

Abstract
Guggulsterone is a plant polyphenol traditionally used to treat obesity, diabetes, hyperlipidemia, atherosclerosis, and osteoarthritis, possibly through an anti-inflammatory mechanism. Whether this steroid has any role in cancer is not known. In this study, we found that guggulsterone inhibits the proliferation of wide variety of human tumor cell types including leukemia, head and neck carcinoma, multiple myeloma, lung carcinoma, melanoma, breast carcinoma, and ovarian carcinoma. Guggulsterone also inhibited the proliferation of drug-resistant cancer cells (e.g., gleevac-resistant leukemia, dexamethasone-resistant multiple myeloma, and doxorubicin-resistant breast cancer cells). Guggulsterone suppressed the proliferation of cells through inhibition of DNA synthesis, producing cell cycle arrest in S-phase, and this arrest correlated with a decrease in the levels of cyclin D1 and cdc2 and a concomitant increase in the levels of cyclin-dependent kinase inhibitor p21 and, p27. Guggulsterone induced apoptosis as indicated by increase in the number of Annexin V- and TUNEL-positive cells, through the down-regulation of anti-apoptototic products. The apoptosis induced by guggulsterone was also indicated by the activation of caspase 8, bid cleavage, cytochrome c release, caspase 9 activation, caspase 3 activation, and PARP cleavage. The apoptotic effects of guggulsterone were preceded by activation of JNK and down-regulation of Akt activity. JNK was needed for guggulsterone-induced apoptosis, inasmuch as inhibition of JNK by pharmacological inhibitors or by genetic deletion of MKK4 (activator of JNK) abolished the activity. Overall, our results indicate that guggulsterone can inhibit cell proliferation and induce apoptosis through the activation of JNK, suppression of Akt, and down-regulation of anti-apoptotic protein expression.
Keywords: Guggulsterone, JNK, caspase, Akt, apoptosis
1. Introduction
Complementary and alternative medicine serves the medicinal needs of almost 80% of the world's population [1]. Use of such formulations can be traced back to millennia-old medical practices like Ayurveda and traditional Chinese medicine. One such traditional medicine, guggulsterone, [4,17(20)-pregnadiene-3,16-dione], is a plant polyphenol obtained from the gum resin of the Commiphora mukul tree; it has been used in oriental medical settings to treat obesity, diabetes, hyperlipidemia, atherosclerosis, and osteoarthritis [2, 3]. The anti-inflammatory activity of gum guggul is well known [4-6], but the molecular mechanisms underlying guggulsterone's action have begun to unfold recently. Guggulsterone can suppress inflammation by inhibiting inducible nitric oxide synthetase (iNOS) [7]. We have shown that this steroid can suppress activation of an inflammatory transcription factor NF-κB induced by various carcinogens and tumor promoters [8]. We have also demonstrated that guggulsterone can inhibit Receptor Activator of NF-κB Ligand (RANKL)-induced osteo-clastogenesis through inhibition of NF-κB activation pathway [9]. Although NF-κB activation is closely linked with carcinogenesis, in part through regulation of apoptosis [10], what role does guggulsterone has in cancer is poorly understood [11, 12].
Most apoptotic agents are known to induce apoptosis through one of two pathways, namely a receptor-mediated or a non-receptor-mediated or chemical-induced pathway [13, 14]. Cytokines, including members of the TNF superfamily, induce apoptosis by interacting with the death receptor, which sequentially recruits TNF receptor-associated death domain; Fas-associated death domain (FADD); FADD-like interleukin-1 converting enzyme (FLICE) (also called caspase-8), and caspase-3; the last then cleaves various substrates leading to apoptosis. In contrast, non-receptor-mediated apoptosis involves cleavage of Bid by activated caspase-8, which causes the release of cytochrome c from the mitochondria, which together with APAF1 activates caspase-9 and which in turn activates caspase-3, resulting in apoptosis. Recent evidence also indicate that apoptosis pathway is regulated by NF-κB [15], by c-Jun N-terminal kinase (JNK) [16], and by GADD 45[17].
Because NF-κB activation has been closely linked with apoptosis, we investigated the role of guggulsterone in modulation of proliferation and apoptosis of a variety of tumor cell types. The effects of this steroid on cells resistant to chemotherapeutic agents like STI 571 (gleevac), doxorubicin and dexamethasone, was also examined. To decipher the mechanism, the role of NF-κB regulated anti-apoptotitic gene products, caspases, JNK, and Akt in regulation of apoptosis was investigated. Overall, our results indicate that guggulsterone can inhibit cell proliferation and induce apoptosis through the activation of JNK, suppression of Akt, and down-regulation of anti-apoptotic protein expression.
2. Materials and Methods
2.1. Materials
Z-Guggulsterone, obtained from Steraloids, Inc. (Newport, RI), was dissolved in dimethyl sulfoxide (DMSO) as a 10 mM stock solution and stored at -20°C. Penicillin, streptomycin, RPMI 1640 medium, and FBS were obtained from Invitrogen (Grand Island, NY). The following polyclonal antibodies were obtained from Santa Cruz Biotechnology Inc. (Santa Cruz, CA): anti-cyclin D1 against amino acids 1-295, which represents full-length cyclin D1 of human origin; anti-MMP-9; anti-polyadenosine ribose polymerase (PARP); anti-IAP1; anti-IAP2; anti-Bcl-2; anti-Bid; anti-Bax, anti-cdc2, anti-cFLIP, anti-JNK, and anti-Bfl-1/A1. Anti-survivin antibody was purchased from R & D systems (Minneapolis, MN). Anti-phosphorylated JNK, anti-phosphorylated Akt (Ser 473), Akt (Thr 308), anti-caspase 3, anti-caspase 9, anti-cleaved caspase 3, anti-cleaved caspase 9, anti-p21, anti-p27, anti-phosphorylated-PDK1(Ser 241), anti-PDK1, anti-phosphorylated-GSK3β (Ser-9), anti- GSK3β, anti-phosphorylated-PI3K, and PI3K were obtained from Cell Signaling Technology (Danvers, MA). Anti-COX-2, and anti-XIAP antibodies were obtained from BD Biosciences (San Diego, CA). Akt inhibitor SH-5 was obtained from Alexis Biochemicals (San Diego, CA).
2.2. Cell lines
The cell lines used in our studies included ones established from chronic myelogenous leukemia (KBM-5), human monocytic leukemia (U937), human melanoma (A375, WM35), human lymphoblastic leukemia (Jurkat), human chronic myelogenous leukemia (K562), human non-small cell lung carcinoma (H1299), human normal bronchial epithelial cells (BEAS-2B), human multiple myeloma (U266, MM1), human head and neck cancer (HN5, SCC4, FADU), human breast cancer (MCF-7), and human ovarian cancer (HEY8, SKOV3). All cell lines were obtained from the American Type Culture Collection (Manassas, VA). MKK4–deleted murine embryonic fibroblasts cells were kindly provided by Dr. Jonathan Kurie (M.D. Anderson Cancer Center). MKK4 cells were cultured in DMEM with 10% FBS, KBM-5 cells were cultured in Iscove's modified DMEM with 15% FBS, BEAS-2B cells were cultured in keratinocyte serum-free media, melanoma cell lines were cultured in RPMI 1640 supplemented with 10% HEPES and 10% FBS, and all other cell lines were cultured in RPMI 1640 medium with 10% FBS. All media were supplemented with 100 U/ml penicillin and 100 μg/ml streptomycin. The mouse embryonic fibroblast (MEF) derived from GSK-3β –/–C57Bl/6J mice and its wild type were kindly provided by Dr. James R. Woodgett (Ontario Cancer Institute, Toronto, Canada). The MEF derived from p65 –/–C57Bl/6J mice and its wild type were kindly provided by Dr. David Baltimore (California Institute of Technology, Pasadena, CA). The MEF derived from IKK-β mice and its wild type were kindly provided by Dr. Inder M. Verma (The Salk Institute for Biological Studies, La Jolla, CA 92037, USA.).
2.3. Cytotoxicity assay
The cytotoxicity of guggulsterone was determined by the MTT uptake method. Briefly, 5000 cells were incubated with guggulsterone in triplicate in a 96-well plate at 37°C. MTT solution was then added to each well. After a 2-hour incubation at 37°C, extraction buffer (20% SDS, 50% dimethylformamide) was added, cells were incubated overnight at 37°C, and the optical density was then measured at 570 nm using a 96-well multiscanner (Dynex Technologies, MRX Revelation, Chantilly, VA).
2.4. Thymidine incorporation assay
The antiproliferative effects of guggulsterone were also monitored by the thymidine incorporation method [18]. For this, 5000 cells were cultured in 100 μL of medium in triplicate in 96-well plates in the presence or absence of guggulsterone for 72 hours. Six hours before the completion of the experiment, cells were pulse treated with 0.5 μCi (0.0185 MBq) [3H]-thymidine, and the uptake of thymidine was monitored by means of a Matrix-9600 β-counter (Packard Instruments, Downers Grove, IL).
