

Abstract
We used a replication-incompetent, single-cycle, alphavirus replicon vector system to produce virus-like replicon particles (VRP) expressing the extracellular domain of human cytomegalovirus (CMV) glycoprotein B or a pp65/IE1 fusion protein. Efficient production methods were scaled to produce pilot lots and clinical lots of each alphavirus replicon vaccine component. The vaccine induced high-titered antibody responses in mice and rabbits, as measured by ELISA and CMV neutralization assays, and robust T-cell responses in mice, as measured by IFN-γ ELISPOT assay. A toxicity study in rabbits showed no adverse effects in any toxicology parameter. These studies support clinical testing of this novel CMV alphavirus replicon vaccine in humans.
Keywords: cytomegalovirus vaccine, manufacturing process, cellular immunity, toxicology testing
1. Introduction
Human cytomegalovirus (CMV) is a major cause of morbidity and mortality in congenitally infected infants and hematopoietic stem cell and solid organ transplant recipients [1–3]. Although antiviral drugs have reduced the risk of CMV disease in transplant recipients, they are associated with potentially serious side effects and do not prevent late-onset CMV disease [4,5], and they have not had an impact on congenital CMV disease. Development of an effective vaccine to protect against CMV disease is recognized as an important public health priority [6].
An optimal CMV vaccine would ideally induce protective humoral and cellular immune responses. Glycoprotein B (gB) is a major target of neutralizing antibodies against CMV, and pp65 and IE1 are principal targets of protective T cell responses [7–10]. In previous studies, we used a replication-incompetent, single-cycle, alphavirus replicon vector system to produce virus-like replicon particles (VRP) expressing CMV genes and showed that immunization with an alphavirus replicon vaccine expressing the guinea pig CMV homolog of pp65 confers protection against congenital CMV disease [11] and immunization of mice with alphavirus replicon vaccines expressing human CMV gB, pp65 and IE1 proteins induce neutralizing antibodies and robust antigen-specific T cell responses [12]. In the present study we describe the construction, process development and preclinical evaluation of a candidate CMV alphavirus replicon vaccine suitable for clinical testing.
2. Materials and methods
2.1. Plasmid construction
Alphavirus replicon plasmids containing a gene encoding the extracellular portion of CMV gB or a pp65/IE1 fusion protein, under control of an EV71 internal ribosome entry site (IRES), were constructed as described by Kamrud et al. [13]. Genes encoding the extracellular portion of gB from CMV strain Towne and an in-frame fusion protein of pp65/IE1 from CMV strain AD169 were separately amplified by PCR from pERK3 alphavirus replicon plasmids [12] and ligated into the BamHI and XbaI sites of the pCDNA3.3/EMCV and pCDNA3.3/EV71 transfer vectors [13]. A region spanning the IRES and the CMV gene was digested from the transfer plasmid using AscI enzyme and cloned into pERK spacer-replicon vectors containing spacers of different sizes [13]. Based on protein expression and replicon packaging titers, a replicon vector containing an EV71 IRES and a spacer of 342 nucleotides in length was selected for both gB and pp65/IE1 (Figure 1). In the pp65/IE1 gene, an internal Not1 restriction endonuclease site in the pp65 native sequence was inactivated by site-directed mutagenesis to introduce a silent mutation (GCGGCCGC to GCGGCCGA) in the Not1 site. DNA sequencing showed no other differences from the published sequences for pp65 and IE1 and identified a single nucleotide difference from the published sequence that resulted in an Ile-156→Val change for gB [12]. This is a conservative amino acid change that is not in either of the recognized antibody-binding sites that are the principal targets of neutralizing antibodies in serum from persons naturally infected with CMV [14,15].
Figure 1.

Maps of the replicon plasmids containing the gene for CMV gB (A) or pp65/IE1 (B). Starting at the T7 promoter and moving clockwise, the solid arrows represent the four alphavirus nonstructural protein (nsP) genes, the 342 bp spacer, the EV71 IRES, the CMV gene and the kanamycin resistance [KN (R)] gene, respectively.
2.2. VRP production
VRP were produced by electroporation of Vero cells with replicon RNA and two helper RNAs, encoding the Venezuelan equine encephalitis (VEE) virus structural proteins, using modifications of previously described methods [16,17]. VRP concentration, expressed as infectious units (IU) per mL, was determined by an immunofluorescence assay in which serial dilutions of VRP were added to Vero cell monolayers, cultured overnight, reacted with goat anti-gB or anti-pp65 antibody [12] followed by fluorescein isothiocyanate-labeled anti-goat antibody to detect cells expressing the CMV protein. After optimal concentrations of the three RNAs were determined for VRP produced at laboratory scale, VRP were produced at pilot scale and clinical scale using the AlphaVax platform process described below.
Purified DNA plasmids were linearized by NotI endonuclease digestion and used as templates for in vitro RNA transcription using RNA Express T7 kits (Promega, Madison, WI). After transcription the reaction mixture was treated with AluI to digest the replicon DNA template, and RNA was purified by size exclusion chromatography and stored at −80°C until use.
