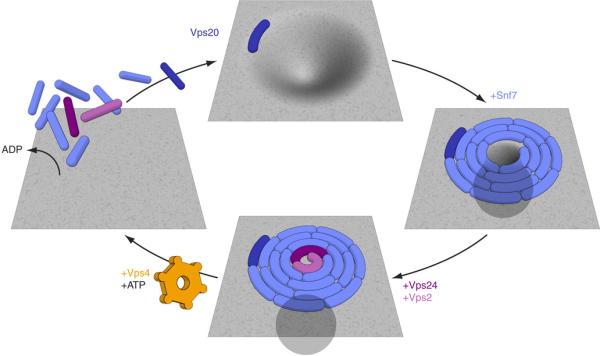
A simple hypothesis for the mechanism of the ESCRT-III-Vps4 membrane scission cycle. The spiral structure is inspired by the observations of Hanson, Heuser, and colleagues 20. ESCRT-III sequesters cargo in MVBs, with Snf7 playing the most important role of the four subunits 35. Any cargo present in the neck near the growing array would be forced out of the neck and further into the bud at this stage, leading to its sequestration. Completion of the spiral would bring the membrane lipids of the neck into critical proximity, allowing fusion. The fused neck would then resolve into the detached vesicle on the interior and the ESCRT-III-coated limiting membrane on the exterior (bottom panel).
In conclusion, ESCRT-III has the instrinsic ability to drive the scission of membrane necks. Vps4 recycles ESCRT-III but does not directly participate in neck closure. Further elaborations of this ESCRT reconstitution should help complete the mechanistic analysis of the pathway.
Methods Summary
Preparation of giant unilamellar vesicles (GUVs)
A thin film of lipid mixtures containing POPC (62 mol%), POPS (10 mol%), cholesterol (25 mol%), PI(3)P (3 mol%) and lissamine-rhodamine-PE (0.1 mol%) was applied to indium-tin oxide covered glass slides and placed into a custom-made Teflon chamber. The glass slides were separated by a 2 mm thin glucose solution (600 mM) and an electric AC-field (1V, 10 Hz) was applied for 4 h at 60° C following the previously described GUV electro-formation protocol34. The GUVs were harvested after cooling to room temperature and immediately used.
ESCRT reactions
A 200 μl observation chamber (Lab-Tek chambered #1.0 Borosilicate) was coated using 1 mg ml−1 BSA to avoid distortion of GUVs upon contact with the bottom of the chamber and rinsed with buffer (50 mM Tris HCl pH 7.4, 300 mM NaCl). 100 μl of GUVs were mixed with this buffer and incubated for 5 min. The buffer and all proteins added to GUVs matched the osmolarity of the GUV solution (∼650 mOsm due to evaporation of ∼10% water during electro-formation at 60° C). Vps20 (full length, residues 1−221) or Vps20ΔC (C-terminal truncation, residues 1−170) were added to the GUV-buffer mixture to yield a final concentration of 160 nM. The solution was stirred by a gentle rotation of the pipet tip, without aspiration. After 5 min of incubation at room temperature, Snf7 (final concentration of 600 nM), Vps24 (200 nM), Vps2 (200 nM), and Vps4 (200 nM) were added in that order, separated by 5 min incubation intervals. For ATP experiments, ATP and MgCl2 (2 mM final concentration) were added to the GUV/protein mixture. To establish uptake of the bulk phase the soluble marker GFP was added as indicated, yielding a final concentration of 1.65 μM. One buffer control was performed for each experiment. Stirring and incubation times were the same for the control and ESCRT-III additions, and GFP was added to the control and ESCRT-III experiments in an identical manner. For Vps20 or Vps20ΔC colocalization experiments a final concentration of 30 nM of Alexa-488 conjugated protein was used, but no other proteins were added. For Vps4 colocalization, the experiment was set up as described above, however, a mixture of unlabeled and labeled Vps4 (5:1) was added to a final total Vps4 concentration of 200 nM.