2.5. Flow cytometric analysis
To determine the effect of guggulsterone on the cell cycle, U937 cells were treated with guggulsterone for different times, washed, and fixed with 70% ethanol [18]. After an overnight incubation at −20°C, cells were washed with PBS and then suspended in staining buffer (Propidium iodide, 10 μg/ml; Tween-20, 0.5%; RNase, 0.1% in PBS). The cells were analyzed using a FACS Vantage flow cytometer that uses CellQuest acquisition and analysis software (Becton Dickinson, San Jose, CA). Gating was set to exclude cell debris, cell doublets, and cell clumps.
2.6. Western blot analysis
Thirty to fifty micrograms of cytoplasmic, nuclear, or whole-cell protein was resolved on 10% SDS–PAGE gel, transferred to a nitrocellulose membrane, blocked with 5% nonfat milk, and probed with specific antibodies as per manufacturer's recommended protocol. The blots were washed, exposed to horse radish peroxidase-conjugated secondary antibodies for 1 hour, and finally detected by ECL reagent (Amersham Pharmacia Biotechnology, Piscataway, NJ). The bands were quantitated using a Personal Densitometer Scan v1.30 using Imagequant software version 3.3 (Molecular Dynamics, Piscataway, NJ).
2.7. RNA analysis and RT-PCR
U937 cells were left untreated or treated with 10 μM guggulsterone for various times, washed, and suspended in Trizol reagent. Total RNA was extracted according to the manufacturer's instructions (Invitrogen, Life Technologies, Grand Island, NY). Two micrograms of total RNA was converted to cDNA by Superscript reverse transcriptase and then amplified by Platinum Taq polymerase using Superscript One Step RT-PCR kit (Invitrogen). The relative expression of COX-2, IL-1β, IL-6, and TNF was analyzed using quantitative RT-PCR with β-actin as an internal control. The RT-PCR reaction mixture contained 25 μl of 2× reaction buffer, 2 μl each of RNA and forward and reverse primers and 1 μl of RT-Platinum Taq in a final volume of 50 μl. The reaction was performed at 50°C for 30 min, 94°C for 2 min, 94°C for 35 cycles of 15 s each, 60°C for 30 s, and 72°C for 1 min with extension at 72°C for 10 min. PCR products were run on 2% agarose gel and then stained with ethidium bromide. Stained bands were visualized under UV light and photographed.
2.8. Annexin V Assay
One of the early indicators of apoptosis is the rapid translocation of the membrane phospholipid phosphatidylserine from the cell's cytoplasmic interface to the extracellular surface and its accumulation there, producing a loss of membrane symmetry that can be detected using annexin V. Briefly, 1 × 106 cells were pretreated with 10 μM guggulsterone for various time points and then subjected to annexin V staining. Cells were washed, stained with FITC-conjugated anti-annexin V antibody, and then analyzed with a flow cytometer (FACSCalibur; BD Biosciences).
2.9. TUNEL Assay
We also assayed apoptosis by the terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate nick end-labeling (TUNEL) method, which examines DNA strand breaks that occur during apoptosis, using an in situ cell death detection reagent (Roche Molecular Biochemicals, Mannheim, Germany). We performed this assay as described previously [19]. Briefly, 5 × 105 cells were incubated with 10 μM guggulsterone for 24 h and then fixed with 4% paraformaldehyde and permeabilized with 0.1% Triton-X 100 in 0.1% sodium citrate. After being washed, the cells were incubated with reaction mixture for 60 min at 37°C. Stained cells were analyzed with a flow cytometer (FACSCalibur; BD Biosciences).
2.10. Live and dead assay
To measure apoptosis, we used the Live and Dead assay (Molecular Probes), which determines intracellular esterase activity and plasma membrane integrity. This assay employs calcein, a polyanionic dye, which is retained within the live cells and provides green fluorescence [20]. It also employs the ethidium monomer dye (red fluorescence), which can enter the cells only through damaged membranes and bind to nucleic acids but is excluded by the intact plasma membrane of live cells. Briefly, 1 × 105 cells were incubated with 10 μM guggulsterone for various times at 37°C. Cells are stained with the Live and Dead reagent (5 μM ethidium homodimer, 5 μM calcein-AM) and then incubated at 37°C for 30 min. Cells were analyzed under a fluorescence microscope (Labophot-2, Nikon, Melville, NY)
2.11. JNK assay
To determine the effect of guggulsterone on the kinase activity of JNK, JNK complex from whole-cell extracts was precipitated with antibody against JNK1, followed by treatment with protein A/G-Sepharose beads (Pierce, Rockford, IL). After 2 h of incubation, the beads were washed with lysis buffer and then assayed in kinase assay mixture containing 50 mM HEPES (pH 7.4), 20 mM MgCl2, 2 mM dithiothreitol, 20 μCi of [γ-32 P] ATP, 10 μM unlabeled ATP, and 2 μg of substrate GST-c-Jun (1-79). The immunocomplex was incubated at 30°C for 30 min and then boiled with SDS sample buffer for 5 min. Finally, the protein was resolved on 10% SDS-PAGE, the gel was dried, and the radioactive bands were visualized using the PhosphorImager. To determine the total amount of JNK1 in each sample, whole-cell extracts were subjected to Western blot analysis using anti-JNK1 antibody.
2.12. Measurement of cytochrome c release
To determine the effect of guggulsterone on cytochrome c release, cells were treated with guggulsterone as indicated and then the cytosolic extracts were prepared as described [21]. Briefly, the cells were washed with PBS, resuspended in the buffer containing 0.25 M sucrose, 30 mM Tris–HCl (pH 7.9), 1 mM EDTA, 1 mM PMSF, 2 mM sodium orthovanadate, 10 mM NaF, 2 μg/ml leupeptin, and 2 μg/ml aprotinin and then homogenized gently with a glass Dounce homogenizer for 20 strokes. The homogenates were centrifuged at 2000 rpm for 10 min to remove nuclei, and the supernatants were centrifuged at 14,000 rpm for 30 min to remove mitochondria and other insoluble fragments. The supernatants were again centrifuged as above to ensure complete removal of mitochondria. Protein (50 μM) was subjected to 15% SDS-PAGE, and then Western blot analysis was performed using anti-cytochrome c antibody.
2.13. PARP cleavage assay
For detection of cleavage products of PARP, whole-cell extracts were prepared by lysing guggulsterone-treated cells in lysis buffer (20 mM Tris, pH 7.4; 250 mM NaCl; 2 mM EDTA, pH 8.0; 0.1 % Triton-X 100; 0.01 μg/ml aprotinin; 0.005 μg/ml leupeptin; 0.4 mM PMSF; and 4 mM NaVO4). Lysates were spun at 14000 rpm for 10 min to remove insoluble material, resolved by 10% SDS PAGE, and probed with PARP antibodies.
3. Results
Guggulsterone inhibited the proliferation of human leukemia, head and neck carcinoma, multiple myeloma, lung carcinoma, melanoma, breast carcinoma, and ovarian cancer cell lines in a dose-dependent manner (Table 1). Guggulsterone also inhibited the proliferation of gleevac-resistant K562 cells, dexamethasone-resistant MM1 cells, and doxorubicin-resistant breast cancer cells. While dexamethasone-resistant and doxorubicin-resistant cells were less sensitive than their parental clone; gleevec-resistant cells were more sensitive than the parental clone. For convenience of cell culture, we selected U937 leukemia cells to investigate the mechanism by which guggulsterone exhibits cytotoxicity.
Table 1. Effect of guggulsterone on viability of tumor cell lines.
| Cell Viability (%) | |||||
|---|---|---|---|---|---|
| Guggulsterone (μM) | 1 | 5 | 10 | 25 | 50 |
| Leukemia | |||||
| Histiocytic leukemia (U937) | 90 | 45 | 19 | 17 | 15 |
| T-cell leukemia (Jurkat) | 61 | 27 | 15 | 14 | 12 |
| Chronic myelogenous leukemia (KBM-5) | 90 | 36 | 16 | 13 | 9 |
| Chronic myelogenous leukemia (K562) | 84 | 84 | 84 | 76 | 70 |
| K562 (STI-resistant) | 91 | 91 | 75 | 73 | 58 |
| Human Multiple Myeloma | |||||
| U266 | 45 | 23 | 22 | 18 | 16 |
| MM1 | 73 | 61 | 61 | 57 | 40 |
| MM1 (Dexamethasone-resistant) | 93 | 86 | 63 | 50 | 45 |
| Human Head and Neck Carcinoma Cells | |||||
| HNSCC 1 (FADU) | 90 | 66 | 21 | 9 | 6 |
| HNSCC from oral cavity (SCC4) | 85 | 66 | 30 | 15 | 13 |
| Head and Neck primary keratinocytes (HN5) | 67 | 40 | 18 | 16 | 13 |
| Human Lung Cells | |||||
| Non-small cell lung carcinoma (H1299) | 82 | 45 | 35 | 24 | 19 |
| Normal bronchial epithelial (BEAS-2B) | 88 | 78 | 27 | 14 | 13 |
| Human Melanoma | |||||
| Metastatic melanoma (A375) | 88 | 65 | 24 | 18 | 12 |
| Non-metastatic melanoma (WM35) | 93 | 59 | 19 | 17 | 11 |
| Human Breast Cancer | |||||
| MCF7 | 100 | 81 | 65 | 42 | 27 |
| MCF7 (Doxorubicin resistant) | 80 | 73 | 68 | 52 | 37 |
| Human Ovarian Cancer | |||||
| Hey 8 | 67 | 67 | 57 | 37 | 28 |
| SKOV3 | 82 | 82 | 59 | 49 | 33 |
| Human Embryonic Kidney | |||||
| 293 | 82 | 55 | 21 | 14 | 12 |
Human Head and Neck Squamous Cell Carcinoma; The results are shown as the mean from triplicate cultures.
