

Abstract
Experimental data accumulated over the past decade show the emerging importance of the late sodium current (I NaL) for the function of both normal and, especially, failing myocardium, in which I NaL is reportedly increased. While recent molecular studies identified the cardiac Na+ channel (NaCh) α subunit isoform (Nav1.5) as a major contributor to I NaL, the molecular mechanisms underlying alterations of I NaL in heart failure (HF) are still unknown. Here we tested the hypothesis that I NaL is modulated by the NaCh auxiliary β subunits. tsA201 cells were transfected simultaneously with human Nav1.5 (former hH1a) and cardiac β1 or β2 subunits, and whole-cell patch-clamp experiments were performed. We found that I NaL decay kinetics were significantly slower in cells expressing α + β1 (time constant τ = 0.73 ± 0.16 s, n = 14, mean ± SEM, P < 0.05) but remained unchanged in cells expressing α + β2 (τ = 0.52 ± 0.09 s, n = 5), compared with cells expressing Nav1.5 alone (τ = 0.54 ± 0.09 s, n = 20). Also, β1, but not β2, dramatically increased I NaL relative to the maximum peak current, I NaT (2.3 ± 0.48%, n = 14 vs. 0.48 ± 0.07%, n = 6, P < 0.05, respectively) and produced a rightward shift of the steady-state availability curve. We conclude that the auxiliary β1 subunit modulates I NaL, produced by the human cardiac Na+ channel Nav1.5 by slowing its decay and increasing I NaL amplitude relative to I NaT. Because expression of Nav1.5 reportedly decreases but β1 remains unchanged in chronic HF, the relatively higher expression of β1 may contribute to the known I NaL increase in HF via the modulation mechanism found in this study.
Electronic supplementary material
The online version of this article (doi:10.1007/s12576-009-0029-7) contains supplementary material, which is available to authorized users.
Keywords: Whole-cell sodium current, Heterologous expression, Human sodium channel subunits
Introduction
Experimental data accumulated over the past decade show the emerging importance of the late sodium current (I NaL) for the function of both normal and, especially, failing myocardium, in which I NaL is reportedly increased [1–3] The importance of the contribution of I NaL to heart failure (HF) mechanisms has been demonstrated in experiments in which “correction” of I NaL in failing cardiomyocytes resulted in:
rescue of normal repolarization;
reduced beat-to-beat action potential duration (APD) variability; and
Accordingly I NaL has recently emerged as a novel possible target for cardioprotection to treat the failing heart [6, 7].
Voltage-clamp studies have identified several types of single Na+ channel activity and whole-cell Na+ currents that could contribute to APD in cardiomyocytes. The variety of Na+ channel activities identified so far has been classified (for review see Ref. [6]) in terms of the late (or persistent) Na+ current i.e. I NaL (or I pNa), and background Na+ currents. In contrast with I NaL, background Na+ currents have been poorly characterized and have no clear molecular identity.
Major biophysical and pharmacological characteristics of the whole-cell I NaL have been studied in great detail in human cardiomyocytes by our research group [3, 8, 9] and can be summarized as follows:
potential-independent slow inactivation and re-activation (~0.5 s);
steady-state activation and inactivation similar to that for I NaT; and
low sensitivity to the specific toxins TTX and STX similar to the cardiac Na+ channel isoform Nav1.5.
A slowly inactivating I NaL with aforementioned biophysical characteristics has been identified in ventricular cardiomyocytes and cardiac Purkinje cells of dogs [1, 3, 5, 10–12], guinea pigs [13–15], rabbits [16], rats [17] and mice [18]. I NaL is also produced by the heterologously expressed cardiac Na+ channel isoform main α-subunit Nav1.5 [7, 19].
Despite explosive interest in this new component of the Na+ current (for recent reviews see Refs. [6, 7, 14, 20]) the mechanisms of I NaL regulation in normal heart and its alterations in HF are not yet understood and are likely to need further collective efforts based on different approaches including detailed biophysical and molecular biology examinations in addition to traditional pharmacological studies. Utilizing antisense inhibition and siRNA technologies our most recent studies explored the molecular identity of I NaL in ventricular cardiomyocytes [7, 21]. These studies suggested the cardiac Na+ channel α-subunit isoform (Nav1.5) was a major contributor to I NaL.
