

Abstract
We have recently identified a new class of compounds that selectively kill cells that express P-glycoprotein (P-gp, MDR1), the ATPase efflux pump that confers multidrug resistance on cancer cells. Several isatin-β-thiosemicarbazones from our initial study have been validated, and a range of analogs synthesized and tested. A number demonstrated improved MDR1-selective activity over the lead, NSC73306 (1). Pharmacophores for cytotoxicity and MDR1-selectivity were generated to delineate the structural features required for activity. The MDR1-selective pharmacophore highlights the importance of aromatic/hydrophobic features at the N4 position of the thiosemicarbazone, and the reliance on the isatin moiety as key bioisosteric contributors. Additionally, a quantitative structure-activity relationship (QSAR) model that yielded a cross-validated correlation coefficient of 0.85 effectively predicts the cytotoxicty of untested thiosemicarbazones. Together, the models serve as effective approaches for predicting structures with MDR1-selective activity, and aid in directing the search for the mechanism of action of 1.
Introduction
Multidrug resistance (MDR) conferred by the ABC transporter family that includes MDR1 (ABCB1, P-glycoprotein, P-gp), presents a significant clinical challenge for drug design and development1. P-gp expression is well-characterized in hematological malignancies, sarcomas, and other solid cancers, and in those tumor types is frequently correlated with poor clinical response to chemotherapy2. Strategies employed to circumvent the reduced drug accumulation conferred by these poly-specific efflux transporters have relied heavily on the development of clinical inhibitors of P-gp for concurrent administration with chemotherapeutics. Although a number of these have shown promise in vitro, translation to the clinic has taken longer than may have been expected1,3, due in part to pharmacokinetic effects caused by inhibition of endogenous function1, and alternative strategies are required.
Early inhibitors were ‘off-the-shelf’ drugs used for other medical conditions that had limited activity in vivo (e.g, verapamil), leading to second-generation inhibitors that were structurally related to first-generation compounds, but altered chemically to improve their affinity for P-gp4. Later, structurally unique third-generation inhibitors were designed specifically for their capacity to inhibit P-gp, yet despite their enhanced efficacy (and in part because of it) the latest generation inhibitors frequently altered the pharmacokinetic profile of the co-administered chemotherapy leading to reduced efficacy and increased side-effects1,3. Given the problems identified above, in addition to problems relating to poor trial design, the clinical benefit of direct P-gp inhibitors remains to be proven.
One strategy to circumvent problems associated with P-gp inhibition and resolve the emergence of clinical MDR is to develop drugs that exploit the expression of P-gp, thus turning a mechanism of drug resistance into a weakness5,6. To this end, we have previously profiled mRNA expression of all 48 known and predicted human ABC transporters in the National Cancer Institute 60 cell line panel (NCI-60), used by the NCI to screen over 100,000 compounds for anticancer activity6. These data were used to identify the individual ABC transporters that conferred multidrug resistance on cells6. Furthermore, bioinformatic correlation of gene expression in the NCI-60 cell lines with cytotoxicity of drugs against the NCI-60 cell lines identified compounds whose activity was potentiated rather than diminished by the expression of P-gp were identified, and recently validated7.
Of the sixty compounds whose activity was inversely related to P-gp expression (MDR1-selective agents), ten possessed a thiosemicarbazone functional group, and seven contained a 1-isatin-3-thiosemicarbazone (isatin-β-thiosemicarbazone) moiety; 1 (NSC73306)8, 2 (NSC658339)8, 3 (NSC716765)8, 4 (NSC716766)8, 5 (NSC716768)8, 6 (NSC716771)8 and 7 (NSC716772)8 shown in Figure 1 6. The remarkable result that seven isostructural compounds would be present in the fifty most statistically significant compounds led us to select 1 as a lead compound to validate its MDR1-selective properties and understand its mechanism of action5. While biochemical assays have shown that 1 does not interact with P-gp as either a substrate or inhibitor, the effectiveness of 1 against MDR cell-lines correlates with their expression of P-gp.5 Importantly, from a clinical perspective, cell lines selected for resistance to 1 show loss of P-gp. As such, 1 represents an exciting prospect for resolving multidrug resistance in the clinic by selectively killing cells that express high levels of P-gp, and re-sensitizing residual cells to conventional chemotherapeutics. This strategy is currently being assessed using P-gp-mediated drug resistant human cancer xenografts in the mouse. The biological activity of thiosemicarbazones has been known for a considerable period of time, both as anticancer (1956)9 and antiviral (1973)10,9 drugs.11, 12 Methisazone (N-methyl-isatin-β-thiosemicarbazone), for example, was effective as prophylaxis against smallpox and vaccinia viruses,11 and 3-aminopyridine-2-carboxaldehyde thiosemicarbazone (9, 3-AP) is currently being evaluated in clinical trials against several malignancies including leukemia (Figure 1).13, 14 At least some of the biological activity of thiosemicarbazones has been shown to involve interaction with metal ions15, 16 and a number of mechanisms of action have been identified including ribonucleotide reductase inhibition, metal dependent radical damage, DNA binding and inhibition of protein synthesis.17–19 The metal chelates of thiosemicarbazones administered to cells are regularly more active than the drug alone.16
Figure 1.

Structures of methisazone, 9, and the seven isatin-β-thiosemicarbazones (1-7) identified in a bioinformatics screen as having activity that is potentiated, rather than inhibited by expression of the multidrug transporter P-gp. 1 is being treated as a lead compound to understand the mechanism of action of the compounds. An overlay of the seven NSC compounds identified in the bioinformatics screen demonstrates the common structural features associated with them.
1 is currently undergoing pre-clinical evaluation. However, its non-optimal aqueous solubility (a feature for which thiosemicarbazones are notorious20) has led to a search for more soluble derivatives, and while 1 is several-fold more active against P-gp-expressing cell lines than their parental lines, we desire to develop a yet more selective derivative based on this compound. This requires an understanding of the relative importance of the key structural features of 1 and its congeners that must be retained to maintain MDR1-selective activity. To this end, we have designed and synthesized a series of compounds, and tested them against a parental HeLa-derived cervical cancer cell line (KB-3-1) and its vinblastine-selected derivative that highly expresses P-gp (KB-V1). Both cytotoxicity and MDR1-selectivity were correlated with structural variations in an effort to construct an active pharmacophore to be used as the basis for further drug design.
Results and discussion
Characterization of compounds from the DTP library
Given that seven isatin-β-thiosemicarbazones were predicted in the initial bioinformatics screen to identify MDR1-selective compounds, they were of primary interest for validation. 1 was selected as a lead compound primarily due to its availability, and the structure was initially believed to incorporate a 5,7-dichloroisatin moiety (mw = 395.3 gmol−1) when submitted to the NCI for testing. However, APCI-MS analysis of the sample from the DTP library revealed it to be the non-substituted isatin derivative, whose structure is correctly shown in Figure 1, and additional quantities were synthesized for subsequent testing.5 Of the seven isatin-β-thiosemicarbazones predicted in the DTP compound library to demonstrate MDR1-selective activity, only 1 and 2 were available for testing, while the remaining five compounds were not publicly available from the DTP for biological testing. However, it was possible to arrange for 1H NMR spectra of these five samples to establish the structure and purity of the compounds as submitted. ESI-MS of the sample of 2 (expected mw = 390.1 gmol−1) revealed a (M+H)+ ion at 221.0, indicating the absence of the ethylmalonate moiety. MS2 confirmed the loss of a -HNC=S fragment revealing the likely structure to be the isatin-β-thiosemicarbazone 3 (Table 1), more of which was subsequently synthesized for this study. Analysis of compounds 3, 4, 5, 6 and 7 by 1H NMR spectra in d6-DMSO revealed that 3, 4 and 6 were relatively pure (~ 95%), however solutions of 5 and 7 returned spectra showing many impurities.
Table 1.
Structures of compounds considered in this study, along with IC50 values determined against the parental KB-3-1 cell line, and the P-glycoprotein expressing cell line KB-V1. The MDR1 selectivity is calculated as the ratio of a compound’s IC50 against KB-3-1 cells divided by its IC50 against KB-V1 cells. A value > 1 indicates that the compound kills P-gp-expressing cells more effectively than parental cells, so-called MDR1-selective activity. A value < 1 indicates that the P-gp expressing cells are resistant to the compound, relative to parental cells, as is normally observed for P-gp substrates. ‘n/a’ denotes compounds, as described in the discussion, that were not tested for their biological activity as they were shown to have been incorrectly identified or impure.
| Compound | IC50, KB 3–1 (μM) | IC50, KB–V1 (μM) | MDR1 selectivitya |
|---|---|---|---|
 1 |
14.2 ± 2.1 | 3.3 ± 1.3 | 4.3 |
 2 |
n/a | n/a | n/a |
 3 |
17.0 ± 6.6 | 34.3 ± 14.2 | 0.5 |
 4 |
14.5 ± 8.1 | 10.2 ± 2.1 | 1.4 |
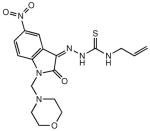 5 |
n/a | n/a | n/a |
 6 |
13.1 ± 4.8 | 5.8 ± 2.5 | 2.3 |
 7 |
28.4 ± 5.2 | 4.0 ± 0.6 | 7.1 |
 8 |
>50 | >50 | - |
 9 |
1.4 ± 2.5 | 5.9 ± 2.6 | 0.2 |
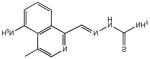 10 |
1.9 ± 0.8 | 2.1 ± 0.3 | 0.9 |
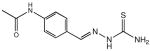 11 |
>50 | >50 | - |
 12 |
>50 | >50 | - |
|
13 |
>50 | >50 | - |
 14 |
>50 | >50 | - |
 15 |
>50 | >50 | - |
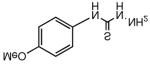 16 |
>50 | >50 | - |
 17 |
n/a | n/a | n/a |
 18 |
39.3 ± 23.3 | 5.2 ± 1.1 | 7.5 |
 19 |
19.6 ± 1.7 | 9.3 ± 2.8 | 2.1 |
 20 |
>50 | >50 | - |
|
21 |
3.4 ± 1.5 | 2.4 ± 0.5 | 1.4 |
 22 |
4.4 ± 3.6 | 30.3 ± 5.9 | 0.1 |
 23 |
26.3 ± 5.5 | 6.7 ± 2.9 | 4.0 |
 24 |
26.1 ± 2.6 | 20.7 ± 7.7 | 1.3 |
 25 |
27.7 ± 3.3 | >50 | - |
|
26 |
>50 | >50 | - |
We synthesized (described below) the correct structures of 1 (Figure 1) and 2 (shown correctly as 3 in Table 1), and 3, 4, 6 and 7 as reported. 5 was not synthesized. Since neither the NCI chemical library nor most other publicly available chemical repositories are validated for structure or purity following submission, our experience strongly suggests that chemicals obtained from these sources must be validated for purity.