3.1. Guggulsterone inhibits proliferation of tumor cells
The 3H-thymidine incorporation method showed that guggulsterone inhibited DNA synthesis in U937 leukemia cells in a dose-dependent manner. Guggulsterone at a concentration of 25 μM was sufficient to inhibit proliferation by 80% within 24 hours (Fig. 1A). In comparison to U937 cells, normal human fibroblasts were found to be relatively resistant to the cytotoxic effects of guggulsterone (Fig. 1B). Because guggulsterone inhibited the proliferation of U937 cells, we asked how this drug affects the progression of these cells through the cell cycle. To answer this question, we treated the cells with 10 μM guggulsterone and performed a cell cycle analysis using flow cytometry. We found that guggulsterone induced a G1/S arrest (Fig. 1C).
Fig. 1. Guggulsterone inhibits proliferation in U937 cells.
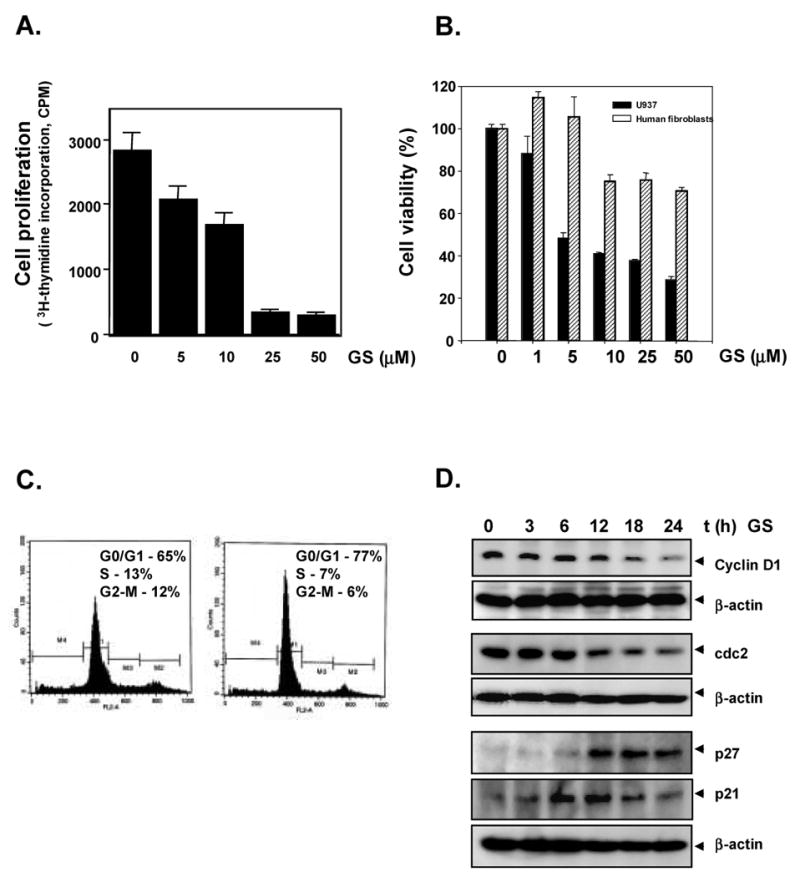
A, U937 cells (5000 cells/0.1 ml) were incubated at 37°C with indicated concentrations of guggulsterone for 72 hours, and the viable cells were assayed using [3H]-thymidine incorporation as described in Materials and Methods. The results are shown as the mean ± s.d. from triplicate cultures. B, Normal human fibroblasts are resistant to guggulsterone-induced cytotoxicity. Normal human fibroblasts and U937 cells (5000 cells/0.1 ml) were incubated at 37°C with indicated concentrations of guggulsterone for 72 h, and the viable cell number was assayed by MTT uptake as described in Materials and Methods. The results are shown as the mean ± s. d. from triplicate cultures. C, Guggulsterone arrests the cells at G1/S phase of the cell cycle. U937 cells (2 × 106 cells/ml) were incubated in the absence or in presence of 10 μM guggulsterone for 48 hours. Thereafter, the cells were washed, fixed, stained with propidium iodide, and analyzed for DNA content by flow cytometry as described in Materials and Methods. D, Guggulsterone modulates cell cycle proteins. Two million U937 cells were treated with guggulsterone (10 μM) for indicated time points and then whole cell extracts were prepared. Sixty micrograms of whole-cell extracts were resolved on 10% SDS-PAGE gel, electrotransferred onto a nitrocellulose membrane, and probed for cyclin D1, cdc2, p27, and p21. The same blots were stripped and reprobed with anti-β-actin antibody to show equal protein loading (lower panel).
3.2. Guggulsterone inhibits the expression of cell cycle regulated proteins
Because D-type cyclins and cyclin-dependent kinases are required for the progression of cells from the G1 phase to the S-phase of the cell cycle [22, 23], we determined the effect of guggulsterone on cyclin D1 expression in U937 cells. We found that guggulsterone inhibited the expression of both cyclin D1 and cdc2 in a time-dependent manner (Fig 1D). The cyclin-dependent kinase inhibitor p21 WAF1/CIP1 and p27 are prototypical member of the Cip/Kip family of cyclin –dependent kinase inhibitors. They negatively modulate cell cycle progression by inhibiting the activity of cyclin/Cdk2 complexes and blocks DNA replication by binding to proliferating cell nuclear antigen [24]. Guggulsterone induced the expression of both p21 and p27 in a time-dependent manner (Fig. 1D).
3.3. Guggulsterone inhibits anti-apoptotic gene products
Because the constitutive expression of the proteins Bfl-1, xIAP, cFLIP, BclXL, Bcl-2, and survivin has been implicated in cell survival and anti-apoptosis [25-33], we examined the effect of guggulsterone on the constitutive expression of these genes. Guggulsterone suppressed the expression of these genes in a time-dependent manner (Fig. 2A). Guggulsterone also downregulated the expression of COX-2 and c-myc, both of which have been closely linked with growth modulation (Fig. 2B).
Fig. 2.
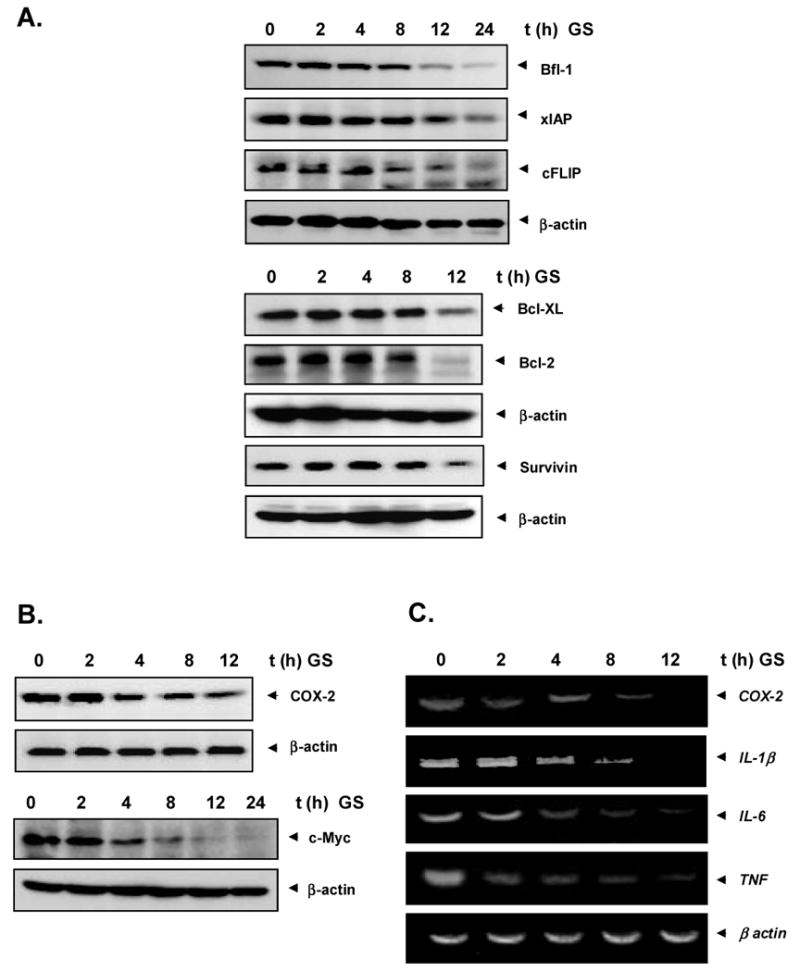
A, Guggulsterone inhibits the expression of anti-apoptotic gene products, Bfl-1/A1, XIAP, cFLIP, Bcl-2, BclXL, and survivin: U937 cells (2 × 106/ml) were left untreated or incubated with 10 μM guggulsterone for different times. Whole-cell extracts were prepared, and 50 μg of the whole-cell lysate was analyzed by Western blotting using antibodies against Bfl-1/A1, XIAP, cFLIP, Bcl-2, BclXL, and survivin as indicated. B, Guggulsterone inhibits COX-2 and c-myc expression. U937 cells (2 × 106/ml) were left untreated or incubated with 10 μM guggulsterone for different times. Whole-cell extracts were prepared, and 80 μg of the whole-cell lysate was analyzed by Western blotting using antibodies against COX-2 and c-myc. C, Guggulsterone inhibits gene expression. Four million U937 cells were treated with guggulsterone (10 μM) for indicated time points, and total mRNA was extracted and examined for expression of COX-2, IL-1β, IL-6, and TNF mRNA by RT-PCR. β-actin mRNA was used as an internal control to show equal RNA loading.