A Vero working cell bank, cryopreserved at passage 142, was thawed and cultured in Eagle’s minimum essential medium (EMEM) with 5% fetal bovine serum (FBS) in 175 cm2 flasks at 37°C, 5% CO2. Culture medium was changed after 24 hr and 72 hr later cells were washed with phosphate buffered saline (PBS), detached by treatment with 0.05% trypsin (Hyclone, Logan, UT) and transferred to 850 cm2 roller bottles. After 72 hr cells were harvested, counted, washed and re-suspended in PBS to a concentration of 1.5–2.0 × 108 cells/mL, mixed with replicon, capsid helper and glycoprotein helper RNA, transferred to 0.4 cm gap cuvettes and electroporated using a Gene Pulser II electroporation unit (BioRad, Hercules, CA). Electroporated cells were resuspended in 100 mL OptiPRO SFM (Invitrogen, Carlsbad, CA) with 4 mM glutamine and cultured at 37°C, 5% CO2 in 850 cm2 roller bottles. After 16–24 hr the medium and cells from all roller bottles were pooled and drawn into a Sartopore 2 capsule filter (Sartorius, Edgewood, NY) pre-wetted with PBS. Cells collected on the filter were washed with PBS and VRP were recovered by washing with a high salt buffer. A portion of the salt wash material (a total of 3 × 108 IU) was tested in a cytopathic effect (CPE) assay to confirm the absence of detectable replication-competent virus. In brief, VRP eluted by salt wash were added to Vero cell culture monolayers in T75 tissue culture flasks at a controlled multiplicity of infection (MOI) of <0.5 and incubated at 37 °C in a 5% CO2 atmosphere for 1 hr. The inoculum was removed and the cells were incubated for 24 hr. The cell culture supernatant from each Passage 1 flask was transferred to a fresh flask of Vero cells and incubated for 1 hr, the inoculum removed and fresh culture medium added. CPE was assessed after incubation for 72 hr.
The salt wash material was concentrated on a Hydrosart 100,000 molecular weight cutoff regenerated cellulose flat-sheet tangential flow filtration (TFF) membrane (Sartorius). The solution was then diafiltered against PBS with 3 mM MgCl2, treated with 50,000 U of Benzonase to degrade contaminating Vero DNA, diluted with 5 M NaCl to a final NaCl concentration of 2 M, and diafiltered against 1 M NaCl in 10 mM phosphate. The TFF pool was filtered through a 0.2 μm filter and loaded on a Cellufine Sulfate column pre-equilibrated with 250 mM NaCl in 10 mM phosphate. The column was sequentially washed with 250 mM NaCl in 10 mM phosphate and 500 mM NaCl in 10 mM phosphate and VRP were eluted with a step gradient to 800 mM NaCl in 10 mM phosphate. Purified VRP were sampled for quality control analysis and formulated as bulk vaccine in an excipient mix that stabilized the VRP during storage at −80°C.
2.3. Quality control testing of VRP
Various process pools were tested for residual protein, DNA and Benzonase concentrations, sodium dodecyl sulfate polyacrilamide gel electrophoresis (SDS-PAGE) and western blot characterization, Southern blot estimation of residual Vero DNA size, and quantitative polymerase chain reaction (qPCR) to determine genome equivalent concentration. Protein was measured by the bicinchoninic acid method using a commercially available kit (Pierce Biotechnology, Rockford, IL) and bovine serum albumin (BSA) as the reference standard. DNA was measured by the picogreen method using a commercially available kit (Invitrogen). Benzonase was measured by ELISA using a commercially available kit (EMD Chemicals, Gibbstown, NJ). SDS-PAGE was performed on NuPAGE 4–12% gradient Bis-tris gels (Invitrogen) under reducing conditions. Western blot analysis was performed as previously described [12] using mouse antibodies against VEE virus envelope and capsid proteins. For Southern blot analysis, samples were treated with proteinase K and DNA extracted on Minelute spin columns (Qiagen), separated by agarose gel electrophoresis, blotted to a nylon membrane and cross-linked. The DNA was hybridized to denatured, psoralen-biotin-labeled, AluI-digested Vero DNA, washed, reacted with streptavidin-labeled alkaline phosphatase (Ambion, Austin, TX) and a chemiluminescent reagent, and the image captured on X-ray film. qPCR was performed on an Applied Biosystems 7500 Fast Real-Time PCR system. Ampliset primers and probes were constructed based on the nsP2 region of the replicon, and RNA was quantified during 40 cycles from a standard curve generated using standards containing from 1 × 103 to 1 × 106 copies of replicon RNA per 5 μL.
2.4. Immunization of mice
For evaluation of antibody responses, groups of BALB/c mice (n=8) were immunized with 1 × 106 IU of gB VRP on study day (SD) 1, 22 and 43 and serum collected on SD -1 and 64. For evaluation of IFN-γ ELISPOT responses, groups of BALB/c mice (n=6) were immunized with 1 × 106 IU of pp65/IE1 VRP on SD 1 and 22 and spleen cells collected on SD 29. Immunizations were performed by intramuscular (IM) injection of 100 μL (50 μL in each hind leg).
2.5. Toxicology
Three groups of 12 New Zealand white rabbits each (6 male and 6 female) were immunized on four occasions at 2-week intervals, on SD 1, 15, 29 and 43. Group 1 animals were treated with 0.5 mL of placebo (formulation buffer) by the SC route injected in the fore limbs and by the IM route injected in the hind limbs. Group 2 animals were treated by the SC route, with 0.5 mL of gB VRP and 0.5 mL of pp65/IE1 VRP injected separately in the fore limbs. Group 3 animals were treated by the IM route, with 0.5 mL of gB VRP and 0.5 mL of pp65/IE1 VRP injected separately in the hind limbs. The target dosage level was 2 × 108 IU in 0.5 mL for each VRP component. On the day of each injection, aliquots of vaccine and placebo were shipped via overnight courier and tested to confirm that shipping and storage conditions did not result in any change in the potency or other characteristics of the products. Toxicity was evaluated by recording mortality/morbidity, body temperature, body weight, food consumption and ophthalmic examinations. Blood samples for clinical pathology (hematology, chemistry and coagulation parameters) were obtained before the first dose, on SD 3 and at termination. Serum for measurement of antibodies was obtained before each dose and at termination. Local reactogenicity was evaluated by examining the injection sites daily for 7 days after each injection and at termination, using a dermal Draize scoring system (none = 0, minimal = 1, mild = 2, moderate = 3, severe = 4).
