

Abstract
Trypanosoma cruzi (TC) causes Chagas disease, which in its chronic stage remains incurable. We have shown recently that specific inhibition of TC sterol 14α-demethylase (TCCYP51) with imidazole derivatives is effective in killing both extracellular and intracellular human stages of TC. An alternative set of TCCYP51 inhibitors has been identified using optical high throughput screening followed by web-database search for similar structures. The best TCCYP51 inhibitor from this search was found to have structural similarity to a class of cyclooxygenase-2-selective inhibitors, the indomethacin-amides. A number of indomethacin-amides were found to bind to TCCYP51, inhibit its activity in vitro and produce strong antiparasitic effects in the cultured TC cells. Analysis of TC sterol composition indicated that the mode of action of the compounds is by inhibition of sterol biosynthesis in the parasite.
Introduction
Chagas disease is an insect-borne parasitic disease threatening millions of lives in Latin America and spreading worldwide as a result of migration (mammalian hosts and insect vectors), HIV-co-infection, blood transfusion and organ transplantation (http://www.cdc.gov/chagas/factsheet.html). For example, a recent American Red Cross study indicates that approximately one of 4,700 blood donors in the United States in 2007 tested positive for Chagas.1 Victims often do not experience specific symptoms during the early stages of the disease, which can either pass unnoticed or be deadly, depending on their immune status to the protozoan parasite Trypanosoma cruzi (TC a). At the chronic stage, when TC infects human tissues and exists predominantly as the intracellular amastigote, the disease is commonly fatal. Serious cardiac and/or intestinal symptoms develop in 20–40% of infected individuals 10 to 20 years later, the probability increasing up to 70% in immunocompromised patients.2
Only acute TC infection can be cured with the two currently available drugs, nifurtimox and benznidazole.3 The drugs are nonspecific, have severe side effects and induce resistance. The demand for new drug candidates has given rise to multiple attempts to use the progress in understanding TC physiology and biochemistry for the development of more specific treatments for Chagas disease. 4–5 Inhibition of the TC sterol biosynthetic pathway is currently amongst the most promising approaches.6 Similar to fungi or plants, TC produces 24-methylated/alkylated (ergosterol-like) sterols which are necessary for membrane formation and cannot be replaced in the parasite membranes by the host cholesterol.7
We are focusing our attention on sterol 14α-demethylase (CYP51). In TC, this cytochrome P450 enzyme catalyzes a three-step reaction of oxidative removal of the 14α-methyl group from 24-methylene dihydrolanosterol.8 We have shown recently that specific inhibition of TCCYP51 with imidazole derivatives is highly effective in killing the parasite.9 In good correspondence with the fact that TCCYP51 has only about 25% amino acid identity to the orthologous fungal enzymes, the structure of the lead compounds we have used differs significantly from the structures of the antifungal imidazole and triazole drugs. However, the compounds still belong to the same class of CYP51 inhibitors, and it is known that azoles currently used as clinical and agricultural fungicides can cause resistance.10 This may be especially crucial for immunocompromised (especially HIV-infected) patients with Chagas as it is very likely for many of them to also have fungal co-infections and to be treated with azoles for a long time.
In order to investigate other options for the development of alternative sets of potential anti-chagastic drugs, optical high throughput screening of TCCYP51 for binding ligands other than azoles has been undertaken. Several compounds producing type 1 (substrate-like) or type 2 (azole-like) spectral responses in the cytochrome P450 Soret band were identified. Following high throughput screening, a web-database search for similar structures revealed additional TCCYP51 ligands, some of them of higher inhibitory potency. The strongest TCCYP51 inhibitor from this search (more than two-fold decrease in activity at equimolar ratio inhibitor/enzyme (I/E2<1 9)) demonstrated a clear antiparasitic effect in the TC cells and was found to have significant structural similarity to indomethacin amide derivatives (cyclooxygenase-2 (COX-2) inhibitors).
Indomethacin (INDO) is a classic non-steroidal anti-inflammatory drug (NSAID) that has been available commercially to treat rheumatoid arthritis since 1963. INDO exerts its anti-inflammatory effect by non-selective inhibition of both cyclooxygenase isoforms, COX-1 and COX-2. Despite the high global sequence homology (>66%) between the COX isoforms, it was discovered that INDO could be converted to a COX-2-selective inhibitor by neutralization of the carboxylic-acid to an ester or amide (INDO-amides).11–12 Additionally, it was found that removing the 2′-methyl group of the INDO scaffold eliminated potency of INDO against both isoforms of COX.13 INDO is an attractive scaffold for drug development because it is relatively inexpensive, readily converted to amide derivatives, and has a long clinical history. We have synthesized a number of amide derivatives of INDO and find that several of them are potent inhibitors of TCCYP51.
Results and Discussion
TCCYP51 ligands found via high throughput screening
Optical high throughput screening (HTS) is based on the P450 property of changing the maximum of the Soret band absorbance upon ligand binding. The changes are connected with alterations in the surrounding environment of the heme iron.14 Heterologously expressed CYP51s are usually purified in the oxidized low-spin ferric form (resting state of the iron-porphyrin complex with the Soret band maximum at 417 nm), the sixth coordinate position of the iron being occupied by a water molecule. Binding of a substrate-like ligand in the active center causes water displacement; the iron becomes penta-coordinated and pops out of the porphyrin plane. This triggers a low-to-high-spin transition in the iron-porphyrin complex, and the Soret band maximum shifts to 390 nm producing a so-called type 1 spectral response in the difference spectra. Alternatively, a type 2 spectral response (red shift in the Soret band maximum to 422–424 nm) reflects direct coordination of a ligand other than water to the low-spin heme iron.
All “substrate-like” ligands identified by HTS produced rather weak spectral responses in TCCYP51. The three best compounds (Figure 1A) revealed apparent dissociation constants in the concentration range of 50–100 μM, which suggests binding affinities at least 50-fold lower than the affinity of the enzyme/substrate complex formation (Kd around 1 μM with the preferred substrate 24-methylenedihydrolanosterol).9 Correspondingly, the inhibitory potencies of these compounds in the reconstituted enzyme reaction were very low (I/E2 >50 (not shown)). We believe this might be connected with the narrowness of the CYP51 substrate specificity15: to preserve their catalytic function through hundreds of million of years of evolution the CYP51 family enzymes developed very strict requirements towards their substrates so that minor alterations in the spatial structure would differentiate a foreign compound and prevent it from being metabolized or interfere with the substrate in the CYP51 active center. The finding supports the notion that the most likely way to design potent substrate-analogous CYP51 inhibitors would be through the subtle modifications of the basic sterol structure in the regions adjusted to the reactive 14α-methyl group.8
Figure 1. Search for TCCYP51 inhibitors.