3.4. Guggulsterone modulates gene expression
To determine whether guggulsterone affects the transcription, the mRNA expression of COX2, IL-6, IL-1β and TNF was examined. The mRNA of all these genes were constitutively expressed and guggulsterone treatment suppressed the expression in a time-dependent manner (Fig 2C). These results suggest that guggulsterone modulates the expression of genes at transcription level.
3.5. Guggulsterone induces apoptosis in tumor cells
We next determined whether guggulsterone induced apoptosis in tumor cells. First of all, we used annexin V staining, which detects an early stage of apoptosis. These results indicated that guggulsterone induced apoptosis in a time dependent manner. The proportion of annexin V positive cells increased from 6% to 87% within 48 hours (Fig. 3A). Similar results were obtained with TUNEL staining (Fig. 3B). The Live and Dead assay, which detects intracellular esterase activity and plasma membrane integrity, also showed that guggulsterone induced apoptosis in a time-dependent manner (Fig. 3C).
Fig. 3. Guggulsterone induces apoptosis.
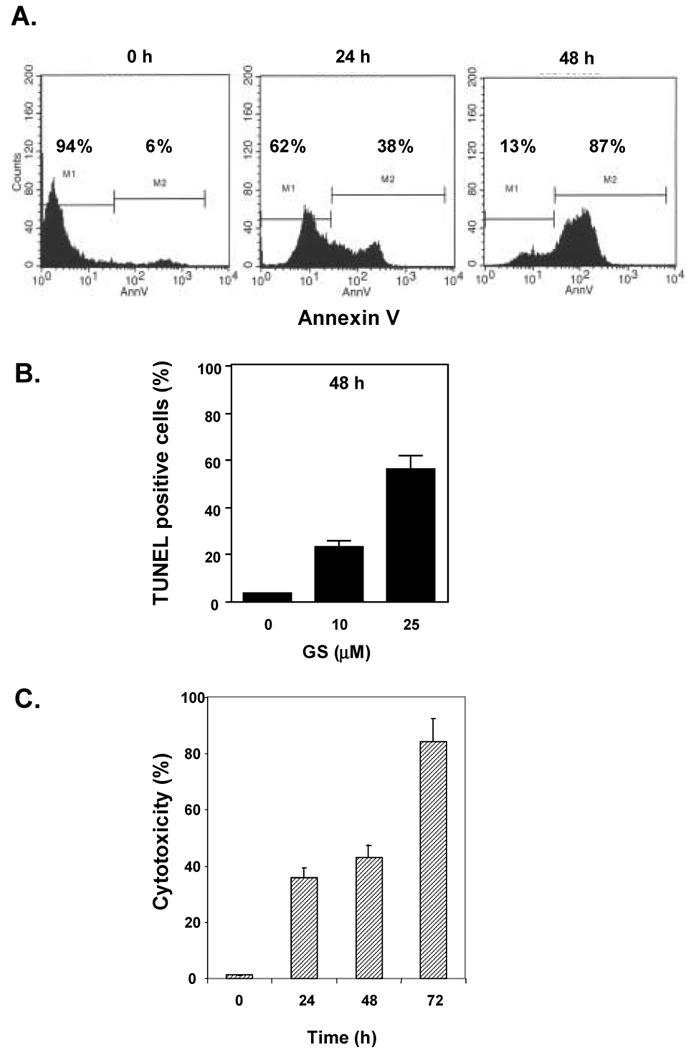
A, Cells were treated with 10 μM guggulsterone for indicated times, incubated with anti-annexin V antibody conjugated with FITC, and analyzed with a flow cytometer for early apoptotic effects. B, U937 cells were treated with different concentrations of guggulsterone for 48 h. Cells were fixed, stained with TUNEL assay reagent, and then analyzed with a flow cytometer for apoptotic effects. The results are shown as the mean ± s.d. from three experiments. C, Cells were treated with 10 μM guggulsterone for indicated times. Cells were stained with Live and Dead assay reagent for 30 min and then analyzed under a fluorescence microscope. The results are shown as the mean ± s.d. from three experiments.
3.6. Guggulsterone activates caspase-8 and induces BID cleavage, Bax expression, and cytochrome c release
Previously it was demonstrated that apoptosis by most agents activates an upstream protease called FLICE, or caspase-8 [13]. Western blotting showed that guggulsterone activated caspase-8 (Fig. 4A, upper panel) and the proapoptotic protein Bax in a time-dependent manner. Because chemotherapeutic agents induce apoptosis through the caspase-8-mediated cleavage of Bid, a proapoptotic member of the bcl-2 family [34], we investigated guggulsterone's effect on Bid cleavage. Western blot analysis demonstrated that guggulsterone also induced Bid cleavage in a time-dependent manner (Fig. 4A, middle panel). Likewise, it induced the release of cytochrome c from the mitochondria (Fig. 4B), another essential step in the apoptosis pathway activated by chemotherapeutic agents [35, 36].
Fig. 4. Guggulsterone induces caspase activation, cytochrome c release and PARP cleavage.
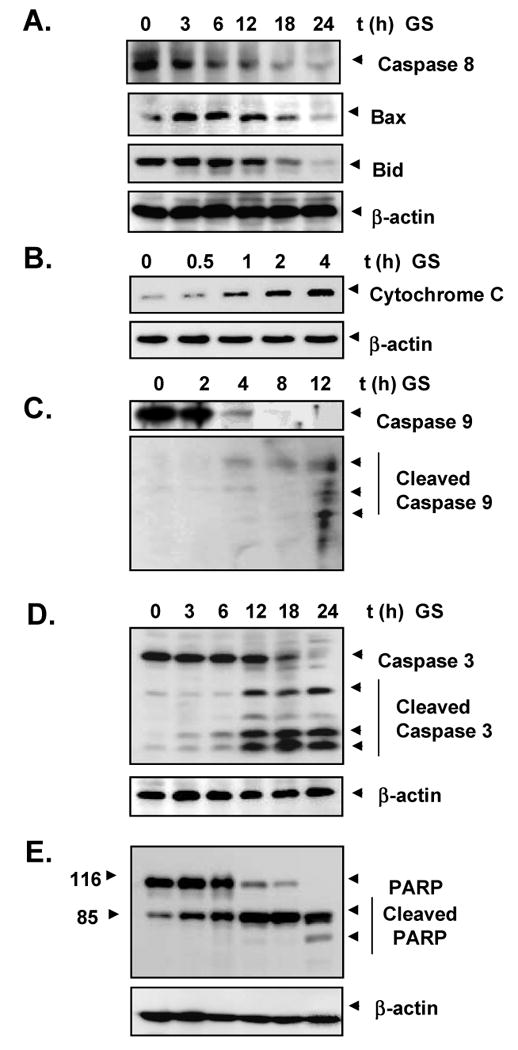
Cells were treated with 10 μM guggulsterone for the indicated times. Whole-cell extracts were prepared and subjected to Western blot analysis using antibodies against A, caspase 8, bax, and bid; B, cytochrome C; C, caspase 9; D, caspase 3; and E, PARP.
3.7. Guggulsterone induces cleavage of procaspase 9, procaspase 3, and PARP
Because procaspase 9, procaspase 3, and PARP are associated with the apoptotic cell death pathway [37], we next examined their levels in guggulsterone-treated U937 cells. Guggulsterone induced the cleavage of procaspase 9 at 4-8 h as seen by the disappearance of the procaspase band and appearance of its cleavage products (Fig. 4C). Treatment with guggulsterone also significantly induced the activation of caspase 3. The cleavage products of procaspase 3 first appeared at 8 hours, and the caspase completely disappeared by 24 hours (Fig. 4D). Guggulsterone induced PARP cleavage at 12 hours, leading to complete cleavage by 24 hours (Fig. 4E). Overall, these results demonstrate that guggulsterone induced the activation of caspase 9 and caspase 3 that led to the cleavage of PARP.
3.8. Guggulsterone suppresses Akt pathway
Because the PKB/Akt pathway provides a survival signal that protects tumor cells from apoptosis induced by various stresses [38], we examined the effect of guggulsterone on Akt activation. As shown in Fig. 5A, Akt was constitutively phosphorylated (i.e., activated) in U937 cells, but Guggulsterone treatment inhibited the Akt activation at 8 hours and completely abrogated it by 12 hours. Our results show that guggulsterone treatment suppressed the phosphorylation of Akt at both serine 473 and threonine 308. Guggulsterone had no effect on Akt protein levels (Fig. 5A1, 5A2).
Fig. 5.