Half of the animals were sacrificed 2 days after the last injection (SD 45) and the other half 2 weeks after the last injection (SD 57). Animals were necropsied as soon as possible following euthanasia. A gross necropsy, which included examination of the external surface of the body, the injection/treatment site, all orifices, the cranial, thoracic, and abdominal cavities and their contents, was conducted. The following organs (sex appropriate) were weighed as soon as possible after dissection: adrenal glands, brain, epididymides, heart, kidneys, liver, lungs (with mainstem bronchi), ovaries, spleen, testes, thymus, and uterus (with cervix). Tissues were collected and preserved in 10% neutral buffered formalin with the exception of the eyes, testes, epididymides, and optic nerves, which were fixed in Modified Davidson’s fixative. The following tissues (sex appropriate) from all groups were histologically evaluated: injection site (skin and underlying muscle), draining lymph nodes (axillary lymph nodes for subcutaneous route and iliac lymph nodes for intramuscular route), mandibular and mesenteric lymph nodes, brain, heart, kidneys, liver, lung, ovaries, spleen, testes, thymus, and gross lesions.
2.6. Enzyme-linked immunosorbent assay (ELISA)
ELISA was used to measure antibody responses to gB and pp65. Briefly, 96-well plates were coated overnight with His-tagged gB or pp65 protein, which had been purified from BHK cells transfected with replicon RNA as previously described [12], and blocked with 3% BSA in PBS. Serial 2-fold dilutions of serum in PBS containing 1% BSA and 0.5% Tween 20 were added to wells and incubated at 30 °C for 1–4 hr. Wells were washed with PBS and detector antibody (alkaline phosphatase-labeled goat anti-mouse or anti-rabbit IgG) added and incubated at 30 °C for 1–4 hr. Wells were washed and alkaline phosphatase substrate (p-nitrophenyl phosphate) added. Absorbance was measured in a VERSAmax plate reader (Molecular Devices, Sunnyvale, CA) approximately every 5 min until the OD405 was ≥ the expected value in positive control wells while remaining <0.2 in negative control wells. Serum endpoint titer was defined as the reciprocal of the maximal dilution for which OD405 was ≥0.2 and the OD405 for the next highest dilution was <0.2.
2.7. CMV neutralization
CMV neutralizing antibodies were measured using a microneutralization assay. A standard amount of sucrose gradient-purified CMV strain AD169 (Advanced Technologies Inc, Columbia, MD) was mixed with RPMI medium 1640 containing 10% heat-inactivated (56 °C, 30 min) FBS and penicillin/streptomycin/amphotericin B, added to an equal volume of serial dilutions of heat-inactivated test serum in RPMI with 10% guinea pig complement (Cedarlane Laboratories, Hornby, Ontario, Canada) and penicillin/streptomycin/amphotericin B, and incubated for 4 hr at 37 °C. The serum/CMV/complement mixtures were added to monolayers of HEL 299 cells (ATCC #CCL137) in 96-well half-area tissue culture plates and cultured overnight at 37 °C in 5% CO2. Wells were fixed with absolute ethanol, reacted with goat ant-IE1 antibody [12] followed by fluorescein isothiocyanate-labeled anti-goat antibody (Invitrogen) and the number of cells expressing IE1 determined by fluorescence microscopy. The endpoint titer was the last serum dilution at which there was at least 80% reduction in the number of IE1-positive cells compared to control wells. For serum samples from nine humans who were CMV seropositive as determined using a commercially available CMV ELISA, the geometric mean titer (GMT) in this assay was 635 (range 200–1600).
2.8. IFN-γ ELISPOT assay
T cell responses to pp65 and IE1 were measured by IFN-γ ELISPOT assay as previously described [12]. In brief, splenic lymphocytes isolated by density gradient centrifugation were stimulated with pools of pp65 or IE1 peptides (15-mers overlapping by 11 amino acids, purchased from SynPep Corp., Dublin, CA) at a final concentration of 1 μg/mL for each peptide, or with no peptide, an irrelevant control peptide, or conconavalin A (4 μg/mL). Cells were added to duplicate wells of ELISPOT assay plates coated with a rat IgG1 anti-mouse IFN-γantibody and left overnight in a 37 °C, 5% CO2 humidified incubator. Wells were washed with PBS containing 0.05% Tween 20, treated with a biotinylated rat IgG1 anti-mouse IFN-γ monoclonal antibody, washed with PBS, and incubated with Avidin-Peroxidase Complex for 1 hr at room temperature. Wells were then washed, incubated with substrate (3-amino-9-ethylcarbazole) for 4 min at room temperature and spot development stopped by distilled water rinse. After drying overnight, plates were shipped to Zellnet Consulting (New York, NY) for spot enumeration by automated analysis with a Zeiss KS ELISPOT system. The mean (± SD) number of spot-forming cells (SFC) per 106 splenic lymphocytes for cells cultured with no peptide was 4.5 ± 3.6. Values for cells cultured with peptides, after subtraction of counts from cells cultured with no peptide, was determined for each animal. A response was considered positive if this value was greater than 20 SFC per 106 splenic lymphocytes. The mean (± SD) response for cells cultured with an irrelevant peptide was 4.2 ± 6.3.
2.9. Statistical analysis
In the toxicology study, body weights, body weight changes, food consumption, body temperature, organ weight, and clinical pathology parameters were analyzed using the Kolmogorov-Smirnov test for normality, the Levene Median test for equal variance, and by one-way Analysis of Variance (ANOVA). If the ANOVA indicated statistically significant differences among experimental groups then the Dunnett’s t-test was used to determine which groups (if any) differed from the control. Statistical analysis was conducted using SigmaStat™ Statistical Software, Version 1 (Jandel Scientific, San Rafael, CA), with a two-tailed probability value of <0.05 as the critical level of significance for all tests.