(A) Ligands (1–6) found via HTS using FMP library of small organic molecules (20,000-compounds selected from ChemDiv, San Diego, CA); (B) inhibitors (7–11) found using web-database search for structural similarity to 5 (http://chemdiv.emolecules.com); (C) 9-like INDO-amide derivatives. Calculated from spectral responses apparent dissociation constants (μM) for the compounds not included in Table 1 are shown in brackets.
The HTS hits which induced type 2 spectral response in TCCYP51 demonstrated higher binding affinities; the most potent inhibitor (5) exhibiting a Kd of 0.44 μM and I/E2 of about 10 (Table 1). Contrary to compounds 4 and 6, which must interact with the TCCYP51 heme iron through their five-membered imidazole ring, 5 is most likely to coordinate through the pyridyl nitrogen. This has been supported by the results of the web-database search using compound 5 (N-(4-pyridyl)-formamide) as a template since similar structures tested (Figure 1B) produced the same spectral shift in the TCCYP51 absorbance (Soret band maximum 422 nm). On the other hand, differences in the calculated apparent binding affinities reflect influence of the configuration of the non-coordinated portion of the ligand molecule, which (similar to azole inhibitors) might enhance the inhibitory effect by forming additional interactions with the amino acid residues in the CYP51 substrate binding cavity. The strongest binding compounds 7 and 9 (Kd = 0.75 and 0.24 μM, respectively) have bulky structures attached to the N-(4-pyridyl)-amide moiety. In 9 this portion of the molecule is larger and more flexible than it is in 7, and this feature must be preferable since in the reconstituted enzyme reaction 9 revealed more than one order of magnitude stronger potency as TCCYP51 inhibitor (I/E2<1, Table 1).
Table 1.
Binding parameters, inhibitory potencies and antiparasitic effects of selected TCCYP51 ligands
| Compound | Spectral Response in TCCYP51 |
Effect on enzymatic activity (I/E2****) |
|||
|---|---|---|---|---|---|
| Type | Soret band maximum, nm | Kd, μM | TCCYP51 | Mouse COX2 | |
| 5 | 2 | 422 | 0.44±0.02 | 10 | |
| 7 | 2 | 422 | 0.75±0.03 | 14 | |
| 9 | 2 | 422 | 0.24±0.01 | <1 | |
| 12 | 2 | 422 | 0.29±0.01 | 9.1 | 2.1 |
| 13 | 2** | 418 | 7.6±0.9 | 13 | >10 |
| 14 | 2 | 422 | 0.54±0.02 | 10 | 10 |
| 15 | 2 | 422 | 0.37±0.05 | 8.5 | 1.7 |
| 16 | 1* | 416 | 3.3±0.48 | 11 | 2.1 |
| 17 | none | 417 | - | 15 | 3.6 |
| 19 | 2** | 420 | 0.56±0.08 | 7.7 | <1 |
| 18 | 2** | 420 | 0.26±0.04 | 4.2 | <1 |
| 20 | 2*** | 422 | 2.31±0.09 | 6.3 | 2.4 |
| 21 | 2** | 419 | 0.36±0.02 | <1 | <1 |
| 22 | 2** | 419 | 0.15±0.01 | <1 | >40 |
weak type 1 response with only ~7% of P450 experiencing low to high spin transition;
modified type 2 response with the smaller shift in the Soret band maximum,
sigmoid curve, Hill coefficient 2.3±0.2 (see Figure 2);
inhibitory potencies of the compounds toward TCCYP51 (minimal final concentration required in the reconstituted reaction 1 μM9) and mouse COX2 (final concentration 0.1 μM) are compared as molar ratios Inhibitor/Enzyme which cause a two-fold decrease in the initial rate of catalysis.
Having an inhibitory effect on TCCYP51 comparable with the effects of antifungal azole drugs fluconazole and ketoconazole9, compound 9 has potential to serve as a lead for the design of novel non-azole CYP51 inhibitors. Because high flexibility of the substrate binding cavity is known to be the basis of the P450 catalysis and ligand binding,16–17 in the absence of TCCYP51 crystal structure, most preferably in a complex with 9, molecular docking and computer modeling are very unlikely to produce reliable information on rational 9 structure modification, especially since sequence identity of TCCYP51 to the closest P450 template of known crystal structure is only 28%. Instead, we continued with investigation of structural similarity and tested as TCCYP51 inhibitors a number of INDO-amide derivatives which share structural features with 9. All these compounds have the same INDO-amide moiety (Figure 1C, shown in square) but differ in the length of the alkyl-linker and position of the nitrogen in the heterocycle (shown in blue); this part of the molecule is of special interest because it is expected to form the sixth coordinate bond with the P450 heme iron.
INDO-amide derivatives as TCCYP51 ligands
While spectral responses of TCCYP51 to the compounds 7–11 were very similar to the responses caused by imidazole and triazole derivatives, INDO-amides induced a variety of spectral changes in the hemoprotein (Figure 2, Table 1). It could be connected with the variations in the location of the nitrogen in the heterocyclic ring. Thus, when the nitrogen is in the para-position (as in compounds 7–11) the Soret band maximum shows a normal shift to 422 nm (14 and 15). The same response is produced if the nitrogen is in the meta-position (12) and the ring does not contain any additional substituents. If it does (18 and 19), the maximum is observed at 420 nm. Taking into account that the same spectral changes are also caused by two compounds which do not carry any nitrogen atoms in the ring (21 and 22), it is not excluded that this modified type 2 response may result from accommodation of the inhibitor in the TCCYP51 active center, which would strengthen the water ligation to the heme iron. This can also be true for 13 (nitrogen in the ortho-position), though in this case the iron coordination is weaker and the absorbance maximum shifts only to 418 nm. Having a shorter arm between the ortho-nitrogen ring and the amide, 16 definitely does not participate in iron coordination as the compound causes very weak substrate-like type 1 spectral response in TCCYP51. Finally, 17 (pyrimidine ring with both nitrogens ortho to the arm) did not induce any spectral changes in TCCYP51.