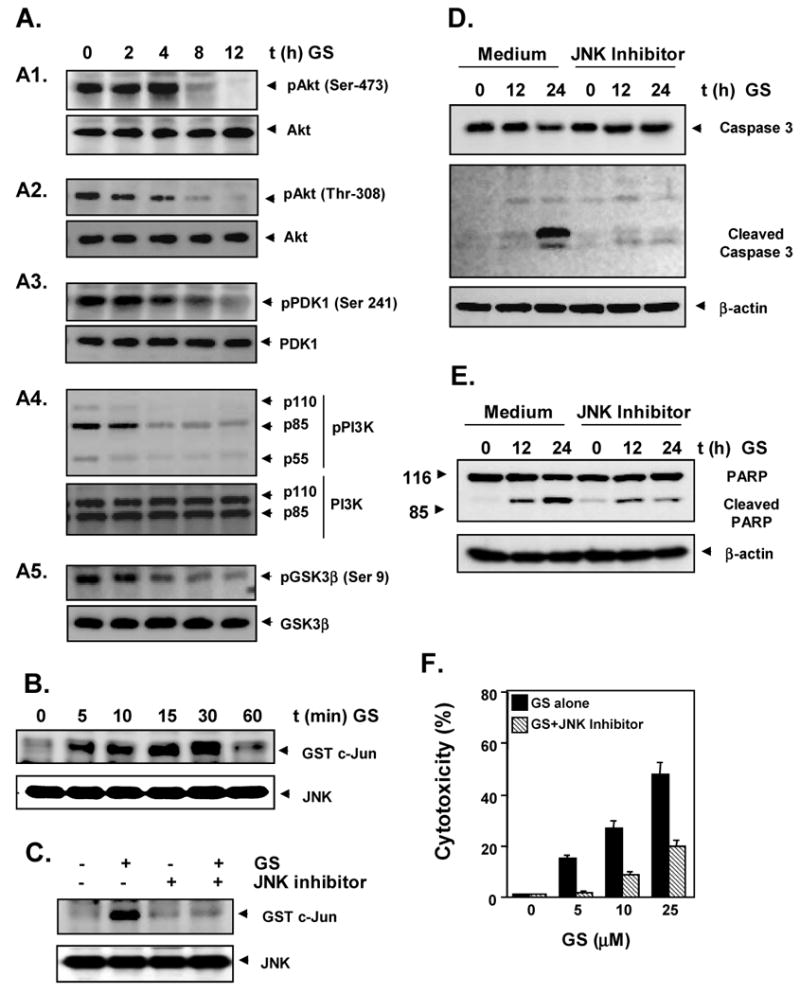
A, Guggulsterone inhibits Akt pathway. U937 cells were incubated with 10 μM guggulsterone for the indicated times. Whole-cell extracts were prepared and analyzed by Western blot analysis using antibodies against phosphorylated Akt (ser 473 and thr 308), Akt, phosphorylated PDK1 (ser 241), PDK1, phosphorylated PI3K, PI3K, phosphorylated GSK3β (ser 9) and GSK3β. B, Guggulsterone induces JNK activation. Cells were incubated with 10 μM guggulsterone for the indicated lengths of time. Whole-cell extracts were immunoprecipitated with an antibody against JNK and analyzed with an immunocomplex kinase assay as described in Materials and Methods. To examine the effect of guggulsterone on the level of expression of JNK proteins, whole-cell extracts were fractionated on SDS-PAGE and examined by Western blot analysis using anti-JNK antibodies. The results shown are representative of three independent experiments. C, D, JNK inhibition blocks caspase 3 activation. U937 cells were pretreated with 10 μM JNK inhibitor (SP600125) for 1 hour and then treated with 10 μM guggulsterone for indicated times. Whole cells extracts were prepared and examined for JNK activation (C) and caspase 3 activation by Western blot (D). E, JNK inhibition blocks PARP cleavage. U937 cells were pretreated with JNK inhibitor (SP600125) for 1 hour and then treated with 10 μM guggulsterone for indicated times, and whole-cell extracts were prepared and examined for PARP cleavage by Western blot. F, JNK inhibition blocks guggulsterone-induced cytotoxicity. U937 (5000 cells/0.1 ml) were incubated at 37°C with indicated concentrations of guggulsterone alone or in combination with JNK inhibitor for 72 h, and the viable cell number was assayed by MTT uptake as described in Materials and Methods. The results are shown as the mean ± s. d. from triplicate cultures.
We then investigated the effect of guggulsterone on the proteins upstream and downstream of Akt in the Akt signaling pathway. Guggulsterone suppressed the phosphorylation of the upstream kinase PDK1 at Serine 241 (Fig. 5A3) and phosphorylation of the p85 subunit of PI3K (Fig. 5A4). Suppression of Akt activation by guggulsterone led to the suppression of phosphorylation at serine 9 of GSK3β, the substrate of Akt (Fig. 5A5).
3.9. Guggulsterone activates JNK
Because JNK activation is linked with apoptosis [16], we measured the effects of guggulsterone on the activation of JNK by an in vitro kinase assay. Guggulsterone induced JNK activation as early as 5 minutes after treatment without altering the levels of JNK protein expression (Fig. 5B). The specific JNK inhibitor, SP600125 completely inhibited guggulsterone-induced JNK activation (Fig. 5C).
3.10. JNK activation is required for guggulsterone-induced apoptosis
We next determined whether suppression of JNK by a specific inhibitor of JNK restrains guggulsterone-induced activation of caspase 3 and cleavage of PARP. Cells were pretreated with SP600125 and then treated with guggulsterone. Western blot analysis showed that SP600125 inhibited the activation of caspase 3 (Fig. 5D) and PARP cleavage (Fig 5E). We next examined the effect of SP600125 on guggulsterone-induced apoptosis. The results in Fig. 5F suggest that suppression of JNK inhibited apoptosis induced by guggulsterone. These results show that guggulsterone most likely induces apoptosis through the activation of JNK.
3.11. Genetic deletion of MKK4 abolishes guggulsterone-induced apoptosis
Because MKK4 phosphorylation has been shown to activate JNK [39], we compared the effect of guggulsterone on JNK activation in mouse embryonic fibroblast from MKK4 gene-deleted mice with its effects on JNK activation in wild-type murine embryonic fibroblasts. Guggulsterone induced JNK activation in wild-type cells but not in MKK4-deleted cells (Fig 6A). To further establish the role of MKK4 in guggulsterone-induced apoptosis, we compared the cytotoxic effect of guggulsterone on MKK4 gene-deleted and wild type cells. We found that guggulsterone was cytotoxic to MKK4 wild-type cells but not mutant cells (Fig. 6B). These results once again suggest that MKK4-induced JNK activation is required for guggulsterone-induced apoptosis.
Fig. 6.
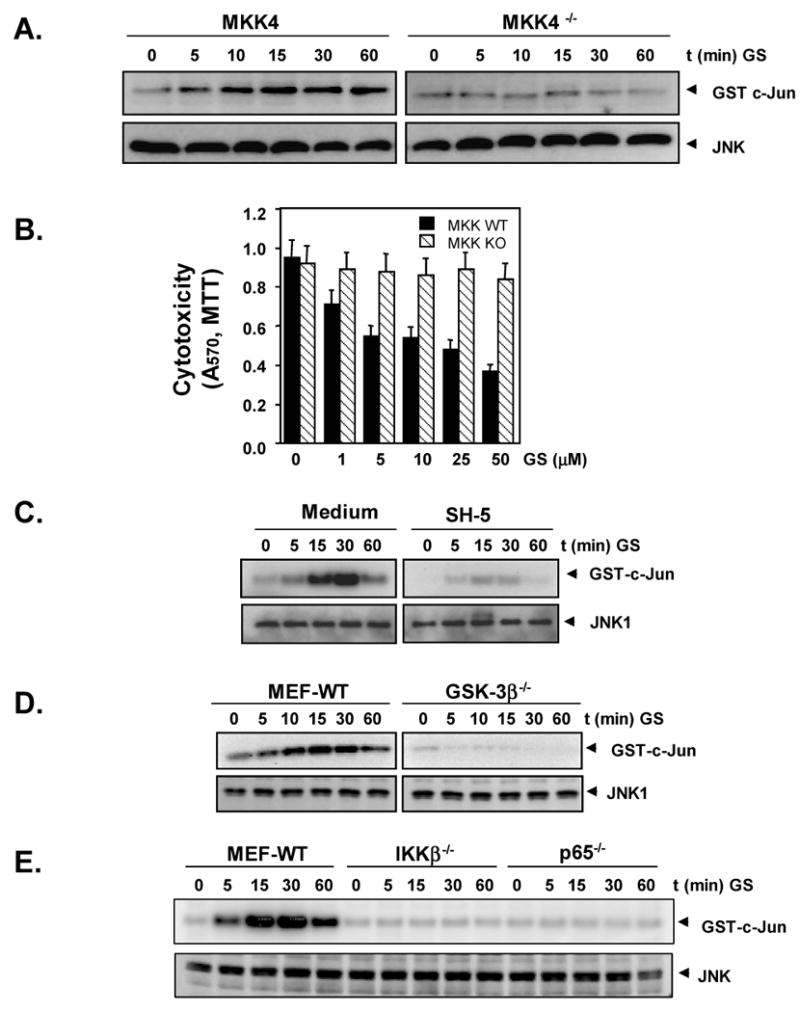
A, MKK4 is required for guggulsterone-induced JNK activation. MKK4 mutants and wild type-cells were treated with 10 μM guggulsterone for indicated times, and a JNK assay was performed as described in Materials and Methods. B, MKK4 is required for guggulsterone-induced cytotoxicity. MKK4 mutants and wild-type cells (1000 cells/0.1 ml) were incubated at 37°C with indicated concentrations of guggulsterone for 72 h, and the viable cell number was assayed by MTT uptake as described in Materials and Methods. The results are shown as the mean ± s. d. of triplicate cultures. C, Akt inhibitor suppresses JNK activation by guggulsterone. U937 cells were pretreated with 5 μM Akt inhibitor (SH-5) for 4 hours and then treated with 10 μM guggulsterone for indicated times. Whole cell extracts were prepared and examined for JNK activation as described in Materials and Methods. D, GSK3β is required for JNK activation by guggulsterone. Wild type and GSK3β-deficient murine embryonic fibroblasts cells were treated with 10 μM guggulsterone for indicated times. Whole cell extracts were prepared and examined for JNK activation as described in Materials and Methods E, p65 and IKK are required for JNK activation by guggulsterone. Wild type-, IKKβ-deficient and p65-deficient murine embryonic fibroblast cells were treated with 10 μM guggulsterone for indicated times. Whole cells extracts were prepared and examined for JNK activation as described in Materials and Methods.