3. Results
3.1. VRP production at laboratory scale
Different concentrations of replicon, capsid and glycoprotein RNA were tested prior to pilot lot and clinical lot manufacture to determine the optimal ratio for VRP production. An RNA concentration of 30 μg per cuvette for replicon, capsid helper and glycoprotein helper was found to be optimal for production of both gB VRP and pp65/IE1 VRP. Western blot analysis of proteins extracted from Vero cells infected with VRP for 18–22 hr showed expression of proteins of the expected molecular weights that were reactive with antibodies specific for pp65 or gB (data not shown).
3.2. Process scale-up for pilot lot and clinical production
Pilot lots of gB VRP and pp65/IE1 VRP were produced from a total of 20 cuvette electroporations per lot and used to provide material for reference standards, stability studies and toxicology studies. Clinical lots were produced from a total of 60 cuvette electroporations per lot using the same platform process as the pilot lots.
The production process was robust and reproducible for both gB VRP and pp65/IE1 VRP during scale-up from pilot lot to clinical manufacturing. The overall process productivity, defined as infectious units produced per cell, and percent yields for infectious units for both gB VRP and pp65/IE1 VRP are shown in Table 1. The yields shown for the harvest by high salt elution (first step in the purification process) were calculated based upon a mass balance of VRP in the salt wash pool and VRP in the medium and PBS wash pools. The yield of VRP in the formulated bulk is the overall process yield after all process sampling and a single freeze thaw on the bulk material. No significant change in titer was observed after the freeze thaw for gB VRP or pp65/IE1 VRP at either process scale.
Table 1.
Process productivity and yields in pilot and clinical lots of CMV VRP vaccines
| gB VRP | pp65/IE1 VRP | |||
|---|---|---|---|---|
| Pilot Lot | Clinical Lot | Pilot Lot | Clinical Lot | |
| Productivity (IU/cell) | 109 | 65 | 104 | 127 |
| % Recovery Harvest | 98.9 | 99.6 | 98.6 | 99.7 |
| % Recovery Formulated Bulk | 39 | 32 | 33 | 45 |
The purity of VRP with respect to protein and DNA was assessed at multiple process steps and was consistent for pilot and clinical scale production operations. Silver-stained SDS-PAGE analysis of purified gB VRP (Figure 2, lanes 8 and 9) and pp65/IE1 VRP (data not shown) demonstrated prominent protein bands that were confirmed by western blot analysis to be alphavirus envelope and capsid proteins (data not shown). As visualized by Southern blot analysis, the size of Vero DNA detected ranged from >10,000 base pairs in upstream process samples to very small fragments <300 base pairs following Benzonase treatment and TFF, and DNA was not detectable in VRP eluted from the Cellufine Sulfate chromatography resin (Figure 3). The absence in the Cellufine Sulfate eluate of DNA detectable by Southern blot analysis was not due to interfering substances, as 1 ng of digested Vero DNA spiked into process samples was clearly visible in the control lane.
Figure 2.

SDS-PAGE analysis of process pools from a pilot lot of gB VRP. Lane 1, molecular weight markers; Lane 2, human serum albumin control; Lane 3, salt wash harvest pool; Lane 4, pool of material at end of tangential flow filtration; Lane 5, tangential flow filtration permeate; Lane 6, Cellufine Sulfate unbound fraction; Lane 7, Cellufine Sulfate wash fraction; Lane 8 VRP elution from Cellufine Sulfate, 2 × 108 IU load; Lane 9 VRP elution from Cellufine Sulfate, 1 × 108 IU load. The positions of molecular weight markers (kDa) are indicated on the left and the positions of the alphavirus envelope (E) and capsid (C) proteins are indicated on the right.
Figure 3.
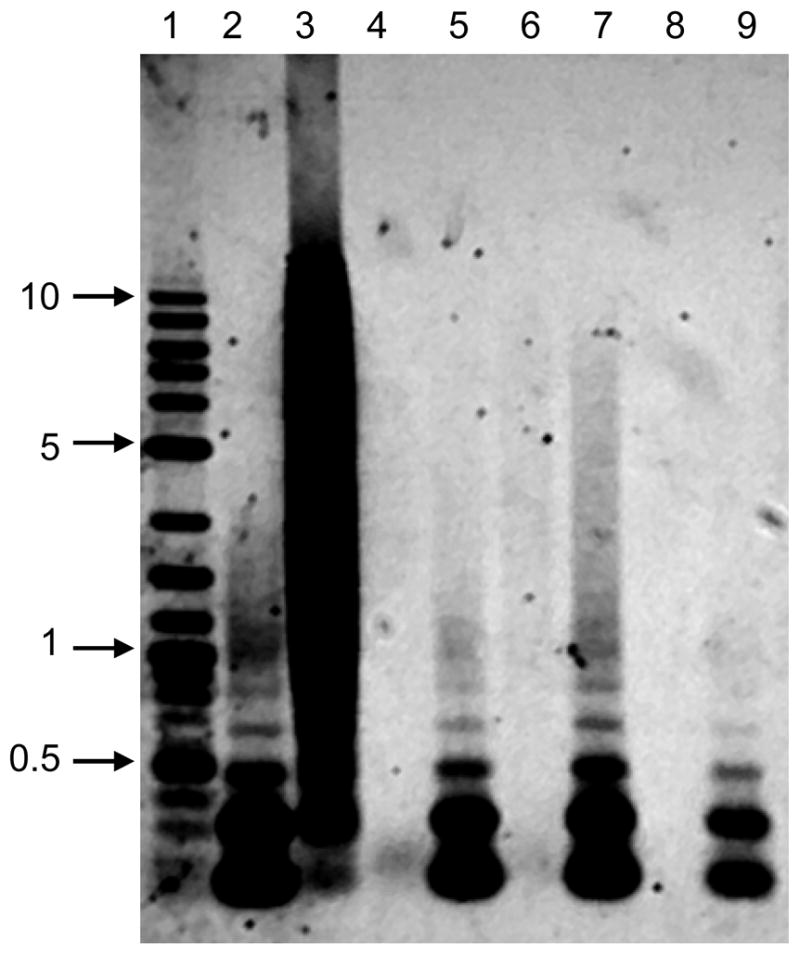
Southern blot analysis of process pools from a pilot lot of pp65/IE1 VRP. Lane 1, DNA size markers; Lane 2, 1 ng of AluI-digested Vero DNA loaded on the gel as a control that underwent no sample processing; Lane 3, Upstream process pool; Lane 4, pool of VRP taken after Benzonase treatment; Lane 5, pool of VRP taken after Benzonase treatment and spiked with 1 ng of AluI-digested Vero DNA; Lane 6, tangential flow filtration pool; Lane 7, tangential flow filtration pool spiked with 1 ng of AluI-digested Vero DNA; Lane 8 VRP elution from Cellufine Sulfate; Lane 9 VRP elution from Cellufine Sulfate spiked with 1 ng of AluI-digested Vero DNA. The positions of DNA size markers (kbp) are indicated on the left.