Figure 2. Spectral responses of TCCYP51 (2 μM) to three INDO-amide derivatives.

(A) 18; (B) 16; (C) 20. Upper spectra: Absolute absorbance in the Soret band region (black line – no ligand, red line – plus ligand. Lower: Difference spectra upon titration with the ligands (0.5 μM steps). Insets: Titration curves.
Though they differ in the location of the Soret band maximum, the majority of the spectral responses upon titration of TCCYP51 with the increasing concentrations of the ligands produced typical hyperbolic curves. The only exception was observed in the case of 20 (Figure 2C). The sigmoid shape of the titration curve was reproducible upon variations in the buffer composition, salt gradient, increase in the concentration of TCCYP51 or use for the titration of more soluble 20-hydrochloride. The Hill coefficient of 2.3 suggested a possibility of simultaneous binding of two inhibitor molecules to the enzyme. This has been tested and confirmed using ESI-mass spectrometry (Supplemental Data, Figure S1). Allosteric effects (non-Michaelis-Menten kinetics) have been reported for several microsomal drug-metabolizing CYPs (e.g 1A2, 2A6, 2C9, 3A4:18) but this is the first example in a CYP51 ortholog and binding of a second molecule may have no inhibitory effect.
Regardless of the quite pronounced differences in the apparent binding affinities calculated from the spectral responses, most of the INDO-amides demonstrated comparably strong inhibitory effects on TCCYP51 (I/E2 around 5–10, reversible) except for 21 and 22 (I/E2<1, functionally irreversible) (Table 1). Altogether this supports our previous observation that, being indicative of binding of a ligand near the heme iron, spectral responses do not necessarily reflect real inhibitory potency9 and the interactions in the regions of the CYP51 substrate binding cavity, remote from the heme iron, might also provide a strong inhibitory effect on the enzyme catalysis and should be considered in the design of new inhibitors.
Antiparasitic effect of CYP51 inhibitors in extracellular and intracellular trypanosomes
TC is an obligate parasite with a complex life cycle that includes four morphologically and metabolically different stages. Proliferative epimastigotes and infective metacyclic trypomastigotes are present in the insect vectors (kissing bugs). Upon infection of mammalian hosts, TC transforms into bloodstream trypomastigotes and proliferative intracellular amastigotes (amastigotes dominate in the chronic stage of Chagas). Pre-treatment of TC trypomastigotes with compounds 9, 13, 18, and 20 produced a clear dose-dependent antiparasitic effect in both human forms of TC (Figure 3). At a 20 μM compound concentration the parasite was practically eliminated from cardiomyocytes. The strongest growth inhibition (ED50<1 μM in TC amastigotes) was observed with 20 which may be at least in part due to its better cellular permeability or the longest lifetime/highest metabolic stability.19 For the comparison, published ED50 values in TC cells for benznidazole, one of the two drugs currently used for clinical treatment of TC infections, were reported to be within 25–50 μM.20–22
Figure 3. Cellular effects of TCCYP51 inhibitors in TC trypomastigotes and amastigotes.
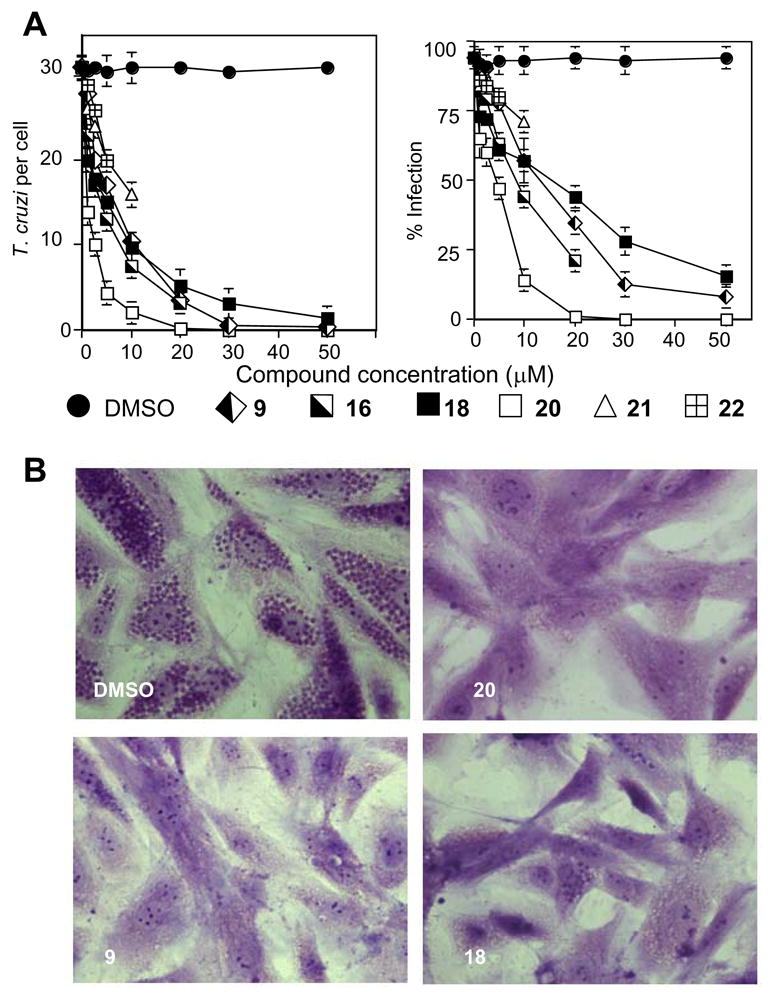
(A) Dose-dependent inhibition of T. cruzi intracellular multiplication and infection in cardiomyocytes by TCCYP51 inhibitors. Trypomastigotes were pre-treated with several concentrations of TCCYP51 inhibitors or mock-treated, exposed to cardiomyocyte monolayers and T. cruzi multiplication was evaluated at 72 h by determining the number of T. cruzi/cell (left panel) and the percent of infection (right panel). The data represent the mean ± standard deviation (SD) of results from triplicate samples. SD did not exceed 10% of the mean. Differences in T. cruzi per cell and % infection between trypomastigotes-mock treated (DMSO) and trypomastigotes treated with each TCCYP51 inhibitor (5–50 μM) are p<0.05 determined by the Student t test. The two most potent TCCYP51 inhibitors (21 and 22) show antiparasitic activities at ≤ 10 μM concentrations) but release cardiomyocyte monolayers at higher concentrations.