Supplementary Material
Acknowledgements
We thank V. Schram and the NICHD imaging core facility for use of the LSM5 LiveDuo microscope, D. Yang and G. Patterson for providing purified Vps4 and GFP, respectively, B. Beach for assistance with subcloning, E. Tyler for generating Fig. 6 and Supplementary Movie 3, Y. Im for assistance with Supplementary Fig. 1, C. Biertümpfel and W. Yang for use of their light scattering instrument, and members of the Hurley and Lippincott-Schwartz labs for discussions. This research was supported by the Intramural Program of the National Institutes of Health, NICHD (J. L.-S.), NIDDK (J. H. H.), and IATAP (J. H. H. and J. L.-S.). T.W. is an EMBO long term fellow.
Online Methods
Cloning, expression and purification of proteins
The ESCRT-III subunits Vps20, Snf7, Vps24 and Vps2 were subcloned into a modified pHis2 expression vector, containing the Escherichia coli maltose binding protein (MBP) gene in between 5’ His6-tag and 3’ TEV protease cleavage site. The C-terminally truncated version of Vps20 (Vps20ΔC, residues 1−179) was generated by the polymerase chain reaction (PCR) using the Vps20 construct above as a template and subcloned as for Vps20. All constructs were transformed into Rosetta pLys cells (Stratagene). N-terminal His6-MBP fusion proteins were expressed at 30° C for 3 hours in LB medium supplemented with ampicillin (100 μg/ml final concentration) after induction with 0.5 mM IPTG at an optical density (OD 600nm) of 0.8. The proteins were affinity purified using Ni-NTA resin (Qiagen) and eluted with 100 mM imidazole. Elution fractions were cleaved using TEV protease and further purified by size exclusion chromatography (SEC; Superdex 200 column) and a second round of chromatography on Ni-NTA resin. Pooled fractions of Snf7 were not concentrated to avoid aggregation of the monomeric protein. The final Snf7 protein concentration was usually between 4 and 8 μM. All other proteins were concentrated until ∼50 μM. Purified proteins were immediately flash frozen in liquid nitrogen and stored at −80° C until use. The monomeric state of all proteins was verified using analytical SEC before usage (Superdex 200 10/30 column; GE Healthcare). Full-length yeast Vps4 was purified from E. coli as a glutathione S-transferase fusion protein. After affinity-purification using glutathione-resin and cleavage with TEV protease, the protein was subjected to SEC, and remnants of GST as well as TEV protease were removed by a second round of chromatography using Ni-NTA and glutathione resin. The purified protein was concentrated to 50 μM and stored at −80° C.
Light scattering analysis
Protein samples (5.5 mg/ml of Vps20 and 1.7 mg ml−1 of Vps20ΔC) were injected onto a TSK-GEL GMPWXL size exclusion column (Tosoh) attached to an Agilent 1200 HPLC system (Agilent Technologies) using a 20 μl sample loop and a flow rate of 0.5 ml min−1. The eluted peaks were analyzed using a Wyatt DAWN HELEOS-II multi-angle light scattering instrument and a Wyatt Optilab rEX differential refractometer. Data were evaluated using ASTRA 5.3.4 software using the Zimm model for static light scattering data fitting. Vps20 and Vps20ΔC were monodisperse and had experimental molecular weights of 29.4 and 24.1 kDa, respectively, as compared to their calculated molecular weights of 26.6 and 21.1 kDa.
Fluorescent labeling of proteins
To label Vps20 or Vps20ΔC, which contain no native Cys residues, the mutant N85C was generated using the QuickChange Mutagenesis Kit (Stratagene). The purified and concentrated protein solution was labeled by incubating Alexa Fluor 488 C5 maleimide (Molecular Probes) at a molar ratio of 1:1 with the protein at 37° C for one hour under a N2 atmosphere. The labeling incorporation was evaluated by running samples on an SDSPA gel and carrying out fluorescence scans of the gel with a Typhoon variable mode imager (GE-Healthcare) using 488 nm excitation and 495 nm detection wavelengths. Quantification of labeling efficiency was achieved by separating excess dye from labeled protein using two consecutive HiTrap Desalting Columns (GE-Healthcare). Molar protein and Alexa488 concentrations of the purified protein were determined using absorption at 280 and 495 nm, respectively. Vps4 contains two cysteine residues at positions 317 and 376, which were used for the labeling reaction. The labeling and purification protocol was similar to that of the Vps20 constructs. Labeling efficiencies for all reactions exceeded 90%.