After Cellufine sulfate chromatography, the purified material had residual protein concentration <1 μg per 108 IU dose, residual DNA concentration <1 ng per 108 IU dose, and no detectable Benzonase (Table 2).
Table 2.
Concentrations of VRP, protein, DNA and Benzonase in purified product eluted from Cellufine Sulfate
| gB VRP | pp65/IE1 VRP | |||
|---|---|---|---|---|
| Pilot Lot | Clinical Lot | Pilot Lot | Clinical Lot | |
| Titer, IU/mL | 1.9 × 1010 | 1.2 × 1010 | 1.7 × 1010 | 2.5 × 1010 |
| Protein, μg/mL (μg/108 IU) | 150 (0.79) | 89.2 (0.74) | 37.9 (0.22) | 157 (0.63) |
| Vero DNA, ng/mL (ng/108 IU) | 36.9 (0.19) | 84.2 (0.70) | 32.7 (0.19) | 76.9 (0.31) |
| Benzonase, ng/mL | <0.125 | <0.125 | <0.125 | <0.125 |
The genome equivalent to infectious unit (GE:IU) ratio was evaluated by qPCR analysis in samples immediately after the salt wash harvest, after Cellufine Sulfate chromatography and in the formulated bulk vaccine. The GE:IU ratio in these samples ranged from 32 to 37 for pilot lots of gB VRP and 11 to 22 for pilot lots of pp65/IE1 VRP. The GE:IU ratio in the formulated bulk produced during clinical scale production was 26 for both gB VRP and pp65/IE VRP.
3.3. Humoral immune responses
Mice immunized with gB VRP developed anti-gB antibodies as measured by ELISA and CMV neutralization assays (Figure 4).
Figure 4.

Antibody response in mice immunized with gB VRP from a pilot lot (open symbols) or a clinical lot (solid symbols) by IM injection on study days 1, 22 and 43. Each data point represents the antibody titer, as determined by gB ELISA or CMV neutralization assay, in serum obtained from individual mice on study day 64. Control values in serum obtained before immunization were <40 for ELISA and <50 for neutralization. The geometric mean titer in this assay was 635 (range 200–1600) for serum samples from nine CMV seropositive humans.
In the rabbit toxicology study, all rabbits injected with pp65/IE VRP and gB VRP had anti-pp65 antibodies as measured by ELISA after one inoculation and anti-gB antibodies after two inoculations, and antibody responses were boosted by repeated immunizations (Figure 5). At SD 57, CMV neutralization titers were <25 in all rabbits that received placebo and the GMT was 1270 (range 800–1600) for rabbits that received vaccine by the SC route, and 1796 (range 800–3200) for rabbits that received vaccine by the IM route.
Figure 5.

Anti-gB and anti-pp65 antibody titers measured by ELISA in rabbits immunized with placebo or with gB VRP and pp65/IE1 VRP by the subcutaneous (SC) or intramuscular (IM) route on study days 1, 15, 29 and 43. Each data point represents the geometric mean ± SEM titerin serum from 6 rabbits (or from 3 rabbits at study days 45 and 57).
3.4. Cellular immune responses
Spleen cells from mice immunized with pp65/IE1 VRP developed robust T cell responses as measured by IFN-γ ELISPOT assay (Figure 6).
Figure 6.

IFN-γ ELISPOT responses in mice immunized with pp65/IE1 VRP from a pilot lot (open symbols) or a clinical lot (solid symbols) by IM injection on study days 1 and 22 and sacrificed on study day 29. Each data point represents the mean number of spot-forming cells (SFC) per 106 spleen cells from individual mice after stimulation with pools of overlapping peptides spanning the amino- or carboxy-terminal half of pp65 (pp65-1 and pp65-2) or the amino- or carboxy-terminal half of IE1 (IE1-1 and IE1-2). Control values from cells stimulated with an irrelevant peptide were 8 ± 6 (mean ± SEM) SFC per 106 spleen cells.
3.5. Results from toxicology testing
Potency testing of samples obtained on the day of each injection demonstrated that animals in Groups 2 and 3 received an average of 1.35 × 108 IU of gB VRP and 0.85 × 108 IU of pp65/IE1 VRP per dose. Multiple inoculations of VRP had no effect on mortality nor did they adversely affect any clinical parameter. Minimal to moderate edema was detected in some subcutaneously-treated animals after three or four doses of vaccine or placebo. Minimal to moderate erythema was detected in some subcutaneously-treated animals after two or three doses of vaccine and minimal to mild erythema after one to four doses of placebo. Minimal edema or erythema was detected in only an occasional intramuscularly-treated animal and did not increase in intensity or frequency with repeated dosing. The mean maximum dermal Draize score at any time point in any group did not exceed 0.67 for edema and did not exceed 1.17 for erythema. In the animals that did develop edema or erythema, the injection sites were generally normal in appearance within 3–5 days after each immunization.