(B). Microscopic observation of the inhibition of T. cruzi multiplication by 20 μM TCCYP51 inhibitors within cardiomyocytes at 72 hr. T. cruzi pre-treated with control DMSO showed high levels of parasite multiplication, whereas cells exposed to trypanosomes pre-incubated with the inhibitors showed a dramatic decrease in the number of intracellular parasites.
Sterol composition of TC cells upon treatment with 20
We have shown recently that treatment of TC cells with a potent TCCYP51 inhibitor profoundly alters their sterol composition.9 While untreated parasites, where sterol biosynthesis is highly efficient, essentially contain only the pathway products (ergosterol and its 24-ethylated analog) and exogenous cholesterol from the medium (Figure 4A), in the inhibitor-treated TC (N-(1-(2,4-dichlorophenyl)-2-(1H-imidazol-1-yl)ethyl)-4-(5-phenyl-1,3,4-oxadiazol-2-yl)benzamide (SDZ-284629), 1μM) the pathway practically stops at the stage of the C14-methylated sterol precursors, predominantly lanosterol, the first cyclized compound of the pathway, and 24-methylenedihydrolanosterol, the preferred TCCYP51 substrate. Treatment of TC with 50 μM 20 produces a very similar effect on cellular sterol composition (Figure 4B) indicating that the mechanism of action of the compound is connected with blocking sterol biosynthesis in the parasite via inhibition of TCCYP51 activity.
Figure 4. Sterols in TC.

(A) Structural formulas of neutral lipids formed upon sterol biosynthesis (1-squalene; 2-squalene epoxide; 3-lanosterol; 4 - 24-methylenedihydrolanosterol; 5 – obtusifoliol; 6- ergosterol; 7- 24- methylergosterol) or up taken from the media (8-cholesterol). 14α-methyl group and formed by CYP51 Δ14–15 double bond are shown in red.
(B) TLC analysis of sterols extracted from TC epimastigotes. St, sterol standards; C, sterols in untreated cells; T, sterols in TC incubated for 24 hours with 50 μM 20.
Summary
Inhibition of sterol biosynthesis in TC is becoming one of the very promising directions in the development of anti-Chagasic drugs. Several antifungal azoles have recently entered clinical trials for anti-trypanosomal chemotherapy.4 However; azole derivatives (at least those currently used as fungicides) have one serious disadvantage: upon long-term treatment they often cause resistance. Experimental development of resistance to fluconazole in the cultured TC cells also has been described.23 Though strong inhibition of TCCYP51 with specific azoles might shorten the treatment time and decrease the probability of resistance, an alternative set of TCCYP51 inhibitors would be highly desirable. The strong antiparasitic effect of indomethacin amide derivatives in both extracellular and intracellular forms of human stages of TC indicates that this group of compounds might find a new application as potential anti-trypanosomal agents. While additional directed modifications, targeted to decrease COX-2 inhibitory potency might be desirable for use as a lead structure 20 in the chronic (especially cardiac) forms of Chagas, this class of compounds is particularly attractive for the acute stage since drugs traditionally aimed to ease non-specific symptoms of inflammation and fever can actually provide treatment to the disease. Finally, HTS of larger libraries of small bioactive molecules in combination with the database search for structural similarity may help to discover other novel CYP51 inhibitors.
Experimental Section
The TCCYP51 gene subcloned into the pCW vector (Nde I/Hind III cloning sites) was expressed in E. coli and purified as previously described.8 The protein was electrophoretically pure, had spectrophotometric index of OD417/OD278 of 1.55 and specific heme content of 17 nmol/mg. P450 concentration was determined from the difference spectra of the reduced carbon monoxide complexes using the extinction coefficient of 91mM−1 cm−1 (450–490 nm)24 or from the absolute absorbance spectra of the ferric water-coordinated TCCYP51 in the Soret band maximum (417 nm) using the extinction coefficient of 117 mM−1 cm−1.8
High throughput screening (HTS) and web search for similar structures
HTS of the FMP library of small organic molecules (20,000-compounds selected from ChemDiv, San Diego, CA) was performed using a pipetting robot (Sciclone 3000; Caliper Lifesciences), a plate reader for absorbance scanning (Safire; Tecan), and various robots for washing and dispensing. Compounds were dissolved in DMSO at 10 mM stock concentrations and distributed in 0.4-μl aliquots into 384-well microtiter plates. Buffer (40 μl of 50 mM potassium-phosphate buffer, pH 7.4, containing 100 mM NaCl) was added to each well to achieve a 100 μM concentration of the tested compounds. Plates were incubated for 15 min at 37°C and then sonicated for 5 min to allow solubilization of suspensions of the most hydrophobic compounds. Compound-specific changes in absorption spectra (350 to 450 nm) were recorded automatically after adding 10 μl of 5 μM TCCYP51 in 50 mM potassium-phosphate buffer, pH 7.4 containing 100 mM NaCl and 0.1 mM EDTA, optical path length 7 mm, and then validated manually by spectral titration. Search for similar structures was done using the ChemDiv database (http://chemdiv.emolecules.com). The examples of TCCYP51 ligands (shown in Figure 1) identified by HTS are: 1, 1-(3-amino-4-chlorophenyl)(thien-2-yl)methanone; 2, 3-(4-aminophenyl)-2H-chromen-2-one; 3, [5-(4-Fluorobenzylidene)-4-oxo-2-thioxo-thiazolidin-3-yl]-acetic acid; 4, 3-thien-2-yl-5,6-dihydroimidazo[2,1-b][1,3]thiazole; 5, 3-(2-furyl)-5,6-dihydroimidazo[2,1-b][1,3]thiazole; 6, 2-phenyl-N-pyridin-4-ylbutanamide. Compound 5-like ligands found via web search, ChemDiv numbers: 5556-0480 (7); 6903-0018 (8); C155-0123 (9); 3124-0167 (10); 000L-5707 (11).