3.12. Suppression of Akt and activation of JNK by guggulsterone are functionally related
Because guggulsterone suppressed Akt activation and upregulated JNK activation, we determined whether JNK upregulation and Akt downregulation are functionally related. We determined the effect of Akt inhibitor SH-5 on guggulsterone-induced JNK activation. We found that suppression of Akt by Akt inhibitor SH-5 led to the suppression of JNK activation (Fig. 6C).
To further confirm the results, we examined the effect of guggulsterone on JNK activation in GSK3β deficient cells. GSK3β is the substrate for Akt and is downstream of Akt in the Akt signaling pathway. The results show that guggulsterone failed to activate JNK in GSK3β-deficient cells (Fig. 6D). Thus it is possible that suppression of Akt and activation of JNK by guggulsterone are related to each other.
We next determined whether guggulsterone-induced activation of JNK and suppression of Akt is mechanistically related to NF-κB. To determine this, we examined the effect of guggulsterone on JNK activation in IKKβ-deficient and p65-deficient murine embryonic fibroblasts. NF-κB activation is impaired in IKKβ-deficient and p65-deficient murine embryonic fibroblasts. Guggulsterone did not induce JNK activation in both IKKβ-deficient and p65-deficient murine embryonic fibroblasts (Fig. 6E). These results suggest that activation of JNK is mechanistically related to NF-κB activation.
4. Discussion
The present report describes the mechanism through which guggulsterone induces apoptosis in tumor cells. Guggulsterone inhibited proliferation of a wide variety of tumor cells, including ones resistant to gleevac, dexamethasone, and doxorubicin. It arrested cells at S-phase through the down-regulation of cyclin D1 and cdc2 and upregulation of p21, and p27, and it inhibited expression of the antiapoptotic genes Bfl-1, xIAP, cFLIP, cMyc, Bcl-2, Bcl-XL, survivin, c-myc, and COX-2. Guggulsterone increased the number of apoptotic cells as demonstrated by annexin V staining, TUNEL, and live-dead assays. Further, it activated caspase-8, induced Bid cleavage, caused mitochondrial cytochrome c release, and induced caspase-3 activation and PARP cleavage. We also found that guggulsterone induced activation of JNK that was mediated through MKK4. Finally, suppression of JNK activation by the specific JNK inhibitor SP600125 or deletion of the MKK4 gene blocked guggulsterone-induced apoptosis.
Our results suggest that guggulsterone blocked proliferation of tumor cells by arresting the cells in S-phase of the cell cycle. Cyclin D1 and cdc2 are required for the progression of cells from the G1 phase to the S-phase of the cell cycle [22, 23], and guggulsterone suppressed the expression of both of them in a time-dependent manner in U937 cells. The expression of the cyclin-dependent kinase inhibitors p21 WAF1/CIP1 and p27, which block cell cycle progression by inhibiting the activity of cyclin/Cdk2 complexes, was also upregulated by guggulsterone.
Our results indicate that guggulsterone treatment downregulates the expression of antiapoptotic gene products Bfl-1, xIAP, cFLIP, cMyc, Bcl-2, Bcl-Xl, survivin, c-myc and COX-2. These observations are in agreement with a report in which guggulsterone was shown to downregulate the expression of Bcl-2 and Bcl-XL in prostate cancer cell lines [11]. Our study also demonstrates that guggulsterone induces apoptosis in part through the activation of caspase-8. However, the mechanism by which guggulsterone activates caspase-8 is not clear. Several reports indicate that auto-activation induced by oligomerization can activate caspase-8 [40, 41]. Thus, it may be possible that guggulsterone induces the oligomerization of caspase-8. Activation of caspase 8 cleaves Bid that then translocates to the mitochondria and stimulates the release of cytochrome c, which in turn activates caspase 9. Our results demonstrate that guggulsterone induced Bid cleavage followed by the release of cytochrome c and the activation of caspase-3 and PARP cleavage. Thus, one possible mechanism by which guggulsterone induces apoptosis is through changes in the mitochondrial membrane potential, which would lead to the release of cytochrome c from the mitochondria, leading to sequential activation of caspase-9 and caspase-3. These observations are in agreement with a report in which guggulsterones were shown to induce a loss in mitochondrial membrane potential, leading to apoptosis [12].
Interestingly, we found that normal human fibroblast cells are relatively resistant to growth inhibition by guggulsterone in comparison to tumor cells. These observations are in agreement with previous reports that guggulsterone inhibits proliferation of PC-3 cells, whereas the normal prostate epithelial cell line is resistant to guggulsterone [11].
The pharmacological activity of guggulsterone has been suggested to be mediated by the nuclear hormone receptor FXR. Guggulsterone has been shown to be an antagonist to ligand for FXR. Previous studies have shown that FXR agonists enhance apoptosis in ovarian cancer cells [42] and vascular smooth muscle cells [43]. FXR is expressed at a higher level in ductal epithelial cells of normal breast and infiltrating ductal carcinoma cells in breast cancer. FXR was also present in the human breast carcinoma cells, MCF-7 and MDA-MB-468. Activation of FXR by high concentrations of ligands induced apoptosis in breast cancer cells [44]. A recent study by De Gottardi et al. shows that overexpression of bile acid receptor FXR in Barrett's esophagus enhances apoptosis by guggulsterone in vitro [45]. Thus, it is possible that this steroid acts through the bile acid receptor.
Our results indicate that besides downregulating antiapoptotic gene products, guggulsterone also mediates its effects through downregulation of Akt pathway. This pathway is closely linked with cell survival [46]. We examined the effect of guggulsterone treatment on Akt phosphorylation and found that guggulsterone suppressed the phosphorylation of Akt at both Serine 473 and Threonine 308 residues. Guggulsterone also suppressed the phosphorylation of the upstream kinase PDK1 at Serine 241. The phosphorylation of the p85 subunit of another upsteam kinase, PI3K was also suppressed upon guggulsterone treatment. Suppression of Akt by guggulsterone led to the suppression of phosphorylation of GSK3β, the substrate of Akt. We also found that guggulsterone activated JNK and that suppression of JNK by its specific inhibitor abolished the activation of caspase 3, PARP cleavage, and cell proliferation. The activation of JNK requires the activation of an upstream kinase MKK4 [21]. Using MKK4-gene deleted murine embryonic fibroblast cells, we found that guggulsterone activated JNK in wild type cells but not in MKK4-deficient mutant cells, thus suggesting that the activation of JNK by guggulsterone requires MKK4. The lack of JNK activation in mutated cells correlated with suppression of guggulsterone-induced apoptosis; again suggesting the critical role of JNK. These results are in agreement with reports that JNK is required for apoptosis induced by TNFα [47], FasL [48], and TRAIL [49, 50]. JNK activation is also needed for apoptosis induced by chemotherapeutic agents [51]. Whether the mechanism of action of guggulsterone is different in various cell types is possible but unlikely as we found that GS activated JNK in both leukemic as well as in epithelial cells.
Guggulsterone treatment suppressed the activation of Akt, the prosurvival signal but induced the JNK activation. The results suggest that suppression of Akt and activation of JNK by guggulsterone are related to each other. Guggulsterone did not induce JNK activation in IKKβ-deficient and p65-deficient murine embryonic fibroblasts, thereby, suggesting that activation of JNK is mechanistically related to NF-κB activation, which has antiapoptotic role.
Because of lack of any known toxicity, guggulsterone should be further explored for its anticancer potential. Whether the concentrations used in our studies can be achieved in vivo, is unclear at present. No data is at present available on the pharmacokinetics, pharmacodynamics, and bioavailability of guggulsterone in animals or human. The hypolipidemic and antoxidant effects have been reported in human with as little as 50 mg of guggulipids, administered twice daily for 24 weeks [52]. How does these doses compare with studies performed here, is not clear. Overall, our results indicate that guggulsterone inhibits the growth of wide variety of cells and induces apoptosis through downregulation of antiapoptotic gene products, modulation of cell cycle proteins, activation of caspases, inhibition of Akt and activation of JNK.