Synthesis
The straightforward synthesis of isatin-β-thiosemicarbazones by the condensation of isatin and a thiosemicarbazide has been known for more than fifty years (Figure 2c).21, 22 The compounds 3, 4, 5, 6 and 7 have previously been reported by Karali and co-workers, in the context of anti-viral and anti-cancer screening results, and were synthesized by such a method23–25, though the preparation of the precursor thiosemicarbazides was not described.
Figure 2.

Scheme showing general synthetic steps in the preparation of isatin-β-thiosemicarbazones. (a) General synthetic method used for the synthesis of isatin-β-thiosemicarbazones, the example here being 1. Equimolar quantites of isatin (or a substituted isatin) and a thiosemicarbazide (here 4-(4-methoxyphenyl)-3-thiosemicarbazide) are stirred in hot ethanol, a few drops of glacial acetic acid are added, and the solution is refluxed for a short period that on cooling resulted in the precipitation of the final product. (b) The phenylthiosemicarbazide is generally reported to be synthesized by the reaction of hydrazine with the substituted phenylisothiocyanate of choice, such as p-methoxyphenyisothiocyanate shown here, yielding 4-(4-methoxyphenyl)-3-thiosemicarbazide. (c) In our hands the reaction between hydrazine and p-halogen phenylisothiocyanates such as p-fluorophenylisothiocyanate did not proceed as expected, but resulted (d) in an insoluble precipitate that was unable to undergo further reaction with isatin. It was identified by mass spectrometry to be a 2:1 combination of thiocyanate and hydrazine (mw = 338 gmol−1) that formed even when hydrazine was added drop-wise to the excess thiocyanate. Using a t-Boc protected form of hydrazine, t-butylcarbazate, which is effectively monofunctional hydrazine, the t-Boc protected thiosemicarbazide could be produced, from which the thiosemicarbazone was recovered under acidic conditions.
A number of preparative approaches are available for thiosemicarbazides, one of the more straightforward being the addition of hydrazine to an isothiocyanate (Figure 2a), which we generally employed in our preparations.26 In synthesizing 6, the preparation of 4-fluorophenylthiosemicarbazide from 4-fluorophenylisothiocyanate and hydrazine yielded an insoluble product that was identified by 1H NMR to involve reaction of 2 mole of thiocyanate with one mole of hydrazine (Figure 2b). Drop-wise addition of hydrazine to an excess of thiocyanate could not ameliorate the formation of this product. In order to prevent this, t-Boc protected hydrazine, t-butylcarbazate27, was reacted with 4-fluorophenylisothiocyanate resulting in t-Boc protected 4-fluorophenyl thiosemicarbazide that could be readily deprotected with acid (Figure 2c). This synthetic route was used subsequently to ensure the ready preparation of pure thiosemicarbazides.
Compounds 12 and 13 in Table 1 were prepared by condensation of 2-indanone and 1-indanone (in lieu of isatin) with 4-methoxyphenyl-3-thiosemicarbazide. Compound 14 was prepared by reacting thiosemicarbazide with isatin. The isatin-β-semicarbazones 15 and 25 were prepared by the reaction of the corresponding isatin with semicarbazide in lieu of their respective thiosemicarbazides. Compounds 18, 19, 20, 22 and 23 were prepared by condensation of the starting material (5-fluoroisatin, N-methyl isatin, sodium isatin-5-sulfonate, benz(g)indole-2,3-dione and 5-nitroisatin, respectively) with 4-methoxyphenyl-3-thiosemicarbazide.
Cytotoxicity
To ascertain which structural features are responsible for the selective cytotoxicity of 1, the structurally diverse compounds described above were designed and synthesized for testing (Table 1). Some of the synthesized compounds were analogs of 1 that incorporated simple substitutions on the isatin framework, whereas others broadly modify the structure to test which structural elements were required for activity. We focused our interest, principally, on understanding two biological properties of 1: (1) The absolute cytotoxicity (measured using the MTT assay) of compounds against parental P-gp-negative KB-3-1 adenocarcinoma cells. This was tested to determine whether compounds were active, and if so, how this activity compared with that of the lead compound 1; (2) MDR1 selectivity, as indicated by greater sensitivity in KB-V1 cells compared to KB-3-1 cells, and test whether greater selectivity for MDR1 expressing cells than that of 1 could be achieved. To this end, compounds were also tested against the KB-V1 cervical adenocarcinoma cell line that expresses high levels of P-gp (the protein product of MDR1). The KB-V1 cell line was originally developed by step-wise selection of KB-3-1 cells in the drug vinblastine.28 MDR1 selectivity is determined by the ratio of IC50 against KB-3-1 cells divided by its IC50 against KB-V1 cells. A value > 1 indicates that the compound kills P-gp expressing cells more effectively than parental cells, resulting in so-called MDR1-selective activity.5 Alternatively, a value < 1 indicates that the P-gp expressing cells are resistant to the compound, relative to parental cells, as is normally observed for drugs effluxed by P-gp.29
Of the five DTP compounds identified in the bioinformatics screen and synthesized as described above, four compounds (1, 4, 6 and 7, Table 1) demonstrated varying levels of MDR1 selectivity, with only 7 showing a greater MDR1 selectivity value than 1 (7.1 and 4.3, respectively). 3, the only DTP compound possessing an alkenyl rather than aryl moiety at the N4 position, was not selective for KB-V1 cells, and 14, which was identified as the true structure of 2 from the DTP collection, was found to be inactive against both cell lines (IC50 > 50 μM).
Two thiosemicarbazones that have been previously tested extensively in vitro and in the clinic are the anti-cancer compounds 3-AP (9, 3-aminopyridine-2-carboxaldehyde thiosemicarbazone) and MAIQ (10, 4-methyl-5-amino-1-formylisoquinoline thiosemicarbazone) and these were chosen to determine whether MDR1 selectivity is a common feature of biologically active thiosemicarbazones (Table 1). Neither compound is substituted at the N4 position, and they possess a pyridine and isoquinoline group, respectively, in lieu of isatin, resulting in an NNS tridentate moiety capable of tightly chelating iron, thereby inhibiting ribonucleotide reductase (RR).30 Neither were found to be MDR1 selective, though their cytotoxicities against the KB-3-1 line were approximately an order of magnitude greater than that of 1 (1.4 and 1.9 versus 14.2 μM). The resistance of KB-V1 cells to these compounds was not surprising given that L1210 murine leukemia cells selected for resistance to 10 (L1210 MQ-580) have been shown to express P-gp31, and demonstrated cross-resistance to 932 (1 is not a P-gp substrate5). The anti-tubercular thiosemicarbazone, thiacetazone (11, 1-(4′-formylacylamine)-3-thiosemicarbazone, Table 1), which is also unsubstituted at the thiosemicarbazone N4 position and contains a phenyl group rather than isatin, was inactive against both cell lines. Isatin itself was also shown to be inactive.
When the p-methoxyphenyl group of 1 was replaced with an alkenyl group in 24 (Table 1), significant activity was lost (14.2 vs 26.1 μM), along with MDR1 selectivity (4.2 versus 1.4) which, along with the observation that 3 was not selective, suggests that an aromatic group at the N4 position improves selectivity. Given that KB-V1 cells were resistant to the RR inhibitors 9 and 10, and that this class of N4 unsubstituted compounds are known to be P-gp substrates, we incorporated a p-methoxyphenyl group at the N4 position of PT (pyridine-2-carboxaldehyde thiosemicarbazone), an analog of 9, forming 21. Not only did compound 21 show superior cytotoxicity to 1 (3.4 versus 14.2 μM), KB-V1 cells were not resistant to it, but were modestly (1.4-fold) sensitive.
Of the compounds synthesized for testing, 12 and 13 contain indanone moieties conjugated to the thiosemicarbazone at positions 1 and 2, lacking the hydrogen bonding potential of the original isatin. Compound 13 removes the lactam moiety of the isatin in 1. Despite minimal structural disruption, both compounds were inactive against both cell lines. The semicarbazone derivatives 15 (structurally analogous to 14), and 25 (based on 1) were designed to test whether the thiosemicarbazone (a strong metal coordination functional group) was essential to activity. Like its thiosemicarbazone counterpart, 24 was inactive against both cell lines, but 25 was weakly active (IC50 = 27.7 μM) only against the KB-3-1 parental line.
A number of substitutions on the isatin group of 1 were explored to examine the effect on activity (the 5,7-dichloro-isatin derivative of 1, 17, was not tested as it was found to be insoluble in the solvents used including DMSO, ethanol, and water). The 5-fluoroisatin (18) analog demonstrated lower activity against the parental KB-3-1 line (39.3 μM) coupled with greater selectivity than 1 for P-gp expressing cells (7.5 vs 4.3). Likewise, the 5-nitroisatin analogue 23 showed slightly lower activity, but similar selectivity, to 1. The 5-isatinsulfonic acid derivative of 1 (20) was prepared to markedly increase aqueous solubility (which it did); however, the compound was inactive, possibly due to the overall negative charge on the molecule in solution resulting in poor cellular uptake. The N-methyl derivative of 1 (analogous to methisazone), 19, showed lower activity and selectivity relative to the parent compound (IC50 = 4.4 μM, selectivity: 2.1). Compound 22 was prepared using benz(g)indole-2,3-dione, and while active (IC50 = 4.4 μM), it was not selective against the V1 line (IC50 = 30.3 μM), suggesting it is a P-gp substrate.