Synthesis and characterization of INDO-amide derivatives
Purity of the compounds was determined using NMR and ESI-MS as described19 and was ≥95%. Materials and instrumentation were as previously described.11 A reaction mixture containing indomethacin (300 mg, 1.0 eq, 0.84 mmol) in 6 mL of anhydrous CH2CL2 was treated with dicyclohexylcarbodiimide (192 mg, 1.1 eq 0.92 mmol), dimethylaminopyridine (10 mg, 0.1 eq, 0.084 mmol), and the appropriate amine (1.1 eq, 0.92 mmol). After stirring at room temperature for 5 h, the reaction mixture was filtered and the filtrate was concentrated in vacuo. The residue was diluted with water (2 × 15 mL) and extracted with CH2CL2 (3 × 15 mL). The combined organic solution was washed with brine (2 × 15 mL), dried over MgSO4, filtered, and concentrated in vacuo. The crude product was purified by trituration with ethanol to give a precipitate. The solid was collected in a fritted funnel. The HPLC analysis was done using a C18 column with UV detection at 235 nm using a gradient of 90:10 A:B to 100% B over 30 min with solvent A= water with 0.05% TFA and B = acetonitrile with 0.05% TFA.
Compound 12 was obtained as an off-white solid (320 mg, 69% isolated yield). Melting point = 183°C, 1H NMR (300 MHz, CDCl3, ppm) δ 8.40 (d, J = 1.4 Hz, 1H) 8.32 (dd, J1 = 4.7 Hz, J2 = 1.4 Hz, 1H), 8.06 (m, 1H,), 7.65 (d, J = 8.6 Hz, 2H), 7.50 (m, 3H), 7.25 (m, 1H), 6.94 (d, J= 2.4 Hz, 1H), 6.86 (d, J = 9.0 Hz, 1H), 6.71 (dd, J1 = 9.0 Hz, J2 = 2.4 Hz, 1H), 3.84 (s, 2H), 3.81(s, 3H), 2.46 (s, 3H); ESI-MS m/z (positive) 434 [C24H20ClN3O3+H]+, HPLC: retention time= 18.1 min.
Compound 13 was obtained as a yellow solid (120 mg, 32% isolated yield). Melting point = 181 °C, 1H NMR (CDCl3) δ 8.39 (d, J = 4.3 Hz, 1H), 7.68 (d, J = 13.1 Hz, 2H), 7.62 (dt, J = 1.8, 7.7 Hz, 1H), 7.47 (d, J = 13.1 Hz, 2H), 7.15 (m, 2H), 6.92 (d, J = 2.4 Hz, 2H), 6.88 (s, 1H), 6.69 (dd, J = 2.4, 9.0 Hz, 1H), 4.52 (d, J = 5.1 Hz, 2H), 3.79 (s, 3H), 3.72 (s, 2H), 3.39 (s, 3H) ESI-MS m/z 469 [C25H22ClN3O3+ Na]+, HPLC: retention time = 18.6 min.
Compound 14 was obtained as an off-white solid (204 mg, 55% isolated yield). Melting point = 178 °C, 1H NMR (CDCl3) δ 8.47 (dd, J = 1.8, 4.3 Hz, 2H), 7.63 (d, J = 13.1 Hz, 2H), 7.47 (d, J = 13.1 Hz, 2H), 7.05 (dd, J = 1.8, 4.3 Hz, 2H), 6.89 (d, J = 2.4 Hz, 1H), 6.84 (d, J = 9.0 Hz, 1H), 6.70 (dd, J = 2.4, 9.0 Hz, 1H), 4.40 (d, J = 6.3 Hz, 2H), 3.80 (s, 3H), 3.74 (s, 2H), 2.41 (s, 3H) ESI-MS m/z 469 [C25H22ClN3O3+ Na]+, HPLC: retention time = 17.2 min.
Compound 15 was obtained as an off-white solid (53 mg, 13% isolated yield). Melting point = 181 °C, 1H NMR (CDCl3) δ 8.48 (dd, J = 1.6, 4.4 Hz, 2H), 7.65 (d, J = 13.1 Hz, 2H), 7.49 (d, J = 13.1 Hz, 2H), 7.04 (dd, J = 1.6, 4.4 Hz, 2H), 6.85 (m, 1H), 6.72 (dd, J = 2.5, 9.0 Hz, 1H), 5.83 (d, J = 7.8 Hz, 1H), 5.10 (quint, J = 7.4 Hz, 1H), 3.78 (s, 3H), 3.68 (s, 2H), 2.40 (s, 3H), 1.35 (d, J = 7.4 Hz, 3H) ESI-MS m/z 483 [C26H24ClN3O3+ Na]+, HPLC: retention time = 16.2 min.
Compound 16 was obtained as a yellow solid (310 mg, 85% isolated yield). Melting point = 163 °C, 1H NMR (CDCl3) δ 8.24 (d, J = 8.4 Hz, 1H), 8.10 (d, J = 5.9 Hz, 1H), 7.95 (s, 1H), 7.51 (d, J = 13.1 Hz, 2H), 7.08 (m, 1H), 6.94 (d, J = 2.4 Hz, 1H), 6.87(d, J = 9.0 Hz, 1H), 3.87 (s, 2H), 3.82 (s, 3H), 2.45 (s, 3H) ESI-MS m/z 455 [C24H20ClN3O3+ Na]+, HPLC: retention time= 17.1 min.
Compound 17 was obtained as a yellow solid (60 mg, 16% isolated yield). Melting point = 167 °C, 1H NMR (CDCl3) δ 8.61 (d, J = 4.9 Hz, 2H), 8.41 (br s, 1H), 7.69 (d, J = 13.1 Hz, 2H), 7.47 (d, J = 13.1 Hz, 2H), 7.02 (t, J = 3.2 Hz, 1H), 6.96 (d, J = 2.4 Hz, 1H), 6.87(d, J = 9.0 Hz, 1H), 6.69 (dd, J = 2.4, 9.0 Hz, 1H), 4.03 (s, 2H), 3.80 (s, 3H), 2.44 (s, 3H), ESI-MS m/z 456 [C23H19ClN4O3 + Na]+, HPLC: retention time = 18.9 min.