Lipids
The following lipids were purchased from Avanti Polar Lipids: 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-L-serine (POPS), 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC), cholesterol, 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine-N-(lissamine rhodamine B sulfonyl) (rhodamine-PE). Phosphatidylinositol 3-phosphate and 3,5-bisphosphate (diC16) were purchased from Echelon. POPS, POPC, cholesterol, and rhodamine-PE were dissolved in chloroform. Phosphoinositides were dissolved in 1:2:0.8 chloroform:methanol:water.
Confocal fluorescence microscopy
Images were taken in multi-tracking mode on a Zeiss LSM510 or LSM5 LiveDuo confocal microscope with a 63× Plan Apochromat 1.4 NA objective and a 488/543 dichroic mirror at a resolution of 512 × 512 pixel. The GFP and Alexa-488 dyes were excited using the 488 nm line and the rhodamine was excited with a 543 nm HeNe laser. GFP and Alexa488 emission was collected with a 505−530 nm bandpass filter. Rhodamine emission was collected with a 560 nm longpass filter. The pinholes for each channel were set for an approximate 1.5 − 2.5 μm optical slice. Laser power was 9 μW for the 543 nm channel and 24, 400, or 810 μW for GFP bulk phase expermiments, Vps20 colocalization or Vps4 colocalization, respectively, in the 488 nm channel. Images were analyzed using the LSM Examiner software.
Enumeration of ILV formation
Randomly chosen field of views were evaluated to reveal the number of ILV per GUV. Reactions were performed as described above and all GUVs in the particular field of view were scanned in the z direction. All ILV per GUV were counted and for each experimental condition 100 GUVs were analyzed. Results were summarized in histograms.
Three-dimensional fluorescence microscopy of GUVs
Z-stacks over time were taken with an LSM5 LiveDuo (Carl Zeiss MicroImaging Inc., Germany) using 63× 1.4 NA Plan-Apochromate oil objective at 90 frames/sec, 1024 × 256 pixels and a piezo focusing motor for fast hyperfine sectioning (0.35 μm). Static and animated 3D visualizations of the data stacks were produced with IMARIS 6.1.0 software package (Bitplane Inc., Switzerland).
List of proteins appearing in the manuscript
Vps20, Snf7, Vps24, Vps2, Vps4
References
- 1.Gruenberg J, Stenmark H. The biogenesis of multivesicular endosomes. Nat. Rev. Mol. Cell Biol. 2004;5:317–323. doi: 10.1038/nrm1360. [DOI] [PubMed] [Google Scholar]
- 2.Russell MRG, Nickerson DP, Odorizzi G. Molecular mechanisms of late endosome morphology, identity and sorting. Curr. Opin. Cell Biol. 2006;18:422–428. doi: 10.1016/j.ceb.2006.06.002. [DOI] [PubMed] [Google Scholar]
- 3.Slagsvold T, Pattni K, Malerod L, Stenmark H. Endosomal and nonendosomal functions of ESCRT proteins. Trends Cell Biol. 2006;16:317–326. doi: 10.1016/j.tcb.2006.04.004. [DOI] [PubMed] [Google Scholar]
- 4.Saksena S, Sun J, Chu T, Emr SD. ESCRTing proteins in the endocytic pathway. Trends Biochem. Sci. 2007;32:561–573. doi: 10.1016/j.tibs.2007.09.010. [DOI] [PubMed] [Google Scholar]
- 5.Piper RC, Katzmann DJ. Biogenesis and function of multivesicular bodies. Annu. Rev. Cell Devel. Biol. 2007;23:519–547. doi: 10.1146/annurev.cellbio.23.090506.123319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Williams RL, Urbe S. The emerging shape of the ESCRT machinery. Nat. Rev. Mol. Cell Biol. 2007;8:355–368. doi: 10.1038/nrm2162. [DOI] [PubMed] [Google Scholar]