Multiple inoculations of gB VRP and pp65/IE1 VRP had no effect on body weights or body weight changes, food consumption or body temperature. Modest but statistically significant effects were observed in selected clinical chemistry, hematologic and coagulation parameters associated with test article administration. Group 2 and 3 female rabbits had higher triglyceride concentration values on SD 3. Minimally higher total protein values in Group 2 and 3 females and higher globulin concentration and lower albumin to globulin ratio in Group 2 and 3 males and females on SD 45 and 57 were considered to be a reflection of immunoglobulin synthesis subsequent to test article administration. Elevated fibrinogen concentration, longer activated partial thromboplastin time, and shorter prothrombin time on SD 45 were considered to be a reflection of acute inflammatory response proteins released from the liver.
Multiple inoculations of gB VRP and pp65/IE1 VRP had no adverse effect on gross pathology and no effect on organ weights, organ-to-body weight or organ-to-brain weight ratios. Treatment-related gross pathology findings were limited to red discoloration, sometimes with gelatinous material, at the injection sites and occasional discoloration of draining lymph nodes in several animals necropsied on SD 45. Microscopic examination of injection sites and draining lymph nodes on SD 45 revealed vasculitis, necrosis, hemorrhage, mineralization, and myodegeneration considered to be secondary to the host inflammatory response to a foreign antigen. Inflammatory cell syncytia formation within the draining lymph nodes was seen with both components of the vaccine. Test article-related findings had resolved or were of much lesser severity by SD 57.
4. Discussion
A number of vaccine approaches designed to induce responses against relevant CMV target antigens have been evaluated in clinical trials [6]. The earliest trials evaluated live attenuated CMV vaccines, initially the AD169 strain and subsequently a more attenuated Towne strain [18,19]. In an effort to find a balance between safety and efficacy, a chimeric wild-type/Towne strain live attenuated CMV vaccine [20] was tested in a Phase 1 clinical trial, but further development of this product has not been pursued, presumably due to persistent concerns regarding the safety of a live virus vaccine capable of causing latent infection. A recombinant gB protein combined with MF59 adjuvant [21] is currently being tested in Phase 2 clinical trials in adolescent females (ClinicalTrials.gov number NCT00133497) and in patients awaiting renal or liver transplants (ClinicalTrials.gov number NCT00299260), and a recombinant gB protein combined with a different adjuvant is being tested in a Phase 1 clinical trial (ClinicalTrials.gov number NCT00435396). A canarypox vector (ALVAC) expressing either gB or pp65 was safe and immunogenic when tested in Phase 1 clinical trials [22,23], but the gB ALVAC vaccine in combination with multiple injections of the gB/MF59 vaccine did not increase the immunogenicity compared to multiple injections of gB/MF59 alone [23]. A trivalent DNA vaccine expressing gB, pp65 and IE1, and a bivalent DNA vaccine expressing gB and pp65 CMV genes, adjuvanted using a proprietary poloxamer compound and designed to elicit T cell responses, have been tested in Phase 1 clinical trials. Both appeared to be safe and well tolerated, but lower T cell responses were observed after immunization with the trivalent vaccine [24] and the bivalent product was prioritized for advancement into Phase 2 clinical trials [25].
Since studies in animal models of CMV disease, and clinical trials with CMV immune globulin and adoptive transfer of CTL, have shown that both humoral and cellular immune responses can provide protection against CMV disease, a preferred vaccine candidate should be capable of stimulating both arms of the adaptive immune system [26] and none of the approaches outlined above have achieved this goal. Development of a trivalent CMV vaccine based on alphavirus replicon vectors may provide opportunities to improve upon previously tested strategies. The high level of gene expression from single-cycle alphavirus RNA replicon vectors that specifically target to dendritic cells [27] may induce better immune responses than other vaccine delivery systems. In addition, alphavirus replicon vaccines contain a single-cycle, replication-incompetent RNA vector that has no DNA intermediate form and amplifies RNA only in the cytoplasm, which may offer a significant safety advantage over DNA vaccines where the vaccine construct can be detected up to one year post-vaccination, increasing the risk of integration into the host genome [28].
Results from the studies presented here demonstrate that an alphavirus replicon vaccine for CMV can be produced using GMP-compliant methods and scaled sufficiently for clinical evaluation. The use of replicons in which expression of a CMV gene is under control of an IRES element [13] enabled us to select replicons that were optimized for expression of the CMV protein, which may be important for inducing robust immune responses, and for VRP productivity, which is important for scalability. Other important factors contributing to the high VRP productivity include the use of a high cell density during electroporation and use of high salt elution to release VRP from cells. For the CMV gB and pp65/IE1 VRP, more than 98% of VRP were recovered in the salt wash harvest. For 13 other clinical scale lots of VRP expressing a variety of antigens that were produced using this platform production and purification process, an average of 99% (range 97.4% to 99.7%) of total VRP were recovered in the salt wash harvest (T. Talarico, unpublished observations).
The platform process also achieved excellent product purity. Based on studies in nonhuman primates with VRP expressing a variety of antigens, and human immunogenicity data with a VRP vaccine expressing HIV Gag [29], we anticipate that a human immunogenic dose of most VRP vaccines will be between 107 and 108 IU. The concentration of residual DNA in the purified material was <1 ng per 1 × 108 IU, which is 10-fold lower than the limits for cellular DNA concentration in viral vaccines recommended by the World Health Organization [30], and its size was less than 300 base pairs. Protein concentration in the purified product was <1 μg per 1 × 108 IU and, based on silver staining of SDS-PAGE gels, the alphavirus capsid and envelope proteins contributed a substantial portion of this amount. Each particle contains 240 copies of the E1, E2 and capsid proteins with molecular weights of 48,000, 46,000 and 31,000, respectively. Assuming that the number of particles is equal to the number of genome equivalents, with a genome equivalent to IU ratio of 25 the capsid and envelope proteins would be expected to contribute 125 ng per 1 × 108 IU, accounting for 15–50% of the total protein measured.