Compound 18 was obtained as an off-white solid (250 mg, 59% isolated yield). Melting point = 177 °C, 1H NMR (300 MHz, CDCl3, ppm) δ 8.15 (d, 1H, J = 2.1 Hz), 7.50 (m, 5H), 7.422 (d, 1H, J = 8.4 Hz), 6.83 (m, 2H),6.69 (dd, J1 = 9 Hz, J2 =2.4 Hz 1H), 6.19 (br t, J = 6 Hz, 1H), 4.37 (d, J = 6Hz, 2H), 3.79 (s, 3H), 3.71(s, 2H), 2.36 (s, 3H), ESI-MS m/z (positive) 482 [C25H21Cl2N3O3]+, HPLC: retention time = 19.1 min.
Compound 19 was obtained as an off-white solid (264 mg, 69% isolated yield). Melting point = 183 °C, 1H NMR (300 MHz, CDCl3, ppm) δ 8.54 (s, 1H), 7.60 (m, 4H), 7.49 (m, 2H), 6.85 (m, 2H), 6.70 (d, J = 8.7 Hz, 1H), 6.12 (br s, 1H), 4.47 (s, 2H), 3.79 (s, 3H), 3.72 (s, 2H), 2.39 (s, 3H), ESI-MS m/z (positive) 516 [C26H21ClF3N3O3]+, HPLC: retention time =20.0 min.
Compound 20 was obtained as a yellow solid (280 mg, 59% isolated yield), Melting point = 188 °C, 1H NMR (300 MHz, CDCl3, ppm) δ 8.40 (d, J = 1.4 Hz, 1H), 8.32 (dd, J1 = 4.7 Hz, J2 = 1.4 Hz, 1H), 8.06 (m, 1H), 7.65 (d, J = 8.6, 2H), 7.48 (d, J = 8.6, 2H), 7.24 (m, 1H), 6.94 (d, J = 2.5 Hz, 1H), 6.86 (d, J = 9.0 Hz, 1H), 6.72 (dd, J1 = 9.0, j2 = 2.5, 1H), 5.61 (br s, 1H), 3.81 (s, 3H), 3.59 (s, 2H), 3.45 (m, 2H), 2.70 (t, J = 6.7 Hz, 2H), 2.04 (s, 3H) ESI-MS m/z (positive) 462 [C26H24ClN3O3+H]+, HPLC: retention time = 15.1 min.
Compound 21 was obtained as an off-white solid (360 mg, 72% isolated yield). Melting point = 191 °C, 1H NMR (300 MHz, CDCl3, ppm) δ 7.65 (d, J = 8.6, 2H), 7.53 (br s, 1H), 7.48 (d, J = 8.6, 2H), 7.30 (m, 2H), 7.18 (dd, J1 = 9.0, j2 = 2.5, 1H), 6.94 (d, J = 2.5 Hz, 1H), 6.86 (d, J = 9.0 Hz, 1H), 6.72 (dd, J1 = 9.0, j2 = 2.5, 1H), 5.61 (br s, 1H), 3.81 (s, 3H), 3.59 (s, 2H), 3.45 (m, 2H), 2.70 (t, J = 6.7 Hz, 2H), 2.04 (s, 3H) ESI-MS m/z (positive) 495 [C27H24Cl2N2O3+H]+, HPLC: retention time =16.1 min.
Compound 22 was obtained as an off-white solid (301 mg, 62% isolated yield) Melting point = 180 °C, 1H NMR (300 MHz, CDCl3, ppm) δ 7.59 (d, J = 8.4 Hz, 2H), 7.47 (d, J = 8.4 Hz,, 2H), 7.13 (m, 3H), 6.90 (m, 4H), 6.71 (dd, J1 = 8.9, j2 = 2.4, 1H), 5.61 (br s, 1H), 3.81 (s, 3H), 3.59 (s, 2H), 3.45 (m, 2H), 2.70 (m, 1H), 2.04 (s, 3H), 1.28 (d, J = 6.8 Hz, 3H), ESI-MS m/z (positive) 475 [C28H27ClN2O3+H]+, HPLC: retention time = 17.3 min.
Spectral titration of TCCYP51
The apparent affinities of ligand binding were evaluated by the spectral changes they induced in the TCCYP51.9 The titration experiments were carried out at 24°C in 2 ml tandem cuvettes, containing 2 μM TCCYP51 in buffer A in the wavelength range 350–500 nm using a Shimadzu UV-2401PC spectrophotometer. The tested compounds (1 or 10 mM stock solutions in DMSO, depending on the affinity of the interaction) were added in 1-μl aliquots to the test cuvette until the maximum in the TCCYP51 spectral response was reached. Equal volumes of DMSO were added to the reference cuvette. The apparent Kd’s were determined from the equilibrium titration curves by plotting absorbance changes against the concentration of free ligand and fitting the data to a rectangular hyperbola using SigmaPlot Statistics.25
ESI-MS analysis of non-covalent interactions in TCCYP51
The experiments were conducted with an ESI-oaTOF mass spectrometer (micrOTOF, Bruker Daltonics, Inc., Billirica, MA) which has been modified for enhanced collisional cooling in the source for the analysis of non-covalent protein complexes. This was completed by the addition of a valve in turbo pump line restricting vacuum. The standard instrument parameters were used with the exception of the following: capillary voltage 3.5 kV, capillary exit 250 V, fore pressure 4.99 mbar, and TOF pressure 6.29 × 10−7 mbar. TCCYP51 was buffer exchanged into 50 mM ammonium acetate, pH 7.0; 20-HCl was dissolved in ethanol and added to the protein solution to give the final concentration of 100 μM. The ESI flow rate was 180 μLh-1. Spectra were acquired in positive polarity mode, externally calibrated, and processed using DataAnalysis software (Bruker Daltonics, Inc.) by smoothing and base-line subtracting.