- 7.Hurley JH. ESCRT Complexes and the Biogenesis of Multivesicular Bodies. Curr. Opin. Cell Biol. 2008;20:4–11. doi: 10.1016/j.ceb.2007.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Morita E, Sundquist WI. Retrovirus budding. Annu. Rev. Cell Devel. Biol. 2004;20:395–425. doi: 10.1146/annurev.cellbio.20.010403.102350. [DOI] [PubMed] [Google Scholar]
- 9.Bieniasz PD. Late budding domains and host proteins in enveloped virus release. Virology. 2006;344:55–63. doi: 10.1016/j.virol.2005.09.044. [DOI] [PubMed] [Google Scholar]
- 10.Fujii K, Hurley JH, Freed EO. Beyond Tsg101: the role of Alix in ‘ESCRTing’ HIV-1. Nat. Rev. Microbiol. 2007;5:912–916. doi: 10.1038/nrmicro1790. [DOI] [PubMed] [Google Scholar]
- 11.Carlton JG, Martin-Serrano J. Parallels between cytokinesis and retroviral budding: a role for the ESCRT machinery. Science. 2007;316:1908–1912. doi: 10.1126/science.1143422. [DOI] [PubMed] [Google Scholar]
- 12.Morita E, et al. Human ESCRT and ALIX proteins interact with proteins of the midbody and function in cytokinesis. EMBO J. 2007;26:4215–4227. doi: 10.1038/sj.emboj.7601850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Samson RY, Obita T, Freund SM, Williams RL, Bell SD. A role for the ESCRT system in cell division in Archaea. Science. 2008;322:1710–1713. doi: 10.1126/science.1165322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lindas AC, Karlsson EA, Lindgren MT, Ettema TJG, Bernander R. A unique cell division machinery in the Archaea. Proc. Natl. Acad. Sci. USA. 2008;105:18942–18946. doi: 10.1073/pnas.0809467105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hinshaw JE. Dynamin and its role in membrane fission. Annu. Rev. Cell Devel. Biol. 2000;16:483–519. doi: 10.1146/annurev.cellbio.16.1.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Babst M, Katzmann DJ, Estepa-Sabal EJ, Meerloo T, Emr SD. ESCRT-III: An endosome-associated heterooligomeric protein complex required for MVB sorting. Dev. Cell. 2002;3:271–282. doi: 10.1016/s1534-5807(02)00220-4. [DOI] [PubMed] [Google Scholar]
- 17.Muziol T, et al. Structural basis for budding by the ESCRT-III factor CHMP3. Dev. Cell. 2006;10:821–830. doi: 10.1016/j.devcel.2006.03.013. [DOI] [PubMed] [Google Scholar]
- 18.Zamborlini A, et al. Release of autoinhibition converts ESCRT-III components into potent inhibitors of HIV-1 budding. Proc. Natl. Acad. Sci. USA. 2006;103:19140–19145. doi: 10.1073/pnas.0603788103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shim S, Kimpler LA, Hanson PI. Structure/Function Analysis of Four Core ESCRT-III Proteins Reveals Common Regulatory Role for Extreme C-terminal Domain. Traffic. 2007;8:1068–1079. doi: 10.1111/j.1600-0854.2007.00584.x. [DOI] [PubMed] [Google Scholar]
- 20.Hanson PI, Roth R, Lin Y, Heuser JE. Plasma membrane deformation by circular arrays of ESCRT-III protein filaments. J. Cell Biol. 2008;180:389–402. doi: 10.1083/jcb.200707031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Babst M, Wendland B, Estepa EJ, Emr SD. The Vps4p AAA ATPase regulates membrane association of a Vps protein complex required for normal endosome function. EMBO J. 1998;17:2982–2993. doi: 10.1093/emboj/17.11.2982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Obita T, et al. Structural basis for selective recognition of ESCRT-III by the AAA ATPase Vps4. Nature. 2007;449:735–739. doi: 10.1038/nature06171. [DOI] [PubMed] [Google Scholar]