Disregarding the anionic sulfonate, it appears that small substitutions at position 5 of the isatin nucleus on 1 do not abolish its MDR1-selectivity, and indeed in the case of 18 the electron-withdrawing fluorine appears to improve its selectivity. N-methylation reduced selectivity, but did not abolish it. The isatin feature is required for selectivity – simply retaining the 6- and 5-membered ring system as in 12 and 13 is not sufficient for activity. Incorporating steric bulk onto the isatin moiety in 22 appears to improve cytotoxicity. However, its MDR1-selective activity is lost. Collectively, these compounds suggest that an isatin is required for MDR1-selective activity, and that greater selectivity may be possible by substituting electron-withdrawing substituents on the isatin (though the 5-nitroisatin substitution in 23 does not enhance activity over 1). A diverse range of substitutions at both the isatin and phenyl ring will be examined in a subsequent study in a quest to uncover optimal selectivity.
Importantly, it seems that the range of activity of current RR inhibitor thiosemicarbazones may be enhanced by substitution at the thiosemicarbazone N4 position with a hydrophobic or aromatic group. Subsequent to our report of the MDR1-selectivity of the thiosemicarbazone 1, Whitnall et al. demonstrated that a dipyridyl thiosemicarbazone with a dimethyl substitution at the N4 position also demonstrated MDR1-selective activity, though the selectivity of other compounds reported was not demonstrated.33
Structure-activity studies
The poor water solubility of otherwise promising thiosemicarbazones is a well known challenge to their clinical use.20 By identifying the key structural features required for MDR1-selectivity, we sought to design and synthesize variants that display a greater selectivity for resistant cells, and that have improved aqueous solubility. Commensurate with observations for 10 and 1, pharmacophore modeling and quantitative structure activity relationships (QSAR) were employed as two quantitative measures used to gauge the structural relationships of the compounds described above with respect to cytoxicity and MDR1-selectivity.
In this study, the generation of a pharmacophore for the cytotoxicity of compounds against the parental line KB-3-1 (AHRX; Figure 3a) allowed us to determine what molecular components were required for cytotoxicity, compared with those additionally required for MDR1-selectivity. The need for a thiosemicarbazone site (X) is apparent, as compounds that lack this feature (15 and 25) proved inactive. The thiosemicarbazone functional group was assigned a single site, X, for two reasons; first, assigning individual bonding features of the thiosemicarbazone would potentially result in a large number of extra sites common to most molecules, and second, it is possible the thiosemicarbazone group is coordinating to metal ions to effect its cytotoxicity and/or MDR1-selectivity as a bidentate ligand, as is the case for other classes of thiosemicarbazones30.
Figure 3.

Pharmacophores that contribute cytotoxicity and selectivity a). The pharmacophore features required for cytotoxicity, indicated by the activity of compounds tested against the parental KB-3-1 cell line. Pink sphere = A, H-bond acceptor, with the sphere located at the projected point of a hydrogen bond donor. Green sphere = H, hydrophobic group. Ring = aromatic ring. Blue sphere = X, thiosemicarbazone functional group. The pharmacophore sites are underlaid with the structure of 10 to aid in relating the pharmacophore features to structural features of the active compounds. b). The pharmacophore features required for selectivity against the P-gp expressing KB-V1 cell line: AH1H2H3R1R2X. Pink sphere = A, H-bond acceptor, with the sphere located at the projected point of a hydrogen bond donor. Green sphere = H, hydrophobic group. Ring = aromatic ring. Blue sphere = X, thiosemicarbazone functional group. Yellow sphere = Exclusion sphere. The pharmacophore sites are underlaid with the structure of 1 to aid in relating the pharmacophore features to structural features of the active compounds.
The hydrogen bond acceptor site (A), corresponding to the lactam oxygen of isatin-β-thiosemicarbazones or the aromatic nitrogen of 9, 10 and 21, was deemed essential for cytotoxicity, along with their associated aromatic ring/hydrophobic sites (R/H). Molecules lacking any one of the features (A, H, R or X) are inactive, and the failure of thiacetazone, 12, 13, 16 and 26 to match the KB-3-1 cytotoxicity pharmacophore can be understood in terms of missing the hydrogen bonding site, A. The use of a projected point for the hydrogen bond acceptor — simulating the corresponding hydrogen bond donor in the receptor — was introduced to allow structurally dissimilar active compounds to form hydrogen bonds to the same location, regardless of their point of origin and directionality. Such is the case with the isatin-β-thiosemicarbazones, 9, 10 and 21, in which the hydrogen bond acceptors originate in different locations of the ring/hydrophobic region, but are still capable of hydrogen bonding to a common site.
Apart from correctly identifying all of the active thiosemicarbazones, the KB-3-1 pharmacophore was used to pre-align the molecules for QSAR analysis. The performance of the single-factor atom-based QSAR model on the training and test set molecules is illustrated in Figure 4a. The scatter plot indicates exceptionally high correlation (r2 = 0.96) and cross-validated correlation (q2 = 0.85) coefficients, indicative of a model with strong predictive power and significance. Figure 4a also compares experimental and predicted pIC50 values for both the training and test set molecules, showing that activity was effectively predicted. This observation further supports the validity of the pharmacophore model, suggesting the spatial arrangement of chemical features, when aligned by the pharmacophore, is indicative of the probable active conformation of the molecule. The KB-3-1 cytoxicity QSAR model is intended as a preliminary effort to quantitatively infer how cytotoxic new compounds might be, and although strong predictive power is evident, the low number of training/test set compounds warrant caution when using this model for this purpose. Of course, the predictive accuracy will improve once more compounds are added to training and test sets.
Figure 4.


Pharmacophores predict cytotoxicity and selectivity a). Scatter plot and comparison of the KB-3-1 cytoxicity QSAR model applied to thirteen active thiosemicarbazones. The training set correlation is characterized by one partial least-square (PLS) factor (SD = 0.12, R2 = 0.96). The test set correlation is characterized by one PLS factor (Root mean-square error (RMSE) = 0.16, q2 = 0.85, Pearson-R = 0.95). Experimental and calculated pIC50 values are shown for the QSAR training and test set. B). Scatter plot and comparison of the KB-V1 cytoxicity QSAR model applied to twelve active thiosemicarbazones. The training set correlation is characterized by one PLS factor (SD = 0.11, R2 = 0.91). The test set correlation is characterized by one PLS factor (RMSE = 0.29, q2 = 0.42, Pearson-R = 0.70). Experimental and calculated pIC50 values are shown for the QSAR training and test set.
In defining the pharmacophore for the MDR1-selective selectivity demonstrated by 1, 4, 6, 7, 18, 19, 21, 23, and 24 (Table 1), three additional features (R2,H2 and H3) were incorporated into the pharmacophore (described above); all represent electron-rich substitution at the N4 position of thiosemicarbazones common to MDR1-selective compounds. These features, which match well with 1 (Figure 3b), resulted in the MDR1-selectivity pharmacophore AH1H2H3R1R2X. Setting a minimum of three sites for matching, corresponding to AH1X, the MDR1-selectivity pharmacophore can identify compounds that are cytotoxic but not necessarily selective (overlaid in Figure 5a). Incorporating further constraints by increasing the minimum number of required sites to the full seven descriptors (AH1H2H3R1R2X), the pharmacophore highlights compounds that show selectivity for KB-V1 cells. Employing all seven sites predicts only compound 21 and the isatin-β-thiosemicarbazones possessing either a p-methoxyphenyl or p-fluorophenyl group at the N4 position (Figure 5b). The performance of the single-factor atom-based QSAR model on the training and test set molecules for prediction of cytotoxicity against the MDR1 expressing KB-V1 line is illustrated in Figure 4b. The scatter plot indicates a high correlation coefficient (r2 = 0.91) although the cross-validated correlation coefficient is lower (q2 = 0.42). Experimental and predicted pIC50 values against KB-V1 cells for both the training and test set molecules are also shown in Figure 4b, showing that activity was effectively predicted.
Figure 5.

Refined pharmacophore predicting sensitivity of KB-V1 cells a). The pharmacophore for activity against KB-V1 cells with overlay of compounds that match three of seven pharmacophore features (AH1X). b). Pharmacophore for activity against KB-V1 cells with overlay of compounds that satisfy all pharmacophore features (AH1H2H3R1R2X). The compounds that meet these requirements are the isatin-β-thiosemicarbazones and compound 21.
Table 2 demonstrates the pharmacophore site match combinations for MDR1 selectivity and the compounds that satisfy each set of criteria. A minimal requirement of three sites (AH1X) satisfies all compounds listed. However, three sites alone cannot discriminate between compounds that do and do not demonstrate selectivity for KB-V1 cells, while including all seven sites misses some compounds with KB-V1 selective activity (>1 fold experimental selectivity). Incorporating all seven sites (AH1H2H3R1R2X) reveals only MDR1-selective compounds, but three (4, 7 and 24) are excluded. A combination of five or six features (AH1H2R2X and AH1H2R1R2X), both incorporating R2 and H2, correctly select all MDR1-selective compounds but 24, the false negative being due to the lack of a phenyl ring at N4. Using the predicted activity of compounds against KB-3-1 (parental) and KB-V1 (P-gp expression) cells, the predicted MDR1 selectivity can be determined (Table 2). The QSAR model correctly predicts the selectivity of 11 of 12 compounds listed, and makes no false positive predictions, though the magnitude of selectivity is not well predicted. The cytotoxicity and MDR1-selective pharmacophores and QSAR models will be employed to aid in pre-synthetic screening of drug design candidates. Based on the AH1H2H3R1R2X pharmacophore, isatin-β-thiosemicarbazones and other highly active thiosemicarbazones with aromatic substitution at the N4 position will be developed, validated, and the pharmacophore further optimized.
Table 2.