Reconstitution of enzymatic activity and inhibition assay
Sterol 14α-demethylase activity of TCCYP51 in vitro was reconstituted with cytochrome P450 reductase (CPR) from Trypanosoma brucei as an electron donor partner26 and 24-methylenedihydrolanosterol (MDL) as a substrate.8 The concentrated proteins were pre-incubated for 10 min at room temperature with dilauroyl-α-phosphatidylcholine (DLPC) at molar ratio TCCYP51:CPR:DLPC=1:2:50. The final reaction mixture contained 1 μM TCCYP51 and 50 μM substrate (unlabeled and [3H]-MDL were mixed to give ~2,000 cpm/nmol and added from 1 mM stock solution in 45%, 2-hydroxypropyl-β-cyclodextrin) in 20 mM MOPS (pH 7.4), 50 mM KCl, 5 mM MgCl2, 10% glycerol, 0.4 mg/ml isocitrate dehydrogenase and 25 mM sodium isocitrate. For the inhibition assay, the reaction was performed in the presence of the increasing concentrations of the compounds tested as TCCYP51 inhibitors (concentration range 1–100 μM). After 5 min of pre-incubation with the inhibitors at 37°C, the reaction was initiated with the addition of NADPH (5 μM). Sterols were extracted with ethyl acetate and analyzed by reverse phase HPLC in the linear gradient of methanol: acetonitrile: H2O=9:9:2 (solution A) and methanol (solution B) (0–100%) using Waters C18 column and β-RAM radioactivity detector. The inhibitory potency was estimated as molar ratio inhibitor/enzyme at which the activity decreased 2-fold (I/E2).9 Inhibitory effect of the compounds on COX-2 enzyme was determined at 100 nM final concentration of mouse cyclooxygenase 2 as previously described19 and the potency was expressed as I/E2 to correspond to the way it is expressed for TCCYP51.
Cell culture and growth inhibition assay in TC cells
Trypomastigotes (106 organisms) were pre-exposed to the CYP51 inhibitors dissolved in DMSO at several concentrations (1–50 μM) or to control DMSO for 30 min. Exposure of different concentrations of CYP51 inhibitors to trypomastigotes for 30 min did not affect their motility. The parasites were then exposed in triplicate to rat cardiomyocyte monolayers at the ratio 10 parasites/cell in Lab Tech chambers for 2h as described.27 To verify that the inhibitory effect was on trypanosomaes and not on cardiomyocytes, in additional experiments excess compound was removed from the trypanosomes prior to infection, producing similar results. After removing the unbound parasites, monolayers were incubated with DMEM supplemented with 10% FBS for 72 hr to allow parasite intracellular multiplication and the number of T. cruzi per 200 cells and the percent of infection were microscopically determined in Giemsa stained monolayers.27
TC cellular sterol analysis
Epimastigotes (plating density 107 cells/ml) were cultured in brain heart infusion supplemented with hemin and 10% calf serum for 120 hours without an inhibitor and in the presence of 20. The inhibitor was added daily to maintain 50 μM concentration. The cell pellet was washed with Hanks’ balanced salt solution without phenol red to remove excess cholesterol from the serum and saponified using 10% KOH in 98% aqueous methanol at reflux temperature for 1 hour. The TC sterols were extracted with hexane and analyzed by silica gel TLC in hexane:ethyl acetate (8:2) as described previously.9
Supplementary Material
Acknowledgments
This work was supported by grants from the American Heart Association (0535121N to G.I.L), the National Institutes of Health (GM067871 to M.R.W and G.I.L., ES00267 to M.R.W and L.J.M., AI080580, AI007281 and HL007737 to F.V. and CA89450 to L.J.M.), Welch Foundation (D-1276 to W.D.N.) and from the Department of Defense (W81XWH-05-01-0179 to R.M.C).
Abbreviations
- TC
Trypanosoma cruzi
- CYP51
sterol 14α-demethylase
- COX2
cyclooxygenase 2
- INDO
indomethacin
- NSAID
non-steroidal anti-inflamatory drug
- CPR
cytochrome P450 reductase
- MDL
24-methylenedihydrolanosterol
- I/E2
molar ratio inhibitor/enzyme which causes a two-fold decrease in the initial rate of catalysis
Footnotes
Supporting Information. ESI-mass spectrometry of TCCYP51, ligand free and compound 20 bound. This material is available free of charge via the Internet at http://pubs.acs.org
References
- 1.Centers for Disease Control and Prevention (CDC) Blood donor screening for Chagas disease-United States, 2006–2007. Morb Mortal Wkly Rep. 56:141–143. [PubMed] [Google Scholar]
- 2.Ferreira MS, Borges AS. Some aspects of protozoan infections in immunocompromised patients-a review. Mem Inst Oswaldo Cruz. 2002;97:443–457. doi: 10.1590/s0074-02762002000400001. [DOI] [PubMed] [Google Scholar]
- 3.Wilkinson SR, Taylor MC, Horn D, Kelly JM, Cheeseman I. A mechanism for cross-resistance to nifurtimox and benznidazole in trypanosomes. Proc Natl Acad Sci U S A. 2008;105:5022–5027. doi: 10.1073/pnas.0711014105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Croft SL, Barrett MP, Urbina JA. Chemotherapy of trypanosomiases and leishmaniasis. Trends Parasitol. 2005;21:508–512. doi: 10.1016/j.pt.2005.08.026. [DOI] [PubMed] [Google Scholar]
- 5.El-Sayed NM, Myler PJ, Blandin G, Berriman M, Crabtree J, Aggarwal G, Caler E, Renauld H, Worthey EA, Hertz-Fowler C, et al. Science. 2005;309:404–409. [Google Scholar]
- 6.Cammerer SB, Jimenez C, Jones S, Gros L, Lorente SO, Rodrigues C, Rodrigues JC, Caldera A, Ruiz Perez LM, da Souza W, Kaiser M, Brun R, Urbina JA, Gonzalez Pacanowska D, Gilbert IH. Quinuclidine derivatives as potential antiparasitics. Antimicrob Agents Chemother. 2007;51:4049–4061. doi: 10.1128/AAC.00205-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Haughan PA, Goad LJ. Lipid Biochemistry of Trypanosomatids. In: Coombs G, North M, editors. Biochemical Protozoology. Taylor & Francis; London: 1991. pp. 312–328. [Google Scholar]