- 23.Stuchell-Brereton M, et al. ESCRT-III recognition by VPS4 ATPases. Nature. 2007;449:740–744. doi: 10.1038/nature06172. [DOI] [PubMed] [Google Scholar]
- 24.Kieffer C, et al. Two distinct modes of ESCRT-III recognition are required for VPS4 functions in lysosomal protein targeting and HIV-1 budding. Dev. Cell. 2008;15:62–73. doi: 10.1016/j.devcel.2008.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 25.Ghazi-Tabatabai S, et al. Structure and disassembly of filaments formed by the ESCRT-III subunit Vps24. Structure. 2008;16:1345–1356. doi: 10.1016/j.str.2008.06.010. [DOI] [PubMed] [Google Scholar]
- 26.Lata S, et al. Helical Structures of ESCRT-III are Disassembled by VPS4. Science. 2008;321:1354–1357. doi: 10.1126/science.1161070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Matsuo H, et al. Role of LBPA and Alix in multivesicular liposome formation and endosome organization. Science. 2004;303:531–534. doi: 10.1126/science.1092425. [DOI] [PubMed] [Google Scholar]
- 28.Trajkovic K, et al. Ceramide triggers budding of exosome vesicles into multivesicular Endosomes. Science. 2008;319:1244–1247. doi: 10.1126/science.1153124. [DOI] [PubMed] [Google Scholar]
- 29.Sens P, Johannes L, Bassereau P. Biophysical approaches to protein-induced membrane deformations in trafficking. Curr. Opin. Cell Biol. 2008;20:476–482. doi: 10.1016/j.ceb.2008.04.004. [DOI] [PubMed] [Google Scholar]
- 30.Romer W, et al. Shiga toxin induces tubular membrane invaginations for its uptake into cells. Nature. 2007;450:670–U3. doi: 10.1038/nature05996. [DOI] [PubMed] [Google Scholar]
- 31.Teo H, Perisic O, Gonzalez B, Williams RL. ESCRT-II, an endosome-associated complex required for protein sorting: Crystal structure and interactions with ESCRT-III and membranes. Dev. Cell. 2004;7:559–569. doi: 10.1016/j.devcel.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 32.Evans WH, Hardison WG. Phospholipid, cholesterol, polypeptide and glycoprotein composition of hepatic endosome subfractions. Biochem. J. 1985;232:33–36. doi: 10.1042/bj2320033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kobayashi T, et al. Separation and characterization of late endosomal membrane domains. J. Biol. Chem. 2002;277:32157–32164. doi: 10.1074/jbc.M202838200. [DOI] [PubMed] [Google Scholar]
- 34.Angelova MI, Dimitrov DS. Liposome electroformation. Faraday Discuss. Chem. Soc. 1986;81:303–311. [Google Scholar]
- 35.Teis D, Saksena S, Emr SD. Ordered Assembly of the ESCRT-III Complex on Endosomes Is Required to Sequester Cargo during MVB Formation. Dev. Cell. 2008;15:578–589. doi: 10.1016/j.devcel.2008.08.013. [DOI] [PubMed] [Google Scholar]
- 36.Nickerson DP, West M, Odorizzi G. Did2 coordinates Vps4-mediated dissociation of ESCRT-III from endosomes. J. Cell Biol. 2006;175:715–720. doi: 10.1083/jcb.200606113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kostelansky MS, et al. Molecular architecture and functional model of the complete yeast ESCRT-I heterotetramer. Cell. 2007;129:485–498. doi: 10.1016/j.cell.2007.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wickner W, Schekman R. Membrane fusion. Nat. Struct. Mol. Biol. 2008;15:658–664. doi: 10.1038/nsmb.1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.