KB-V1 pharmacophore site matches for compounds examined, and the site feature combinations for which they satisfy selectivity for KB-V1 cells. Setting a minimal requirement for matching, such as AH1X satisfies all compounds listed, but cannot discriminate between compounds that do and do not demonstrate selectivity for KB-V1 cells, while setting all seven sites misses some compounds with KB-V1 selective activity. The AH1H2X and AH1H2R2X site combinations both predict for all KB-V1 selective compounds, though each with one false positive. The QSAR model correctly predicts the selectivity of 11 of 12 compounds listed, though it performs less well at predicting by how much a compound will select for KB-V1.
| Compound | Pharmacophore Site Matches | Fold Selectivity | >1 Fold Selectivity | ||||||
|---|---|---|---|---|---|---|---|---|---|
| AH1X | AH1H2X | AH1H2R2X | AH1H2R1R2X | AH1H2H3R1R2X | Experimental | Calculated | Experimental | Calculated | |
| Compound 1 | x | x | x | x | x | 7.6 | 7.4 | Yes | Yes |
| Compound 6 | x | x | x | x | x | 4.3 | 2.2 | Yes | Yes |
| Compound 18 | x | x | x | x | x | 3.6 | 2.6 | Yes | Yes |
| Compound 19 | x | x | x | x | x | 2.1 | 2.0 | Yes | Yes |
| Compound 21 | x | x | x | x | x | 2.3 | 3.1 | Yes | Yes |
| Compound 23 | x | x | x | x | x | 1.4 | 1.8 | Yes | Yes |
| Compound 4 | x | x | x | x | 7.1 | 5.4 | Yes | Yes | |
| Compound 7 | x | x | x | x | 1.4 | 2.6 | Yes | Yes | |
| Compound 3 | x | x | 1.3 | 0.9 | Yes | No | |||
| Compound 24 | x | x | 0.5 | 1.0 | No | No | |||
| Compound 9 | x | 0.9 | 0.4 | No | No | ||||
| Compound 10 | x | 0.2 | 0.2 | No | No | ||||
Given that the pharmacophore was developed based on an initial observation of structural commonality from a cohort of 60 compounds identified by a bioinformatics screen, the pharmacophore site features were queried against the remaining 53 compounds in the library (other than the isatin-β-thiosemicarbazones) to ascertain whether there was an underlying structural commonality to them. Querying the geometry-optimized structures of the bioinformatics hits for those that satisfy at least AH1X identified the seven isatin-β-thiosemicarbazones (Figure 1) from which the pharmacophore was partially derived, as well as the three remaining thiosemicarbazones in the set: NSC669341 (27, 6 site matches, AH1H2H3R2X), NSC695331 (28, 4 sites, AH1R2X) and NSC695333 (29, 6 site matches, AH1H2H3R2X). Each geometry optimized structure is overlaid with the MDR1-selective pharmacophore in Figure 6, and the structures show phenyl substitution at the N4 position (with an electron withdrawing substituent in each case), as well as hydrogen bond acceptors (A1) with a hydrophobic region (H1) flanking the thiosemicarbazone (X) functional group. One of the three thiosemicarbazones was available, and while it was not highly active (~20 μM) its cytotoxicity was effectively equipotent against both the multidrug resistant and parental lines (Figure 6a) as expected from the pharmacophore.
Figure 6.

The pharmacophore for activity against KB-V1 cells was tested against the 60 compounds previously identified in a bioinformatic analysis for compounds that were predicted to be selectively active against P-gp expressing cells. The activities of two available compounds, the thiosemicarbazone 27, and 30, are given. a). Querying the structural database for compounds that satisfy at least AH1X (X = thiosemicarbazone) identified the seven NSC isatin-β-thiosemicarbazones shown in Figure 1 from which the pharmacophore was partially derived, and the three remaining thiosemicarbazones in the set: 27, 28 and 29, shown over the site features demonstrating the good fit of structural features of these molecules with the pharmacophore. b). Querying the structural database for compounds that satisfy at least three out of seven sites, including AH1R2, but not the thiosemicarbazone feature (X), gave a further 13 non-thiosemicarbazone NSC compounds from our dataset that appear to satisfy the structural criteria for selectively killing KB-V1 cells. Two examples are shown; 30 and 31.
Querying the structural database for matches that are not thiosemicarbazones, by excluding X from the search criteria but requiring at least three other site matches, gave a further 13 non-thiosemicarbazone NSC compounds from our dataset that appear to satisfy the structural criteria for MDR1 selectivity. Two examples are shown in Figure 6b; NSC168468 (30, 2-hydroxy-3-(hydroxy(oxido)amino)benzaldehyde-2-quinolinylhydrazone, 5 site matches, AH1H2R1R2) and NSC621481 (31, 3-(2-chlorophenyl)-1-(1,4-dihydroxy-3-methyl-15,45-quinoxalin-2-yl)-2-propen-1-one, 5 site matches, AH1H2R1R2), and the remaining structures are given in the supplementary information. As with the thiosemicarbazones described above, aromatic regions connected by a linker are common to many of the structures, often with further hydrogen bonding potential in the linker. Validation of the available non-thiosemicarbazone 30 revealed an MDR1 selectivity ratio of 3.3, reinforcing the pharmacophore’s potential applicability outside thiosemicarbazones.
The additional features on the MDR1-selectivity pharmacophore and the exciting fact that non-thiosemicarbazones from our dataset of MDR1-selective compounds satisfy the pharmacophore developed here suggests that the selectivity is not mediated by the thiosemicarbazone functional group. While thiosemicarbazones are known for the metal-chelation mediated mechanisms of action, the isatins also have a diverse range of biological properties such as tyrosine kinase inhibition (TKI), particularly when conjugated as indolin-2-ones (as is the case here) 34.
It has not escaped our attention that 5-halogen substituted indolin-2-one moieties are reported to be efficacious TKIs and demonstrate greater cytotoxicity than their unhalogenated analogs35 (as is the case for the isatins36). For example, sunitinib (5-[5-fluoro-2-oxo-1,2-dihydroindol-(3Z)-ylidenemethyl]-2,4-dimethyl-1H-pyrrole-3-carboxylic acid (2-diethylaminoethyl)amide) is a rationally designed multi-targeted TKI approved for the treatment of gastrointestinal stromal cancers and advanced renal-cell carcinoma37, 38 that possesses a 5-fluoro-indolin-2-one moiety (like that of 18). Sunitinib’s TKI activity against VEGF-R2 and PDGF is an order of magnitude greater than that of the non-fluorinated analog (as are the 5-chloro and 5- bromo examples35) and its activity is mediated by the adenine mimic properties of the substituted isatin that binds to the kinase active site39. Meijer and co-workers have also examined a series of indirubin natural products (indirubins contain an indone-2-one moiety) and shown that derivatives with 5- and 6- halogenated indolin-2-one moieties demonstrate better kinase inhibitory activity (CDK1, CDK5, GSK-3) than their non-substituted counterparts40, 41, and co-crystallization studies showed that the indone-2-one group is binding in the active site41.
Given these precedents, the kinase inhibitory activity of 1 was examined against a representative panel of 50 kinases. While the positive control staurosporine demonstrated strong inhibition of most kinases in the panel, 1 showed no inhibitory activity at the highest dose concentration of 20 μM against the majority of kinases (Supplementary Table S1), and an IC50 could only be calculated for RSK1 (Ribosomal S6 kinase 1) with a relatively high value of 34.9 μM. The cytotoxicity of sunitinib itself was examined against the cell line pair employed here, and found not to display MDR1-selective activity (IC50 KB-3-1 = 2.3 μM, IC50 KB-V1 = 4.1 μM, RR = 0.6), reinforcing that kinase inhibitory activity alone is not adequate for conferring MDR1-selective cytotoxicity.
Unlike high-throughput screens that identify molecules with activity against a specific biological assay, bioinformatics screens based on correlation with gene expression identify candidates whose activity can be validated, but is not indicative of the mechanisms of action of hits, which can be diverse. While QSAR studies cannot reveal mechanism of action, they aid in understanding the structural features required for biological activity. In future studies, the pharmacophore identified in this report, along with the underlying understanding that isatin, conjugated as indolin-2-one to thiosemicarbazones, can result in compounds that selectively kill multidrug resistant (P-gp expressing) cells, will be used to methodically produce improved analogs of 1. Substitution at the 5 and 6 positions of isatin and around the phenyl ring should allow us to obtain maximal selectivity for P-gp expressing cells.
Additionally, incorporating an amine into the system would theoretically help to solubilize the drug as a hydrochloride salt; while no reliable pharmacophore for substrates of P-gp exists, it generally transports neutral and cationic organic molecules42. Thiosemicarbazones such as 9 and 10 that are P-gp substrates (discussed above) are not MDR1-selective, and 1 cannot be effluxed by P-gp and shows MDR1-selective activity. While the site of action of 1 has not yet been identified, this suggests that MDR1-selective compounds cannot be P-gp substrates, and that thiosemicarbazones such as 14 and 22 are inactive or non-selective because chemical substitution has sensitized them to detection and efflux by P-gp. This may mean that small substitutions on a MDR1-selective pharmacophore will reverse the selectivity, and a comprehensive library of compounds will allow us to probe this feature. Taken together, the information presented within this report should facilitate the design of 1 analogs that are both more selective and soluble, thereby accelerating their transition from bench-to-bed.
Conclusions
The models developed here, the first of their kind for predicting cytotoxicity and MDR1 selectivity, constitute valuable tools that will aid in the design of new MDR1-selective cytotoxins based on isatin-β-thiosemicarbazones. The necessity for isatin, and enhancement with halogenation to achieve greater P-gp-targeted selectivity, will serve as the foundation in the search for potential drug targets. Of great interest to those devoted to overcoming P-gp-mediated drug resistance, the pharmacophore we describe suggests that α-N-heterocyclic carboxaldehyde thiosemicarbazones such as 3-AP could be substantially improved. Here we have laid a foundation upon which to build more soluble and P-gp-selective analogs of 1. Evolution of this chemical family offers the promise of curing those cancers most resistant to chemotherapy.