- 8.Lepesheva GI, Zaitseva NG, Nes WD, Zhou W, Arase M, Liu J, Hill GC, Waterman MR. CYP51 from Trypanosoma cruzi: a phyla-specific residue in the B′ helix defines substrate preferences of sterol 14alpha-demethylase. J Biol Chem. 2006;281:3577–3585. doi: 10.1074/jbc.M510317200. [DOI] [PubMed] [Google Scholar]
- 9.Lepesheva GI, Ott RD, Hargrove T, Kleshchenko Y, Schuster I, Nes WD, Hill G, Villalta F, Waterman MW. Sterol 14α-Demethylase as a Potential Target for Antitrypanosomal Therapy: Enzyme Inhibition and Parasite Cell Growth. Chem Biol. 2007;14:1283–1293. doi: 10.1016/j.chembiol.2007.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Espinel-Ingroff A. Mechanisms of resistance to antifungal agents: Yeasts and filamentous fungi. Rev Iberoam Micol. 2008;2:101–106. doi: 10.1016/s1130-1406(08)70027-5. [DOI] [PubMed] [Google Scholar]
- 11.Kalgutkar AS, Marnett AB, Crews BC, Remmel RP, Marnett LJ. Ester and amide derivatives of the nonsteroidal antiinflammatory drug, indomethacin, as selective cyclooxygenase-2 inhibitors . J Med Chem. 2000;43:2860–2870. doi: 10.1021/jm000004e. [DOI] [PubMed] [Google Scholar]
- 12.Kalgutkar AS, Crews BC, Rowlinson SW, Marnett AB, Kozak KR, Remmel RP, Marnett LJ. Biochemically based design of cyclooxygenase-2 (COX-2) inhibitors: facile conversion of nonsteroidal antiinflammatory drugs to potent and highly selective COX-2 inhibitors. Proc Natl Acad Sci U S A. 2000;97:925–930. doi: 10.1073/pnas.97.2.925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Prusakiewicz JJ, Felts AS, Mackenzie BS, Marnett LJ. Molecular basis of the time-dependent inhibition of cyclooxygenases by indomethacin. Biochemistry. 2004;43:15439–15445. doi: 10.1021/bi048534q. [DOI] [PubMed] [Google Scholar]
- 14.Ortiz de Montellano PR, Correia MA. Inhibition of cytochrome P450 enxzymes. In: Ortiz de Montellano Pr., editor. Cytochrome P450: Structure, Mechanism, and Biochemistry. New York: Plenum Publishing Corp; 1995. pp. 305–364. [Google Scholar]
- 15.Lepesheva GI, Waterman MR. Sterol 14alpha-demethylase cytochrome P450 (CYP51), a P450 in all biological kingdoms. Biochim Biophys Acta. 2007;1770:467–477. doi: 10.1016/j.bbagen.2006.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Poulos TL. Cytochrome P450 flexibility. Proc Natl Acad Sci US A. 2003;100:13121–13122. doi: 10.1073/pnas.2336095100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lepesheva GI, Seliskar M, Knutson CG, Stourman NV, Rozman D, Waterman MR. Conformational dynamics in the F/G segment of CYP51 from Mycobacterium tuberculosis monitored by FRET. Arch Biochem Biophys. 2007;464:221–227. doi: 10.1016/j.abb.2007.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Atkins WM. Non-Michaelis-Menten kinetics in cytochrome P450-catalyzed reactions. Annu Rev Pharmacol Toxicol. 2005;45:291–310. doi: 10.1146/annurev.pharmtox.45.120403.100004. [DOI] [PubMed] [Google Scholar]
- 19.Remmel RP, Crews BC, Kozak KR, Kalgutkar AS, Marnett LJ. Studies on the metabolism of the novel, selective cyclooxygenase-2 inhibitor indomethacin phenethylamide in rat, mouse, and human liver microsomes: identification of active metabolites. Drug Metab Dispos. 2004;32:113–122. doi: 10.1124/dmd.32.1.113. [DOI] [PubMed] [Google Scholar]
- 20.Faundez M, Pino L, Letelier P, Ortiz C, López R, Seguel C, Ferreira J, Pavani M, Morello A, Maya JD. Buthionine sulfoximine increases the toxicity of nifurtimox and benznidazole to Trypanosoma cruzi. Antimicrob Agents Chemother. 2005;49:126–130. doi: 10.1128/AAC.49.1.126-130.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.de Castro SL, de Meirelles MN. Effect of drugs on Trypanosoma cruzi and on its interaction with heart muscle cell “in vitro”. Mem Inst Oswaldo Cruz. 1987;82:209–218. doi: 10.1590/s0074-02761987000200009. [DOI] [PubMed] [Google Scholar]
- 22.Saraiva J, Vega C, Rolon M, da Silva RE, Silva ML, Donate PM, Bastos JK, Gomez-Barrio A, de Albuquerque S. In vitro and in vivo activity of lignan lactones derivatives against Trypanosoma cruzi. Parasitol Res. 2007;100:791–795. doi: 10.1007/s00436-006-0327-4. [DOI] [PubMed] [Google Scholar]
- 23.Buckner FS, Wilson AJ, White TC, Van Voorhis WC. Induction of resistance to azole drugs in Trypanosoma cruzi. Antimicrob Agents Chemother. 1998;42:3245–3250. doi: 10.1128/aac.42.12.3245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Omura T, Sato R. The carbon monoxide-binding pigment of liver microsomes. Ii. Solubilization, purification, and properties. J Biol Chem. 1964;239:2379–2385. [PubMed] [Google Scholar]
- 25.Lepesheva GI, Virus C, Waterman MR. Conservation in the CYP51 family. Role of the B′ helix/BC loop and helices F and G in enzymatic function. Biochemistry. 2003;42:9091–9101. doi: 10.1021/bi034663f. [DOI] [PubMed] [Google Scholar]
- 26.Lepesheva GI, Nes WD, Zhou W, Hill GC, Waterman MR. CYP51 from Trypanosoma brucei is obtusifoliol-specific. Biochemistry. 2004;43:10789–10799. doi: 10.1021/bi048967t. [DOI] [PubMed] [Google Scholar]
- 27.Nde PN, Simmons KJ, Kleshchenko Y, Pratap S, Lima MF, Villalta F. Silencing of the laminin γ-1 gene blocks Trypanosoma cruzi infection. Infect Immun. 2006;74:1643–1648. doi: 10.1128/IAI.74.3.1643-1648.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.