Results of GLP toxicology testing showed no adverse effects in any toxicology parameter. Local reactogenicity was observed, which on clinical evaluation was generally minimal to moderate with SC injections and none to minimal with IM injections, and there were changes in serum chemistry and coagulation parameters consistent with the expected host inflammatory response to a foreign antigen.
Immunization of mice and rabbits with gB VRP and pp65/IE1 VRP induced robust humoral and cellular immune responses. The CMV neutralization titers were similar to or higher than those in naturally infected humans. The strongest cellular immune responses were observed after stimulation with peptide pool pp65-1, which contains overlapping peptides that span the amino-terminal half of the pp65 protein and contains an immunodominant MHC H2-Dd T cell epitope, LGPISGHVL [12]. Responses were also seen after stimulation with pools of peptides that span the carboxy-terminal half of the pp65 protein or the amino- or carboxy-terminal half of the IE1 protein. The magnitude of the T cell responses were similar or higher than those seen after immunization with a VRP vaccine in which a pp65/IE1 fusion protein was expressed from a 26S subgenomic promoter [12].
In summary, these results support clinical evaluation of this novel CMV alphavirus replicon vaccine in humans. Phase 1 testing of this product was initiated in April, 2007.
Acknowledgments
We thank Pam Norberg, Chad Cecil, Randy Lamm, Kevin Williams, Tim Wagner, Deepa Patel, Renee Doggett and Holly Stone for performing analytical assays, Bryan Rivers for providing purified antigens and goat antisera, Ian Caley for critical review of the manuscript, Jeff Davis at Integrated Laboratory Systems, Research Triangle Park, NC, for immunizations and sample collection in mice, and Bridge GPS (formerly Gene Logic), Gaithersburg, MD, for conducting the toxicology study. Studies involving the use of animals complied with all relevant federal guidelines and institutional policies and were approved by an Institutional Animal Care and Use Committee. This research was supported in part by a Small Business Innovative Research grant (R4 AI060060) from the National Institute of Allergy and Infectious Diseases, National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Staras SA, Dollard SC, Radford KW, Flanders WD, Pass RF, Cannon MJ. Seroprevalence of cytomegalovirus infection in the United States, 1988–1994. Clin Infect Dis. 2006;43(9):1143–1151. doi: 10.1086/508173. [DOI] [PubMed] [Google Scholar]
- 2.Hebart H, Einsele H. Clinical aspects of CMV infection after stem cell transplantation. Hum Immunol. 2004;65(5):432–436. doi: 10.1016/j.humimm.2004.02.022. [DOI] [PubMed] [Google Scholar]
- 3.Rowshani AT, Bemelman FJ, van Leeuwen EM, van Lier RA, ten Berge IJ. Clinical and immunologic aspects of cytomegalovirus infection in solid organ transplant recipients. Transplantation. 2005;79(4):381–386. doi: 10.1097/01.tp.0000148239.00384.f0. [DOI] [PubMed] [Google Scholar]
- 4.Boeckh M, Fries B, Nichols WG. Recent advances in the prevention of CMV infection and disease after hematopoietic stem cell transplantation. Pediatr Transplant. 2004;8 (Suppl 5):19–27. doi: 10.1111/j.1398-2265.2004.00183.x. [DOI] [PubMed] [Google Scholar]
- 5.Limaye AP, Bakthavatsalam R, Kim HW, et al. Late-onset cytomegalovirus disease in liver transplant recipients despite antiviral prophylaxis. Transplantation. 2004;78(9):1390–1396. doi: 10.1097/01.tp.0000145989.22373.03. [DOI] [PubMed] [Google Scholar]
- 6.Arvin AM, Fast P, Myers M, Plotkin S, Rabinovich R. Vaccine development to prevent cytomegalovirus disease: report from the National Vaccine Advisory Committee. Clin Infect Dis. 2004;39(2):233–239. doi: 10.1086/421999. [DOI] [PubMed] [Google Scholar]
- 7.Britt WJ, Vugler L, Stephens EB. Induction of complement-dependent and -independent neutralizing antibodies by recombinant-derived human cytomegalovirus gp55–116 (gB) J Virol. 1988;62(9):3309–3318. doi: 10.1128/jvi.62.9.3309-3318.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Speckner A, Glykofrydes D, Ohlin M, Mach M. Antigenic domain 1 of human cytomegalovirus glycoprotein B induces a multitude of different antibodies which, when combined, results in incomplete virus neutralization. J Gen Virol. 1999;80(Pt 8):2183–2191. doi: 10.1099/0022-1317-80-8-2183. [DOI] [PubMed] [Google Scholar]
- 9.Walter EA, Greenberg PD, Gilbert MJ, et al. Reconstitution of cellular immunity against cytomegalovirus in recipients of allogeneic bone marrow by transfer of T-cell clones from the donor. N Engl J Med. 1995;333(16):1038–1044. doi: 10.1056/NEJM199510193331603. [DOI] [PubMed] [Google Scholar]
- 10.Bunde T, Kirchner A, Hoffmeister B, et al. Protection from cytomegalovirus after transplantation is correlated with immediate early 1-specific CD8 T cells. J Exp Med. 2005;201(7):1031–1036. doi: 10.1084/jem.20042384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schleiss MR, Lacayo JC, Belkaid Y, et al. Preconceptual administration of an alphavirus replicon UL83 (pp65 homolog) vaccine induces humoral and cellular immunity and improves pregnancy outcome in the guinea pig model of congenital cytomegalovirus infection. J Infect Dis. 2007;195(6):789–798. doi: 10.1086/511982. [DOI] [PubMed] [Google Scholar]