Experimental Section
Materials and methods
Synthetic materials were sourced from Aldrich unless otherwise noted. Triapine (9, 3-AP, 3-aminopyridine-2-carboxaldehyde thiosemicarbazone) was generously provided by Vion Pharmaceuticals, CT. MAIQ (10, NSC246112, 2-[(5-amino-4-methyl-1-isoquinolinyl)methylene]) was provided by the Developmental Therapeutics Program (DTP), National Institutes of Health. Thiacetazone (11) was purchased from Sigma. Sunitinib was purchased from Toronto Chemicals, Toronto, Canada. Stock solutions of compounds for biological assays were prepared in DMSO and stored in frozen aliquots until use. The kinase inhibitory activity of 1 against a panel of 50 kinases was measured by Reaction Biology Corp, Malvern, PA.
Synthesis of thiosemicarbazones
The thiosemicarbazones were prepared by combining equimolar quantities of the isatins and thiosemicarbazides dissolved in large amounts of ethanol with addition of a few drops of acetic acid to initiate the reaction. On heating the mixture to boiling the thiosemicarbazone often crystallized; if it did not, water was added to encourage it. The best solvent for recrystallization of thiosemicarbazones was invariably DMSO with small amounts of water. With one exception (below), the (M+H)+ and (M−H)− ions lost the elements of the corresponding RNCS on MS2 although (M)− ions were also detected. In the case of isatin allylthiosemicarbazone (Compound 24), the (M+H)+ ion lost the elements of allyl thiourea. The (M−H)− ion lost allylisothiocyanate as expected. Low resolution mass spectra (LRMS) were collected on a Thermo LCQ Classic spectrometer (Madison, WI) and high resolution mass spectra (HRMS) with a Waters LCT Premier spectrometer (Milford, MA). NMR spectra were taken at 300 MHz in deuterated dimethylsulfoxide using a Varian 300 Gemini spectrometer (Palo Alto, CA). In each case, the purity of recrystallized compounds described below was confirmed to be ≥95% by the GC/MS gas chromatograph trace displaying only a single peak during analysis. Furthermore, in each case 1H NMR spectra revealed no impurities.
Compound 1
1-Isatin-4-(4′methoxyphenyl)thiosemicarbazone was prepared by reacting isatin with 4-(4′methoxyphenyl)-3-thiosemicarbazide. Yield 87%, light yellow needles, mp 220–248°C dec., 1H NMR (DMSO-d6) δ = 3.79 (3H, s), 6.98 (2H, dt), 6.98 (2H,dt), 7.45 (2H,dt), 6.95 (1H, d), 7.37 (1H,dt), 7.11 (1H, dt), 7.76 (1H, d), 12.7 (1H,s), 10.7 (1H,s), 11.25 (1H,s). LRMS m/z 327 (M+H)+, m/z 162 (MS2 of 327); m/z 326 (M)−, m/z 325 (M−H)− m/z 160 (MS2 of 325); HRMS m/z 327.0917 (M+H)+, calc. C16H15N4O2S 327.0916.
Comopund 3
1-(5′-Nitroisatin)-4-allyl-3-thiosemicarbazone was prepared by reacting 5-nitroisatin with 4-allyl-3-thiosemicarbazide. Yield 49%, light brown plates, mp 226–230°C dec. 1H NMR (DMSO-d6) δ = 4.28 (2H, t), 5.18 (1H, d), 5.19 (1H, d), 5.92 (1H, m), 7.12 (1H, d), 8.27 (1H, dd), 8.57 (1H, d), 9.78 (1H, t), 11.83 (1H, bs), 12.38 (1H, s). LRMS m/z 306 (M+H)+, m/z 270 (MS2 of 306), m/z 304 (M−H)−, m/z 205 (MS2 of 304); HRMS m/z 306.0649 (M+H)+, calc. C12H12N5O3S 306.0661.
Compound 4
1-(5′-Nitroisatin)-4-phenyl-3-thiosemicarbazone was prepared by reacting 5-nitroisatin with 4-phenyl-3-thiosemicarbazide. Yield 70%, light brown plates, mp 250–252°C dec. 1H NMR (DMSO-d6) δ = 7.31 (1H, t), 7.45 (2H, t), 7.59 (2H, d), 7.17 (1H, d), 8.28 (1H, d), 8.70, 1H, d), 11.1 (1H, s), 12.55 (1H, s), 11.86 (1H, br s); LRMS m/z 342 (M+H)+, m/z 207 (MS2 of 342, m/z 340 (M−H)−, m/z 205 (MS2 of 340); HRMS m/z 342.0663 (M+H)+, calc. C15H12N5O3S 342.0661.
1-(t-Butoxycarbonyl)-4-(4′-fluorophenyl)-3-thiosemicarbazide (Figure 2c) was prepared by reaction of t-butylcarbazate with 4-fluorophenylisothiocyanate. Yield 90%, colorless prisms, m.p. 158–160°C. HRMS m/z 286.1019 (M+H)+, calc. C12H17N3O2FS 286.1026. The t-Boc group was removed by acid hydrolysis, and the resulting 4-(4′fluorophenyl)-3-thiosemicarbazide was used for the synthesis of 6. Reaction of the simple hydrazine with 4-fluorophenylisothiocyanate yielded 1,2-Bis-(4′-fluorophenylthiocyanato)hydrazine (Figure 2b). Yield 89%, colorless prisms, mp 204–205°C. HRMS m/z 339.0557 (M+H)+, calc. C14H13N4F2S2 339.0550; m/z 337.0388 (M−H)−, calc. C14 H11N4F2S2 337.0393.
Compound 6
1-(5′-Bromoisatin)-4-(4′-fluorophenyl)-3-thiosemicarbazone was prepared by reacting 5-bromoisatin with 4-(4′-fluorophenyl)-3-thiosemicarbazide. Yield 97%, orange needles, mp 238–240°C dec. 1H NMR (DMSO-d6) δ = 6.91(1H, d), 7.53 (1H, dd), 7.97 (1H, d), 7.59 (2H, m), 7.28 (2H, t), 10.89 (1H, s), 11.36 (1H, s), 12.61 1H, s); HRMS m/z 392.9821 (M+H)+, calc. C15H11N4OFSBr 392.9821.
Compound 7
1-(5′-Bromoisatin)-4-(4′-nitrophenyl)-3-thiosemicarbazone was prepared by reacting 5-bromoisatin with 4-(4′-nitrophenyl)-3-thiosemicarbazide. Yield 83%, orange needles, mp 270–275°C dec., 1H NMR (DMSO-d6) δ = 6.92 (1H, d), 7.55 (1H, dd), 8.00 (1H, d), 8.07 (2H, d), 8.31 (2H, d), 11.14 (1H, s), 11.41 (1H, s), 12.82 (1H, s); HRMS m/z 417.9608 (M+H)+, calc. C15H9N5O3SBr 417.9609.
Compound 12
1-(2′-Indanone)-4-(4′-methoxyphenyl)-3-thiosemicarbazone was prepared by reacting 2-indanone with 4-(4′-methoxyphenyl)-3-thiosemicarbazide. Yield 76%, white needles, browning in air, mp 152–4°C. 1H NMR (DMSO-d6) δ = 3.75 (3H, s), 3.87 (4H, d), 6.82 (2H, d), 7.29 (2H, m), 7.32 (2H, m), 7.41 (2H, d), 9.81 (1H, s), 10.38 (1H, s); HRMS m/z 312.1179 (M+H)+, calc. C17H17N3OS 312.1171.
Compound 13
1-(1′-Indanone)-4-(4′-methoxyphenyl)-3-thiosemicarbazone was prepared by reacting 1-indanone with 4-(4′-methoxyphenyl)-3-thiosemicarbazide. Yield 96%, white needles, mp 185–6°C. 1H NMR (DMSO-d6) δ = 2.95 (2H, m), 3.08 (2H, m), 3.79 (3H, s), 6.92 (2H, d), 7.30 (1H, m), 7.38 (1H, d), 7.40 (1H, m), 7.41 (2H, d), 8.04 (1H, d), 9.99 (1H, s), 10.53 (1H, s); HRMS m/z 312.1179 (M+H)+, calc. C17H17N3OS 312.1171.
Compound 14
1-Isatin-3-thiosemicarbazone was prepared by reacting isatin with 3-thiosemicarbazide. Yield 92%, fine yellow needles, mp 210–240°C dec. 1H NMR (DMSO-d6) δ = 6.93 (1H,d), 7.09 (1H, t), 7.36 (1H, t), 7.65 (1H, d), 8.68 (1H, s), 9.03 (1H, s), 11.21 (1H, s), 12.47 (1H, s); LRMS m/z 221 (M+H)+, m/z 160 (MS2 of 221), m/z 219, (M−H)−, m/z 160 (MS2 of 219); HRMS m/z 219.0331 (M+H)+, calc. C9H7N4SO 219.0340.
Compound 15
1-Isatin-3-semicarbazone was prepared by reacting isatin with 3-semicarbazide. Yield 85%, light yellow needles, mp C dec., 1H NMR (DMSO-d6) δ = 6.89 (1H, d), 6.91 (1H, s), 7.03 (1H, t), 7.34 (1H, t), 8.06 (1H, d), 10.18 (1H, s), 10.73 (1H, s); HRMS m/z 205.0707 (M+H)+, calc. C9H9N4O2 205.0726.
Compound 16
4-(4′-Methoxyphenyl)-3-thiosemicarbazide was obtained from Trans World Chemical (Rockville, MD). mp 154–156°C. 1H NMR (DMSO-d6) δ 3.73 (3H, s), 4.76, (2H, br s), 6.86 (2H, d), 7.44 (2H, d), 8.72 (1H, br s), 9.50 (1H, br s).
Compound 17
1-(4′,7′-Dichloroisatin)-4-(4′-methoxyphenyl)-3-thiosemicarbazone was prepared by reacting 4,7-dichloroisatin with 4-(4′-methoxyphenyl)-3-thiosemicarbazide. Yield 89%, bright orange needles, mp 280°C dec., 1H NMR (DMSO-d6) δ = 7.03 (2H, d), 7.55 (2H, d), 7.50 (1H, s), 7.19 (1H, s), 10.1 (2H, br s), 12.0 (1H, s); HRMS m/z 395.0142 (M+H)+, calc. C16H13N4O2SCl2 395.0136.