Acknowledgments
We would like to thank Walter Pagel for a careful review of the manuscript. Dr. Aggarwal is a Ransom Horne Jr. Professor of Cancer Research. This work was supported in part by the Odyssey Program and the Theodore N. Law Award for Scientific Achievement at The University of Texas M. D. Anderson Cancer Center (to SS).
Grant support: Supported by the Clayton Foundation for Research (to BBA), a Department of Defense US Army Breast Cancer Research Program grant (BC010610, to BBA), a PO1 grant (CA91844) from the National Institutes of Health on lung chemoprevention (to BBA), and a P50 Head and Neck SPORE grant from the National Institutes of Health (to BBA).
Abbreviations
- NF-κB
nuclear factor-kappa B
- COX-2
cyclooxygenase-2
- MMP-9
matrix metalloproteinase-9
- FBS
fetal bovine serum
- SDS
sodium dodecyl sulfate
- TUNEL
terminal deoxyuridine triphosphate nick end-labeling
- JNK
cJun N-terminal kinase
- MKK
mitogen-activated protein kinase kinase
- PARP
poly (ADP-ribose) polymerase activation
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Engel LW, Straus SE. Development of therapeutics: opportunities within complementary and alternative medicine. Nat Rev Drug Discov. 2002;1:229–37. doi: 10.1038/nrd750. [DOI] [PubMed] [Google Scholar]
- 2.Urizar NL, Moore DD. GUGULIPID: a natural cholesterol-lowering agent. Annu Rev Nutr. 2003;23:303–13. doi: 10.1146/annurev.nutr.23.011702.073102. [DOI] [PubMed] [Google Scholar]
- 3.Sinal CJ, Gonzalez FJ. Guggulsterone: an old approach to a new problem. Trends Endocrinol Metab. 2002;13:275–6. doi: 10.1016/s1043-2760(02)00640-9. [DOI] [PubMed] [Google Scholar]
- 4.Gujral ML, Sareen K, Tangri KK, Amma MK, Roy AK. Antiarthritic and anti-inflammatory activity of gum guggul (Balsamodendron mukul Hook) Indian J Physiol Pharmacol. 1960;4:267–73. [PubMed] [Google Scholar]
- 5.Sharma JN. Comparison of the anti-inflammatory activity of Commiphora mukul (an indigenous drug) with those of phenylbutazone and ibuprofen in experimental arthritis induced by mycobacterial adjuvant. Arzneimittelforschung. 1977;27:1455–7. [PubMed] [Google Scholar]
- 6.Singh BB, Mishra LC, Vinjamury SP, Aquilina N, Singh VJ, Shepard N. The effectiveness of Commiphora mukul for osteoarthritis of the knee: an outcomes study. Altern Ther Health Med. 2003;9:74–9. [PubMed] [Google Scholar]
- 7.Meselhy MR. Inhibition of LPS-induced NO production by the oleogum resin of Commiphora wightii and its constituents. Phytochemistry. 2003;62:213–8. doi: 10.1016/s0031-9422(02)00388-6. [DOI] [PubMed] [Google Scholar]
- 8.Shishodia S, Aggarwal BB. Guggulsterone inhibits NF-kappaB and IkappaBalpha kinase activation, suppresses expression of anti-apoptotic gene products, and enhances apoptosis. J Biol Chem. 2004;279:47148–58. doi: 10.1074/jbc.M408093200. [DOI] [PubMed] [Google Scholar]
- 9.Ichikawa H, Aggarwal BB. Guggulsterone inhibits osteoclastogenesis induced by receptor activator of nuclear factor-kappaB ligand and by tumor cells by suppressing nuclear factor-kappaB activation. Clin Cancer Res. 2006;12:662–8. doi: 10.1158/1078-0432.CCR-05-1749. [DOI] [PubMed] [Google Scholar]
- 10.Aggarwal BB. Nuclear factor-kappaB: the enemy within. Cancer Cell. 2004;6:203–8. doi: 10.1016/j.ccr.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 11.Singh SV, Zeng Y, Xiao D, Vogel VG, Nelson JB, Dhir R, Tripathi YB. Caspase-dependent apoptosis induction by guggulsterone, a constituent of Ayurvedic medicinal plant Commiphora mukul, in PC-3 human prostate cancer cells is mediated by Bax and Bak. Mol Cancer Ther. 2005;4:1747–54. doi: 10.1158/1535-7163.MCT-05-0223. [DOI] [PubMed] [Google Scholar]
- 12.Samudio I, Konopleva M, Safe S, McQueen T, Andreeff M. Guggulsterones induce apoptosis and differentiation in acute myeloid leukemia: identification of isomer-specific antileukemic activities of the pregnadienedione structure. Mol Cancer Ther. 2005;4:1982–92. doi: 10.1158/1535-7163.MCT-05-0247. [DOI] [PubMed] [Google Scholar]
- 13.Ashkenazi S, Cleary TG. A method for detecting Shiga toxin and Shiga-like toxin-I in pure and mixed culture. J Med Microbiol. 1990;32:255–61. doi: 10.1099/00222615-32-4-255. [DOI] [PubMed] [Google Scholar]
- 14.Sun XM, MacFarlane M, Zhuang J, Wolf BB, Green DR, Cohen GM. Distinct caspase cascades are initiated in receptor-mediated and chemical-induced apoptosis. J Biol Chem. 1999;274:5053–60. doi: 10.1074/jbc.274.8.5053. [DOI] [PubMed] [Google Scholar]
- 15.Nakanishi C, Toi M. Nuclear factor-kappaB inhibitors as sensitizers to anticancer drugs. Nat Rev Cancer. 2005;5:297–309. doi: 10.1038/nrc1588. [DOI] [PubMed] [Google Scholar]
- 16.Varfolomeev EE, Ashkenazi A. Tumor necrosis factor: an apoptosis JuNKie? Cell. 2004;116:491–7. doi: 10.1016/s0092-8674(04)00166-7. [DOI] [PubMed] [Google Scholar]
- 17.de Vente JE, Kukoly CA, Bryant WO, Posekany KJ, Chen J, Fletcher DJ, Parker PJ, Pettit GJ, Lozano G, Cook PP, et al. Phorbol esters induce death in MCF-7 breast cancer cells with altered expression of protein kinase C isoforms. Role for p53-independent induction of gadd-45 in initiating death. J Clin Invest. 1995;96:1874–86. doi: 10.1172/JCI118233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shishodia S, Amin HM, Lai R, Aggarwal BB. Curcumin (diferuloylmethane) inhibits constitutive NF-kappaB activation, induces G1/S arrest, suppresses proliferation, and induces apoptosis in mantle cell lymphoma. Biochem Pharmacol. 2005;70:700–13. doi: 10.1016/j.bcp.2005.04.043. [DOI] [PubMed] [Google Scholar]
- 19.Takada Y, Murakami A, Aggarwal BB. Zerumbone abolishes NF-kappaB and IkappaBalpha kinase activation leading to suppression of antiapoptotic and metastatic gene expression, upregulation of apoptosis, and downregulation of invasion. Oncogene. 2005;24:6957–69. doi: 10.1038/sj.onc.1208845. [DOI] [PubMed] [Google Scholar]
- 20.Takada Y, Khuri FR, Aggarwal BB. Protein farnesyltransferase inhibitor (SCH 66336) abolishes NF-kappaB activation induced by various carcinogens and inflammatory stimuli leading to suppression of NF-kappaB-regulated gene expression and up-regulation of apoptosis. J Biol Chem. 2004;279:26287–99. doi: 10.1074/jbc.M400963200. [DOI] [PubMed] [Google Scholar]
- 21.Yang D, Tournier C, Wysk M, Lu HT, Xu J, Davis RJ, Flavell RA. Targeted disruption of the MKK4 gene causes embryonic death, inhibition of c-Jun NH2-terminal kinase activation, and defects in AP-1 transcriptional activity. Proc Natl Acad Sci U S A. 1997;94:3004–9. doi: 10.1073/pnas.94.7.3004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Matsushime H, Roussel MF, Ashmun RA, Sherr CJ. Colony-stimulating factor 1 regulates novel cyclins during the G1 phase of the cell cycle. Cell. 1991;65:701–13. doi: 10.1016/0092-8674(91)90101-4. [DOI] [PubMed] [Google Scholar]
- 23.Enoch T, Nurse P. Mutation of fission yeast cell cycle control genes abolishes dependence of mitosis on DNA replication. Cell. 1990;60:665–73. doi: 10.1016/0092-8674(90)90669-6. [DOI] [PubMed] [Google Scholar]
- 24.Luo Y, Hurwitz J, Massague J. Cell-cycle inhibition by independent CDK and PCNA binding domains in p21Cip1. Nature. 1995;375:159–61. doi: 10.1038/375159a0. [DOI] [PubMed] [Google Scholar]
- 25.Zhu L, Fukuda S, Cordis G, Das DK, Maulik N. Anti-apoptotic protein survivin plays a significant role in tubular morphogenesis of human coronary arteriolar endothelial cells by hypoxic preconditioning. FEBS Lett. 2001;508:369–74. doi: 10.1016/s0014-5793(01)03084-8. [DOI] [PubMed] [Google Scholar]