- 12.Reap EA, Dryga SA, Morris J, et al. Cellular and humoral immune responses to alphavirus replicon vaccines expressing cytomegalovirus pp65, IE1 and gB proteins. Clinical and Vaccine Immunology published ahead of print on 18 April 2007. 2007 doi: 10.1128/CVI.00037–07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kamrud KI, Custer M, Dudek JM, et al. Alphavirus replicon approach to promoterless analysis of IRES elements. Virology. 2007;360(2):376–387. doi: 10.1016/j.virol.2006.10.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wagner B, Kropff B, Kalbacher H, et al. A continuous sequence of more than 70 amino acids is essential for antibody binding to the dominant antigenic site of glycoprotein gp58 of human cytomegalovirus. J Virol. 1992;66(9):5290–5297. doi: 10.1128/jvi.66.9.5290-5297.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Meyer H, Sundqvist VA, Pereira L, Mach M. Glycoprotein gp116 of human cytomegalovirus contains epitopes for strain-common and strain-specific antibodies. J Gen Virol. 1992;73(Pt 9):2375–2383. doi: 10.1099/0022-1317-73-9-2375. [DOI] [PubMed] [Google Scholar]
- 16.Pushko P, Parker M, Ludwig GV, Davis NL, Johnston RE, Smith JF. Replicon-helper systems from attenuated Venezuelan equine encephalitis virus: expression of heterologous genes in vitro and immunization against heterologous pathogens in vivo. Virology. 1997;239(2):389–401. doi: 10.1006/viro.1997.8878. [DOI] [PubMed] [Google Scholar]
- 17.Talarico T, Maughan M, Pancorbo B, Ruiz J, Graham A. Development and manufacture of alphavaccines. Bioprocessing. 2006 Fall;:8–14. [Google Scholar]
- 18.Elek SD, Stern H. Development of a vaccine against mental retardation caused by cytomegalovirus infection in utero. Lancet. 1974;1(7845):1–5. doi: 10.1016/s0140-6736(74)92997-3. [DOI] [PubMed] [Google Scholar]
- 19.Plotkin SA, Starr SE, Friedman HM, et al. Effect of Towne live virus vaccine on cytomegalovirus disease after renal transplant. A controlled trial. Ann Intern Med. 1991;114(7):525–531. doi: 10.7326/0003-4819-114-7-525. [DOI] [PubMed] [Google Scholar]
- 20.Kemble G, Duke G, Winter R, Spaete R. Defined large-scale alterations of the human cytomegalovirus genome constructed by cotransfection of overlapping cosmids. J Virol. 1996;70 (3):2044–2048. doi: 10.1128/jvi.70.3.2044-2048.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pass RF, Duliege AM, Boppana S, et al. A subunit cytomegalovirus vaccine based on recombinant envelope glycoprotein B and a new adjuvant. J Infect Dis. 1999;180(4):970–975. doi: 10.1086/315022. [DOI] [PubMed] [Google Scholar]
- 22.Berencsi K, Gyulai Z, Gonczol E, et al. A canarypox vector-expressing cytomegalovirus (CMV) phosphoprotein 65 induces long-lasting cytotoxic T cell responses in human CMV-seronegative subjects. J Infect Dis. 2001;183(8):1171–1179. doi: 10.1086/319680. [DOI] [PubMed] [Google Scholar]
- 23.Bernstein DI, Schleiss MR, Berencsi K, et al. Effect of previous or simultaneous immunization with canarypox expressing cytomegalovirus (CMV) glycoprotein B (gB) on response to subunit gB vaccine plus MF59 in healthy CMV-seronegative adults. J Infect Dis. 2002;185(5):686–690. doi: 10.1086/339003. [DOI] [PubMed] [Google Scholar]
- 24.Kaslow D. Multi-valent CMV DNA vaccines for use in transplant settings; 10th International CMV/Betaherpesvirus Workshop, abstract 10.08; Williamsburg, VA. 24–28 Apr 2005. [Google Scholar]
- 25.Selinsky C, Luke C, Wloch M, et al. A DNA-based vaccine for the prevention of human cytomegalovirus-associated diseases. Hum Vaccin. 2005;1(1):16–23. doi: 10.4161/hv.1.1.1335. [DOI] [PubMed] [Google Scholar]
- 26.Plotkin SA. Is there a formula for an effective CMV vaccine? J Clin Virol. 2002;25 (Suppl 2):S13–21. doi: 10.1016/s1386-6532(02)00093-8. [DOI] [PubMed] [Google Scholar]
- 27.MacDonald GH, Johnston RE. Role of dendritic cell targeting in Venezuelan equine encephalitis virus pathogenesis. J Virol. 2000;74(2):914–922. doi: 10.1128/jvi.74.2.914-922.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kowalski J, Adkins K, Gangolli S, et al. Evaluation of neurovirulence and biodistribution of Venezuelan equine encephalitis replicon particles expressing herpes simplex virus type 2 glycoprotein D. Vaccine. 2007;25(12):2296–2305. doi: 10.1016/j.vaccine.2006.11.063. [DOI] [PubMed] [Google Scholar]
- 29.Chulay J, Burke D, Karim SSA, et al. AIDS Vaccine 06. Amsterdam: 29 Aug - 1 Sep, 2006. Safety and immunogenicity of an alphavirus replicon HIV Gag vaccine (AVX101) in healthy HIV-uninfected adults; pp. P11–09. [Google Scholar]
- 30.Knezevic I, Griffiths E. WHO Expert Committee on Biologcial Standardization. WHO Technical Report Series. 2004;926:1–109. [PubMed] [Google Scholar]