Compound 18
1-(5′-Fluoroisatin)-4-(4′-methoxyphenyl)-3-thiosemicarbazone was prepared by reacting 5-fluoroisatin with 4-(4′-methoxyphenyl)-3-thiosemicarbazide. Yield 61%, orange needles, mp 238–240°C dec., 1H NMR (DMSO-d6) δ = 3.79 (3H, s), 6.94 (1H, dd), 6.99 (2H, d), 7.21 (H, dt), 7.46 (2H, d), 7.62 (1H, dd), 10.78 (1H, s), 11.25 (1H, s), 12.63 (1H, s); LRMS m/z 345 (M+H)+, m/z 180 (MS2 of 345), m/z 343, (M−H)−, m/z 178, MS2 of 343; HRMS m/z 345.0830 (M+H)+, calc. C16H14N4O2FS 345.0822.
Compound 19
1-(N-methylisatin)-4-(4′-methoxyphenyl)-3-thiosemicarbazone was prepared by reacting N-methylisatin with 4-(4′-methoxyphenyl)-3-thiosemicarbazide. Yield 87%, short yellow prisms, mp 216–17°C, 1H NMR (DMSO-d6) δ = 3.78 (3H, s), 3.24, (3H, s), 6.98 (2H, d), 7.46 (2H, d), 7.16 (1H, d), 7.46 (1H, t), 7.17 (1H, t), 7.80 (1H, t), 10.75 (1h, s), 12.68 (1H, s); LRMS m/z 341 (M+H)+, m/z 176 (MS2 of 341), m/z 339 (M−H)−, m/z 174 (MS2 of 339). HRMS m/z 341.1078 (M+H)+, calc. C17H17N4O2S 341.1072.
Compound 20
1-(Isatin 5′-sulfonic acid)-4-(4′-methoxyphenyl)-3-thiosemicarbazone was prepared by reacting isatin-5-sulfonic acid with 4-(4′-methoxyphenyl)-3-thiosemicarbazide. Yield 98%, dark yellow needles, mp > 300°C. 1H NMR (DMSO-d6) δ = 3.79 (3H, s), 6.97 (2H, dd), 7.44 (2H, dd), 6.86 (1H, d), 7.62 (1H, dd), 8.13 (1H, br s), 10.95 (1H, s), 11.28 (1H, s), 12.70 (1H, s); LRMS no + or − ions. HRMS m/z 407.0469 (M+H)+, calc. C16H15N4O5S2 407.0469.
Compound 21
1-(2′-Pyridinecarboxaldehyde)-4-(4′-methoxyphenyl)-3-thiosemicarbazone was prepared by reacting 2-pyridinecarboxaldehyde with 4-(4′-methoxyphenyl)-3-thiosemicarbazide. Yield 66%, mp 173–4°C, 1H NMR (DMSO-d6) δ = 3.76, (3H, s), 6.95 (2H, d), 7.39 (3H, d,m), 7.84 (1H, t), 8.18 (1H, s), 8.43 (1H,d), 8.58 (1H, d), 10.17 (1H, S), 11.95 (1H, s); HRMS m/z 287.0968 (M+H)+, calc. C14H15N4OS 287.0967.
Compound 22
1-(1H′-Benz[g]indole-2′,3′-dione)-4(4′-methoxyphenyl)-3-semicarbazone was prepared by reacting 1H-Benz[g]indole-2,3-dione with 4-(4′-methoxyphenyl)-3-thiosemicarbazide. Yield 66%, dark red prisms, mp 240–268°C dec., 1H NMR (DMSO-d6) δ = 3.79, (3H, s), 6.99 (2H, d), 7.48 (2H, m), 7.60 (2H, d), 7.66 (1H, d). 7.88 (1H, d), 7.96 (1H, dd), 8.12 (1H, dd) 10.78 (1H, s), 11.97 (1H, s), 12.78 (1H, s); LRMS m/z 377 (M+H)+, m/z 212 (MS2 of 377), m/z 375 (M−H)−, m/z 210 (MS2 of 375); HRMS m/z 377.1073 (M+H)+, calc. C20H17N4O2S 377.1072.
Compound 23
1-(5′-Nitroisatin)-4-(4′-methoxyphenyl)-3-thiosemicarbazone was prepared by reacting 5-nitroisatin with 4-(4′-methoxyphenyl)-3-thiosemicarbazide. Yield 63%, light yellow needles, mp 220–248°C dec., 1H NMR (DMSO-d6) δ = 3.79 (3H, s), 7.00 (2H, d), 7.14 (1H, d), 7.44 (2H, d), 8.28 (1H, dd), 8.69 (1H, d), 11.01 (1H, s), 11.86 (1H, s), 12.51 (1H, s); LRMS m/z 372 (M+H)+, m/s 207 (MS2 of 372), m/s 370 (M−H)−, m/s 205 (MS2 of 370). HRMS m/z 372.0764 (M+H)+, calc. C16H14N5O4S 372.0767.
Compound 24
1-Isatin-4-allyl-3-thiosemicarbazone was prepared by reacting isatin with 4-allyl-3-thiosemicarbazide. Yield 90%, light yellow needles, mp 210°C dec. 1H NMR (DMSO-d6) δ = 4.25 (2H, t), 5.92 (1H, t), 5.18 (1H, m), 5.19 (1H, d), 6.93 (1H,dd), 7.35 (1H, dt), 7.09 (1H, dt), 7.67 (1H, dd), 9.45 (1H, t), 11.21 (1H, s), 12.60 (1H, s); LRMS no (M+H)+ ion, m/z 145 (loss of NHCSNHCH2CH(CH3)), m/z 259 (M−H)−, m/z 160 (MS2 of 259). HRMS m/z 261.0808 (M+H)+, calc. C12H13N4OS 261.0810.
Compound 25
1-Isatin-4-(4′-methoxyphenyl)-3-semicarbazone was prepared by reacting isatin with 4-(4′-methoxyphenyl)-3-semicarbazide. Yield 35%, yellow needles, mp 182–6°C, 1H NMR (DMSO-d6) δ = 3.74 (3H, s), 6.92 (2h, d), 7.49 (2H, d), 6.92 (1H, d), 7.39 (1H, m), 7.06 (1H, m), 8.09 (1H, d), 10.8 (1H, s); LRMS m/z 311 (M+H)+, m/z 162 (MS2 of 311), m/z 309 (M−H)−, m/z 160 (MS2 of 309); HRMS m/z 311.1138 (M+H)+, calc. C16H15N4O3 311.1144.
Compound 26
1-(4′-Acetylaminobenzaldehyde)-4-(4′-methoxyphenyl)-3-thiosemicarbazone was prepared by reacting 4-acetylaminobenzaldehyde with 4-(4′-methoxyphenyl)-3-thiosemicarbazide. Yield 98%, light yellow needles, mp 208–9°C, 1H NMR (DMSO-d6) δ = 2.05 (3H, s), 3.77 (3H, s), 6.92 (2H, d), 7.39 (2H, d), 7.63 (2H, d), 7.82 (2H, d), 9.93 (1H, s), 10.11 (1H, s), 11.68 (1H, s); HRMS m/z 343.1230 (M+H)+, calc. C17H19N4O2S 343.1229.
Cell lines and MTT cytotoxicity assay
KB 3-1 cells (a HeLa deriviative) and its MDR derivative (KB-V1) were grown as previously described5. Cell survival was measured by the MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay as previously described5. Briefly, cells were seeded in 100 μL of growth medium at a density of 5000 cells/well in 96-well plates and allowed to establish for 24 h, at which time serially diluted drugs were added in an additional 100 μL growth medium. Cells were then incubated for 72 h at 37 °C in humidified 5% CO2, at which time the growth media was drawn, and replaced with MTT in IMDM growth media and incubated for 4 h. The MTT solution was then drawn from the wells, and 100 μL acidified ethanol solution was added to each well and after 15 minutes absorption at 560 nm was measured. IC50 cytotoxicity values were determined as the drug concentration that reduced the absorbance to 50% of that in untreated control wells, and derived from at least three separate experiments.
Pharmacophore perception and 3D-QSAR
Table 1 shows the compounds tested for the generation of two three-dimensional (3-D) pharmacophore models in order to rationalize chemical structure with the observed cytotoxicity and selectivity of the isatin-β-thiosemicarbazones. In the development of a pharmacophore for cytotoxicity against the parental KB-3-1 cell line, compounds were divided into an active class (13 compounds) and an inactive class (8 compounds) based on an arbitrary activity threshold of 50 μM. The pharmacophore relating structure to MDR1-selective cytotoxicity against the P-gp expressing KB-V1 cell line was similarly created with the above thresholds, though 22 was removed from the active class and placed into the inactive class given that it was inactive (IC50 > 50 mM) against KB-V1 cells. It is worth stating that the inactivity of a molecule may be due to a range of reasons including a large desolvation penalty, excessive entropy loss upon receptor binding, and poor membrane permeability or active efflux43. As a result some molecules will fail to show any activity while containing all the necessary features in identical spatial arrangements (sites) as the active compounds. Consequently, their inclusion in pharmacophore creation may unfairly jeopardize the selection of the correct features and sites. Thus, inactive compounds 14 and 20 were excluded in this pharmacophore study. Other compounds were excluded as biological data could not be determined for reasons of purity and solubility as described below: 2, 5 and 17. Other structurally diverse compounds that were inactive were included in pharmacophore creation as the members of the inactive class of drugs.