- 26.Schwenzer R, Siemienski K, Liptay S, Schubert G, Peters N, Scheurich P, Schmid RM, Wajant H. The human tumor necrosis factor (TNF) receptor-associated factor 1 gene (TRAF1) is up-regulated by cytokines of the TNF ligand family and modulates TNF-induced activation of NF-kappaB and c-Jun N-terminal kinase. J Biol Chem. 1999;274:19368–74. doi: 10.1074/jbc.274.27.19368. [DOI] [PubMed] [Google Scholar]
- 27.Chu ZL, McKinsey TA, Liu L, Gentry JJ, Malim MH, Ballard DW. Suppression of tumor necrosis factor-induced cell death by inhibitor of apoptosis c-IAP2 is under NF-kappaB control. Proc Natl Acad Sci U S A. 1997;94:10057–62. doi: 10.1073/pnas.94.19.10057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.You M, Ku PT, Hrdlickova R, Bose HR., Jr ch-IAP1, a member of the inhibitor-of-apoptosis protein family, is a mediator of the antiapoptotic activity of the v-Rel oncoprotein. Mol Cell Biol. 1997;17:7328–41. doi: 10.1128/mcb.17.12.7328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stehlik C, de Martin R, Kumabashiri I, Schmid JA, Binder BR, Lipp J. Nuclear factor (NF)-kappaB-regulated X-chromosome-linked iap gene expression protects endothelial cells from tumor necrosis factor alpha-induced apoptosis. J Exp Med. 1998;188:211–6. doi: 10.1084/jem.188.1.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Catz SD, Johnson JL. Transcriptional regulation of bcl-2 by nuclear factor kappa B and its significance in prostate cancer. Oncogene. 2001;20:7342–51. doi: 10.1038/sj.onc.1204926. [DOI] [PubMed] [Google Scholar]
- 31.Grumont RJ, Rourke IJ, Gerondakis S. Rel-dependent induction of A1 transcription is required to protect B cells from antigen receptor ligation-induced apoptosis. Genes Dev. 1999;13:400–11. doi: 10.1101/gad.13.4.400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zong WX, Edelstein LC, Chen C, Bash J, Gelinas C. The prosurvival Bcl-2 homolog Bfl-1/A1 is a direct transcriptional target of NF-kappaB that blocks TNFalpha-induced apoptosis. Genes Dev. 1999;13:382–7. doi: 10.1101/gad.13.4.382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kreuz S, Siegmund D, Scheurich P, Wajant H. NF-kappaB inducers upregulate cFLIP, a cycloheximide-sensitive inhibitor of death receptor signaling. Mol Cell Biol. 2001;21:3964–73. doi: 10.1128/MCB.21.12.3964-3973.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Anto RJ, Mukhopadhyay A, Denning K, Aggarwal BB. Curcumin (diferuloylmethane) induces apoptosis through activation of caspase-8, BID cleavage and cytochrome c release: its suppression by ectopic expression of Bcl-2 and Bcl-xl. Carcinogenesis. 2002;23:143–50. doi: 10.1093/carcin/23.1.143. [DOI] [PubMed] [Google Scholar]
- 35.Luo X, Budihardjo I, Zou H, Slaughter C, Wang X. Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell. 1998;94:481–90. doi: 10.1016/s0092-8674(00)81589-5. [DOI] [PubMed] [Google Scholar]
- 36.Gross A, Yin XM, Wang K, Wei MC, Jockel J, Milliman C, Erdjument-Bromage H, Tempst P, Korsmeyer SJ. Caspase cleaved BID targets mitochondria and is required for cytochrome c release, while BCL-XL prevents this release but not tumor necrosis factor-R1/Fas death. J Biol Chem. 1999;274:1156–63. doi: 10.1074/jbc.274.2.1156. [DOI] [PubMed] [Google Scholar]
- 37.Hakem R, Hakem A, Duncan GS, Henderson JT, Woo M, Soengas MS, Elia A, de la Pompa JL, Kagi D, Khoo W, Potter J, Yoshida R, Kaufman SA, Lowe SW, Penninger JM, Mak TW. Differential requirement for caspase 9 in apoptotic pathways in vivo. Cell. 1998;94:339–52. doi: 10.1016/s0092-8674(00)81477-4. [DOI] [PubMed] [Google Scholar]
- 38.Marte BM, Downward J. PKB/Akt: connecting phosphoinositide 3-kinase to cell survival and beyond. Trends Biochem Sci. 1997;22:355–8. doi: 10.1016/s0968-0004(97)01097-9. [DOI] [PubMed] [Google Scholar]
- 39.Papa S, Zazzeroni F, Bubici C, Jayawardena S, Alvarez K, Matsuda S, Nguyen DU, Pham CG, Nelsbach AH, Melis T, De Smaele E, Tang WJ, D'Adamio L, Franzoso G. Gadd45 beta mediates the NF-kappa B suppression of JNK signalling by targeting MKK7/JNKK2. Nat Cell Biol. 2004;6:146–53. doi: 10.1038/ncb1093. [DOI] [PubMed] [Google Scholar]
- 40.Muzio M, Stockwell BR, Stennicke HR, Salvesen GS, Dixit VM. An induced proximity model for caspase-8 activation. J Biol Chem. 1998;273:2926–30. doi: 10.1074/jbc.273.5.2926. [DOI] [PubMed] [Google Scholar]
- 41.Srinivasula SM, Ahmad M, Fernandes-Alnemri T, Alnemri ES. Autoactivation of procaspase-9 by Apaf-1-mediated oligomerization. Mol Cell. 1998;1:949–57. doi: 10.1016/s1097-2765(00)80095-7. [DOI] [PubMed] [Google Scholar]
- 42.Alberts DS, Hallum AV, 3rd, Stratton-Custis M, Garcia DJ, Gleason-Guzman M, Salmon SE, Santabarbara P, Niesor EJ, Floret S, Bentzen CL. Phase I pharmacokinetic trial and correlative in vitro phase II tumor kinetic study of Apomine (SR-45023A), a novel oral biphosphonate anticancer drug. Clin Cancer Res. 2001;7:1246–50. [PubMed] [Google Scholar]
- 43.Bishop-Bailey D, Walsh DT, Warner TD. Expression and activation of the farnesoid X receptor in the vasculature. Proc Natl Acad Sci U S A. 2004;101:3668–73. doi: 10.1073/pnas.0400046101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Swales KE, Korbonits M, Carpenter R, Walsh DT, Warner TD, Bishop-Bailey D. The farnesoid X receptor is expressed in breast cancer and regulates apoptosis and aromatase expression. Cancer Res. 2006;66:10120–6. doi: 10.1158/0008-5472.CAN-06-2399. [DOI] [PubMed] [Google Scholar]
- 45.De Gottardi A, Dumonceau JM, Bruttin F, Vonlaufen A, Morard I, Spahr L, Rubbia-Brandt L, Frossard JL, Dinjens WN, Rabinovitch PS, Hadengue A. Expression of the bile acid receptor FXR in Barrett's esophagus and enhancement of apoptosis by guggulsterone in vitro. Mol Cancer. 2006;5:48. doi: 10.1186/1476-4598-5-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002;296:1655–7. doi: 10.1126/science.296.5573.1655. [DOI] [PubMed] [Google Scholar]
- 47.Deng Y, Ren X, Yang L, Lin Y, Wu X. A JNK-dependent pathway is required for TNFalpha-induced apoptosis. Cell. 2003;115:61–70. doi: 10.1016/s0092-8674(03)00757-8. [DOI] [PubMed] [Google Scholar]
- 48.Le-Niculescu H, Bonfoco E, Kasuya Y, Claret FX, Green DR, Karin M. Withdrawal of survival factors results in activation of the JNK pathway in neuronal cells leading to Fas ligand induction and cell death. Mol Cell Biol. 1999;19:751–63. doi: 10.1128/mcb.19.1.751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Herr I, Wilhelm D, Meyer E, Jeremias I, Angel P, Debatin KM. JNK/SAPK activity contributes to TRAIL-induced apoptosis. Cell Death Differ. 1999;6:130–5. doi: 10.1038/sj.cdd.4400467. [DOI] [PubMed] [Google Scholar]
- 50.Vivo C, Liu W, Broaddus VC. c-Jun N-terminal kinase contributes to apoptotic synergy induced by tumor necrosis factor-related apoptosis-inducing ligand plus DNA damage in chemoresistant, p53 inactive mesothelioma cells. J Biol Chem. 2003;278:25461–7. doi: 10.1074/jbc.M302161200. [DOI] [PubMed] [Google Scholar]
- 51.Koo MS, Kwo YG, Park JH, Choi WJ, Billiar TR, Kim YM. Signaling and function of caspase and c-jun N-terminal kinase in cisplatin-induced apoptosis. Mol Cells. 2002;13:194–201. [PubMed] [Google Scholar]
- 52.Singh RB, Niaz MA, Ghosh S. Hypolipidemic and antioxidant effects of Commiphora mukul as an adjunct to dietary therapy in patients with hypercholesterolemia. Cardiovasc Drugs Ther. 1994;8:659–64. doi: 10.1007/BF00877420. [DOI] [PubMed] [Google Scholar]