A maximum of 100 conformers were generated for each of the 21 molecules, sampling the conformational space that may represent possible active structures. Conformers not within 5 kJ/mol of the global minimum were automatically discarded. Visual inspection and careful analysis of chemical features were conducted to guide the pharmacophore development process. Particular attention was paid to compounds 10 and 18, as they represent the optimal reference structures for KB-3-1 cytoxicity and selective MDR1-selective cytoxicity, respectively, based on their activity (Table 1). Individual features that compose the thiosemicarbazone functional group were excluded and replaced with a single, custom group-feature (X) representing this functional group. Pharmacophore variants considered for cytotoxicity were composed of four sites with the following chemical features – hydrogen-bond acceptor (A, pink sphere), aromatic ring (R, ring), hydrophobic (H, green sphere) and thiosemicarbazone group (X, blue sphere). Model variant AHRX was pre-selected as it was visually apparent that all active compounds possessed these features.
In the search for a pharmacophore predictive of cytotoxicity against KB-3-1 cells, common pharmacophores exhibiting AHRX were searched in at least 3 of the 13 active structures with a final box size and minimum intersite distance of 1.0 Å. Several hypotheses were generated of which the best that mapped onto 10, based on the PHASE scoring functions 43, 44, was chosen as the final KB-3-1 pharmacophore for use in subsequent screening and QSAR investigations. Similarly, pharmacophore variants considered for MDR1 selectivity consisted of the same above features; however, the number of sites increased to seven—covering the additional phenyl ring and methoxy group as H2/R2 and H3 features, respectively - to place greater weight on mapping the isatin-β-thiosemcarbazones, specifically that of 18 for the reason outlined previously. Common pharmacophore features from at least 3 of the 12 active compounds were searched, revealing several hypotheses of variant AH1H2H3R1R2X. The hypothesis that best mapped onto 18 was chosen. A single exclusion-volume of radius 1.5 Å was added to prohibit 22 from matching the final MDR1 selectivity pharmacophore.
Quality of the Pharmacophoric Hypotheses
The quality of each pharmacophore is first measured in three ways based on the alignments to the input structures: (1) the alignment score, which is the root-mean-squared deviation (RMSD) in the site-point positions; (2) the vector score, which is the average cosine of the angles formed by corresponding pairs of vector features (acceptors, donors, and aromatic rings) in the aligned structures; and (3) a volume score based on the overlap of van der Waals models of the non-hydrogen atoms in each pair of structures. For details see Dixon et al43. We also assigned higher scores to hypotheses that match a greater number of the chosen actives.
Pharmacophore Screening – Finding Matches to Hypothesis
Successful alignments of the compounds in Table 1 to either of the two pharmacophores, based on KB-3-1 cytotoxicity or MDR1 selectivity, require compounds to match a minimum of 3 sites corresponding to AH1X. Alternatively, a 3 site minimum requirement of AH1R2 for matching non-thiosemicarbazones as potential MDR1-selective compounds can also be used (as discussed). This substitution of X for R2 is warranted in light of the fact that all compounds selective for MDR1 identified in this study possess both sites; moreover, replacing AH1X with AH1R2 successfully matches the MDR1-selective thiosemicarbazones. Further specifications for the search criteria implemented here include distance matching tolerances of 2.0 Å and feature matching tolerances for hydrogen bond acceptors of 2.0 Å, with hydrophobic, aromatic ring and thiosemicarbazone tolerances each set to 1.5 Å.
QSAR Modeling
Attempts to statistically analyze three-dimensional molecular fields by means of a QSAR analysis were made, correlating chemical structure with either the KB-3-1 cytotoxicity or MDR1 selectivity data. The QSAR models for KB-3-1 cytotoxicity and MDR1 selectivity were developed using molecules capable of matching either of the two pharmacophores. Molecules were placed into training and test sets once aligned onto the pharmacophore with an effort to minimize structural redundancy while maximizing coverage of the experimental activities. Figure 5 summarizes the compounds investigated. Atom based QSAR models were built for the two pharmacophores using a grid spacing of 0.5 Å and one partial least-square (PLS) factor.
Computational Details
All molecules investigated in this study were built with the Maestro 7.5 graphical interface 45. Each structure was subsequently minimized using the OPLS_200546 with a constant dielectric of 1.0. Rapid torsional sampling was achieved using MacroModel 9.1 45 using the OPLS_2005 force field with minimum atom deviation of 0.5 Å and a distant-dependent dielectric solvation treatment. The maximum relative energy difference was set to 10.0 kcal/mol. Pharmacophore development and subsequent searches, site point superpositions, vector characteristics alignment, molecular volume overlap, penalty scoring and QSAR model building were carried out using Phase 2.043, 44. All calculations were performed in Maestro 7.5 on an AMD64+ workstation using the SUSE10.1 Linux operating system.
Supplementary Material
Acknowledgments
DEH acknowledges Australian Research Council for funding. NKS acknowledges Faculty of Pharmacy, University of Sydney for award of a scholarship. JLH acknowledges the Howard Hughes Medical Institute for award of a HHMI Fellowship. GS is an EMBO SDIG fellow and a Special Fellow o the Leukemia and Lymphoma Society. The authors wish to thank Mr. Edward Sokoloski and Mr. Noel Whittaker for assistance in obtaining NMR and mass spectra. This research was supported by the Intramural Research Program of the National Institutes of Health, National Cancer Institute.
Footnotes
Abbreviations: APCI-MS: Atmospheric Pressure Chemical Ionization Mass Spectrometry, ABC: 3-AP: 3-aminopyridine-2-carboxaldehyde thiosemicarbazone, A: Hydrogen bond acceptor, ATP-Binding Cassette protein, DMSO: dimethylsulfoxide, ESI-MS: Electrospray Ionization Mass Spectrometry, H: Hydrophobic domain, MAIQ: 4-methyl-5-amino-1-formylisoquinoline thiosemicarbazone, MDR: Multidrug resistance, NCI: National Cancer Institute, NSC: National Service Center, R: Phenyl ring, RR: ribonucleotide reductase, TKI: tyrosine kinase inhibitor, X: Thiosemicarbazone.
Supporting Information Available: Kinome inhibition data and addition structures are included as described in the text above. This material is available free of charge via the Internet at http://pubs.acs.org.
Supporting Information Available: Structures of compounds that matched pharmacophore screen, and results of kinase inhibition screen of 1 included. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Szakacs G, Paterson JK, Ludwig JA, Booth-Genthe C, Gottesman MM. Targeting multidrug resistance in cancer. Nat Rev Drug Discov. 2006;5:219–234. doi: 10.1038/nrd1984. [DOI] [PubMed] [Google Scholar]
- 2.Gottesman MM, Fojo T, Bates SE. Multidrug resistance in cancer: role of ATP-dependent transporters. Nat Rev Cancer. 2002;2:48–58. doi: 10.1038/nrc706. [DOI] [PubMed] [Google Scholar]
- 3.Fox E, Bates SE. Tariquidar (XR9576): a P-glycoprotein drug efflux pump inhibitor. Expert Rev Anticancer Ther. 2007;7:447–459. doi: 10.1586/14737140.7.4.447. [DOI] [PubMed] [Google Scholar]
- 4.Leonard GD, Polgar O, Bates SE. ABC transporters and inhibitors: new targets, new agents. Curr Opin Investig Drugs. 2002;3:1652–1659. [PubMed] [Google Scholar]
- 5.Ludwig JA, Szakacs G, Martin SE, Chu BF, Cardarelli C, Sauna ZE, Caplen NJ, Fales HM, Ambudkar SV, Weinstein JN, Gottesman MM. Selective toxicity of NSC73306 in MDR1-positive cells as a new strategy to circumvent multidrug resistance in cancer. Cancer Res. 2006;66:4808–4815. doi: 10.1158/0008-5472.CAN-05-3322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Szakacs G, Annereau JP, Lababidi S, Shankavaram U, Arciello A, Bussey KJ, Reinhold W, Guo Y, Kruh GD, Reimers M, Weinstein JN, Gottesman MM. Predicting drug sensitivity and resistance: profiling ABC transporter genes in cancer cells. Cancer Cell. 2004;6:129–137. doi: 10.1016/j.ccr.2004.06.026. [DOI] [PubMed] [Google Scholar]
- 7.Turk D, Hall MD, Chu BF, Ludwig JA, Fales HM, Gottesman MM, Szakacs G. Identification of MDR1-inverse agents that selectively kill multidrug resistant cancer cells. 2009 doi: 10.1158/0008-5472.CAN-09-2422. submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.National Cancer Institute. Developmental Therapeutics Program. http://dtp.nci.nih.gov.
- 9.Brockman RW, Thomson JR, Bell MJ, Skipper HE. Observations on the antileukemic activity of pyridine-2-carboxaldehyde thiosemicarbazone and thiocarbohydrazone. Cancer Res. 1956;16:167–170. [PubMed] [Google Scholar]
- 10.Levinson W, Faras A, Woodson B, Jackson J, Bishop JM. Inhibition of RNA-dependent DNA polymerase of Rous sarcoma virus by thiosemicarbazones and several cations. Proc Natl Acad Sci U S A. 1973;70:164–168. doi: 10.1073/pnas.70.1.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bauer DJ. A history of the discovery and clinical application of antiviral drugs. Br Med Bull. 1985;41:309–314. doi: 10.1093/oxfordjournals.bmb.a072069. [DOI] [PubMed] [Google Scholar]
- 12.Beraldo H, Gambino D. The wide pharmacological versatility of semicarbazones, thiosemicarba-zones and their metal complexes. Mini Rev Med Chem. 2004;4:31–39. doi: 10.2174/1389557043487484. [DOI] [PubMed] [Google Scholar]
- 13.Giles FJ, Fracasso PM, Kantarjian HM, Cortes JE, Brown RA, Verstovsek S, Alvarado Y, Thomas DA, Faderl S, Garcia-Manero G, Wright LP, Samson T, Cahill A, Lambert P, Plunkett W, Sznol M, DiPersio JF, Gandhi V. Phase I and pharmacodynamic study of Triapine, a novel ribonucleotide reductase inhibitor, in patients with advanced leukemia. Leuk Res. 2003;27:1077–1083. doi: 10.1016/s0145-2126(03)00118-8. [DOI] [PubMed] [Google Scholar]
- 14.Yen Y, Margolin K, Doroshow J, Fishman M, Johnson B, Clairmont C, Sullivan D, Sznol M. A phase I trial of 3-aminopyridine-2-carboxaldehyde thiosemicarbazone in combination with gemcitabine for patients with advanced cancer. Cancer Chemother Pharmacol. 2004;54:331–342. doi: 10.1007/s00280-004-0821-2. [DOI] [PubMed] [Google Scholar]
- 15.Antholine W, Knight J, Whelan H, Petering DH. Studies of the reaction of 2-formylpyridine thiosemicarbazone and its iron and copper complexes with biological systems. Mol Pharmacol. 1977;13:89–98. [PubMed] [Google Scholar]
- 16.Finch RA, Liu MC, Cory AH, Cory JG, Sartorelli AC. Triapine (3-aminopyridine-2-carboxaldehyde thiosemicarbazone; 3-AP): an inhibitor of ribonucleotide reductase with antineoplastic activity. Adv Enzyme Regul. 1999;39:3–12. doi: 10.1016/s0065-2571(98)00017-x. [DOI] [PubMed] [Google Scholar]
- 17.Dean RT, Nicholson P. The action of nine chelators on iron-dependent radical damage. Free Radic Res. 1994;20:83–101. doi: 10.3109/10715769409147506. [DOI] [PubMed] [Google Scholar]
- 18.Kalinowski DS, Richardson DR. The evolution of iron chelators for the treatment of iron overload disease and cancer. Pharmacol Rev. 2005;57:547–583. doi: 10.1124/pr.57.4.2. [DOI] [PubMed] [Google Scholar]
- 19.Pillai CK, Nandi US, Levinson W. Interaction of DNA with anti-cancer drugs: copper-thiosemicarbazide system. Bioinorg Chem. 1977;7:151–157. doi: 10.1016/s0006-3061(00)80065-4. [DOI] [PubMed] [Google Scholar]
- 20.Agrawal KC, Sartorelli AC. The chemistry and biological activity of alpha -(N)-heterocyclic carboxaldehyde thiosemicarbazones. Prog Med Chem. 1978;15:321–356. doi: 10.1016/s0079-6468(08)70259-5. [DOI] [PubMed] [Google Scholar]
- 21.da Silva JFM, Garden SJ, Pinto AC. The Chemistry of Isatins: a Review from 1975 to 1999. J Braz Chem Soc. 2001;12:273–324. [Google Scholar]
- 22.Popp FD. The Chemistry of Isatin. Adv Hetercyc Chem. 1975;18:1–59. [Google Scholar]
- 23.Karali N. Synthesis and primary cytotoxicity evaluation of new 5-nitroindole-2,3-dione derivatives. Eur J Med Chem. 2002;37:909–918. doi: 10.1016/s0223-5234(02)01416-2. [DOI] [PubMed] [Google Scholar]
- 24.Karali N, Terzioglu N, Gursoy A. Synthesis and primary cytotoxicity evaluation of new 5-bromo-3-substituted-hydrazono-1H-2-indolinones. Arch Pharm (Weinheim) 2002;335:374–380. doi: 10.1002/1521-4184(200211)335:8<374::AID-ARDP374>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 25.Terzioglu N, Karali N, Gursoy A, Pannecouque C, Leysen P, Paeshuyse J, Neyts J, De Clercq E. Synthesis and primary antiviral activity evaluation of 3-hydrazono-5-nitro-2-indolinone derivatives. ARKIVOC. 2006;1 [Google Scholar]
- 26.Klayman DL, Bartosevich JF, Griffin TS, Mason CJ, Scovill JP. 2-Acetylpyridine thiosemicarbazones. 1. A new class of potential antimalarial agents. J Med Chem. 1979;22:855–862. doi: 10.1021/jm00193a020. [DOI] [PubMed] [Google Scholar]
- 27.Carpino LA. Oxidative Reactions of Hydrazines. IV. Elimination of Nitrogen from 1,1-Disubstituted-2-arenesulfonhydrazides1–4. Journal of the American Chemical Society. 1957;79:4427–4431. [Google Scholar]
- 28.Shen DW, Cardarelli C, Hwang J, Cornwell M, Richert N, Ishii S, Pastan I, Gottesman MM. Multiple drug-resistant human KB carcinoma cells independently selected for high-level resistance to colchicine, adriamycin, or vinblastine show changes in expression of specific proteins. J Biol Chem. 1986;261:7762–7770. [PubMed] [Google Scholar]
- 29.Gottesman MM. Mechanisms of cancer drug resistance. Annu Rev Med. 2002;53:615–627. doi: 10.1146/annurev.med.53.082901.103929. [DOI] [PubMed] [Google Scholar]
- 30.Liu MC, Lin TS, Sartorelli AC. Chemical and biological properties of cytotoxic alpha-(N)-heterocyclic carboxaldehyde thiosemicarbazones. Prog Med Chem. 1995;32:1–35. doi: 10.1016/s0079-6468(08)70451-x. [DOI] [PubMed] [Google Scholar]
- 31.Cory JG, Cory AH, Lorico A, Rappa G, Sartorelli AC. Altered efflux properties of mouse leukemia L1210 cells resistant to 4-methyl-5-amino-1-formylisoquinoline thiosemicarbazone. Anticancer Res. 1997;17:3185–3193. [PubMed] [Google Scholar]
- 32.Rappa G, Lorico A, Liu MC, Kruh GD, Cory AH, Cory JG, Sartorelli AC. Overexpression of the multidrug resistance genes mdr1, mdr3, and mrp in L1210 leukemia cells resistant to inhibitors of ribonucleotide reductase. Biochem Pharmacol. 1997;54:649–655. doi: 10.1016/s0006-2952(97)00210-4. [DOI] [PubMed] [Google Scholar]
- 33.Whitnall M, Howard J, Ponka P, Richardson DR. A class of iron chelators with a wide spectrum of potent antitumor activity that overcomes resistance to chemotherapeutics. Proc Natl Acad Sci U S A. 2006;103:14901–14906. doi: 10.1073/pnas.0604979103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kingston DG, Newman DJ. The search for novel drug leads for predominately antitumor therapies by utilizing mother nature’s pharmacophoric libraries. Curr Opin Drug Discov Devel. 2005;8:207–227. [PubMed] [Google Scholar]
- 35.Sun L, Liang C, Shirazian S, Zhou Y, Miller T, Cui J, Fukuda JY, Chu JY, Nematalla A, Wang X, Chen H, Sistla A, Luu TC, Tang F, Wei J, Tang C. Discovery of 5-[5-fluoro-2-oxo-1,2- dihydroindol-(3Z)-ylidenemethyl]-2,4- dimethyl-1H-pyrrole-3-carboxylic acid (2-diethylaminoethyl)amide, a novel tyrosine kinase inhibitor targeting vascular endothelial and platelet-derived growth factor receptor tyrosine kinase. J Med Chem. 2003;46:1116–1119. doi: 10.1021/jm0204183. [DOI] [PubMed] [Google Scholar]
- 36.Vine KL, Locke JM, Ranson M, Benkendorff K, Pyne SG, Bremner JB. In vitro cytotoxicity evaluation of some substituted isatin derivatives. Bioorg Med Chem. 2007;15:931–938. doi: 10.1016/j.bmc.2006.10.035. [DOI] [PubMed] [Google Scholar]
- 37.Atkins M, Jones CA, Kirkpatrick P. Sunitinib maleate. Nat Rev Drug Discov. 2006;5:279–280. doi: 10.1038/nrd2012. [DOI] [PubMed] [Google Scholar]
- 38.Chow LQ, Eckhardt SG. Sunitinib: from rational design to clinical efficacy. J Clin Oncol. 2007;25:884–896. doi: 10.1200/JCO.2006.06.3602. [DOI] [PubMed] [Google Scholar]
- 39.Laird AD, Vajkoczy P, Shawver LK, Thurnher A, Liang C, Mohammadi M, Schlessinger J, Ullrich A, Hubbard SR, Blake RA, Fong TA, Strawn LM, Sun L, Tang C, Hawtin R, Tang F, Shenoy N, Hirth KP, McMahon G, Cherrington SU6668 is a potent antiangiogenic and antitumor agent that induces regression of established tumors. Cancer Res. 2000;60:4152–4160. [PubMed] [Google Scholar]
- 40.Beauchard A, Ferandin Y, Frere S, Lozach O, Blairvacq M, Meijer L, Thiery V, Besson T. Synthesis of novel 5-substituted indirubins as protein kinases inhibitors. Bioorg Med Chem. 2006;14:6434–6443. doi: 10.1016/j.bmc.2006.05.036. [DOI] [PubMed] [Google Scholar]
- 41.Meijer L, Skaltsounis AL, Magiatis P, Polychronopoulos P, Knockaert M, Leost M, Ryan XP, Vonica CA, Brivanlou A, Dajani R, Crovace C, Tarricone C, Musacchio A, Roe SM, Pearl L, Greengard P. GSK-3-selective inhibitors derived from Tyrian purple indirubins. Chem Biol. 2003;10:1255–1266. doi: 10.1016/j.chembiol.2003.11.010. [DOI] [PubMed] [Google Scholar]
- 42.Chang C, Ekins S, Bahadduri P, Swaan PW. Pharmacophore-based discovery of ligands for drug transporters. Adv Drug Deliv Rev. 2006;58:1431–1450. doi: 10.1016/j.addr.2006.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dixon SL, Smondyrev AM, Knoll EH, Rao SN, Shaw DE, Friesner RA. PHASE: a new engine for pharmacophore perception, 3D QSAR model development, and 3D database screening: 1. Methodology and preliminary results. Journal of Computer-Aided Molecular Design. 2006;20:647–671. doi: 10.1007/s10822-006-9087-6. [DOI] [PubMed] [Google Scholar]
- 44.Schrodinger L. Phase, version 2.0 User Manual. New York, NY: 2005. [Google Scholar]
- 45.Schrodinger L. MacroModel, version 9.1. New York, NY: 2005. [Google Scholar]
- 46.Schrodinger L. MacroModel, version 9.1 User Manual. Schrodinger, LLC; New York, NY: 2005. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.