Although most recent studies have shown that I NaL is strongly and differently modulated by intracellular Ca2+ in the cardiomyocytes of normal and failing hearts [18, 22], Na+ channels operate not in isolation but within macromolecular complexes [23, 24], which are critical attributes of Na+ channel function (in addition to membrane voltage and ion concentrations). The macromolecular complexes include auxiliary β-subunits, phospholipids and elements of the cytoskeleton, each of which can modulate Na+ channel function including I NaL (for review see Ref. [7]). The β-subunit gene family has four members—β1 (SCN1B), β2 (SCN2B), β3 (SCN3B), and β4 (SCN4B) (for review see Ref. [24]). Despite high homology between β1 and β3, and β2 and β4 the different functional role of these newly discovered isoforms (β3 and β4) could not be ruled out. In addition there is a splice variant β1A of SCN1B that is expressed in embryonic brain and adult heart in rat [25]. All five β-subunits are expressed in rodent heart and are differently localized to specific sub-cellular domains and cell types. The β1 subunit is non-covalently attached to the α subunit, and the β2 subunit is covalently linked to the α subunit by a disulfide bond [26]. Numerous studies indicate a possible role of β auxiliary subunits in modulating Na+ channel expression and function (for review see Ref. [24]), but the possible implication of β-subunits in I NaL modulation has not been studied in detail, especially in HF. Our previous studies using the canine chronic HF model showed that in the state of HF the protein level of Nav1.5 is reduced but remains unchanged for β1 and β2 subunits, making these β subunits relatively upregulated [27].
Thus, an intriguing possibility could be that differential expression of α- and β-subunits in normal and failing hearts can contribute, at least in part, to I NaL alterations observed in HF. Accordingly, in this study, using a heterologous expression system, we specifically tested whether I NaL is modulated by β1 or β2 co-expression with Nav1.5. Our experiments show that β1 substantially and significantly affects I NaL, whereas the effects of β2 were insignificant. More specifically, the β1 subunit modulates I NaL by two mechanisms one of which slows I NaL decay and the other of which increases I NaL amplitude relative to the peak transient (I NaT) current.
Methods
Human kidney epithelial cells tsA201 were transiently transfected by Nav1.5 alone and/or simultaneously with human cardiac β1 or β2 subunits (tagged by GFP or fivefold excess of β-encoding cDNA, as previously suggested [28]). We chose tsA201 cells because there was no expression of endogenous α or β1 and β2 subunits compared with that of some HEK293 cell lines (our unpublished data). Whole-cell recordings were made using the conventional patch-clamp technique. The heterologous expression system provides an unique possibility of measuring the peak transient current (I NaT) and I NaL at the same physiological [Na+]o, = 140 mM. This is impossible in cardiomyocytes because of voltage-control problems. The I NaL amplitude was measured as averaged current within 200–220 ms after the onset of depolarization (2 s duration) and was normalized to the peak I NaT (see examples in Fig. 3a, b). The I NaL decay was evaluated by the single-exponential fit starting 200 ms after the onset of depolarization, as previously suggested [8]. The data points of the peak current in the current–voltage relationships were fitted to the function [29]:
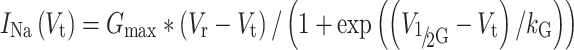 |
1 |
where G max is a normalized maximum Na+ conductance, V r is a reversal potential, V t is the testing voltage, and V 1/2G and kG are the midpoint and slope of the respective Boltzmann functions underlying the NaCh steady-state activation (SSA). The data points were fitted to model equations by use of nonlinear regression (Origin 7.0 software, Microcal Software, MA, USA).
Fig. 3.

Effects of the β subunits on the I NaL produced by the heterologously expressed human cardiac NaCh isoform α subunit (Nav1.5). a Representative examples of superimposed current traces recorded in tsA201 cells transfected with cDNA encoding human cardiac NaCh α-subunit alone or together with β1 (α + β1) or β2 (α + β2). Shown are averaged (10–20) currents with a single-exponential fit to I NaL decay (solid lines) starting 200 ms after the onset of depolarization. The time constant (τ) values are given in the panel. The current amplitudes are relative to the peak I NaT. The voltage-clamp procedure given in the inset. b, c Statistical analysis of the I NaL/I NaT ratio (b) and the decay time course (c) changes in response to coexpression of α with β subunits. The statistically significant difference (P) in panels b and c was evaluated by ANOVA followed by the Bonferroni’s post hoc test. Bars in panels b and c represent means ± SE, n number of cells. There was no significant difference between α + β1-GFP compared with α + β1 (1:5 cDNA ratio) or for α alone compared with α + β2-GFP for both b and c panels
The steady-state availability (or inactivation, SSI) terms (V 1/2A, the midpoint and k A, the slope of the relationship) were measured by a standard double-pulse procedure with the 2-s-duration pre-pulse (V p) ranging from −130 to −40 mV. The data points of I NaT normalized to I NaT measured at −30 and −130 mV prepulse were fitted to a Boltzmann function A (V p):
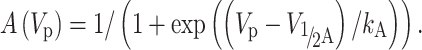 |
2 |
Full details can be found in an extended “Materials and methods” section in the online supplement linked with the online version of this article.
Results
The reporter GFP gene expression monitored the transient transfection of the β-subunits (Fig. 1). Evidently both β1 (Fig. 1a) and β2 (Fig. 1b) can be detected both perinuclear and, importantly, in membrane compartments of the cells in the optical slices under the confocal microscope. Our confocal imaging thus validated the transfection procedure, ensuring that β subunits are located in the cell membrane together with functional Na channel α subunits. It is also important to note that we did not make any attempt to quantify the β-subunits expression level based on these results because our objective was to study β subunit effects on I NaL in all-or-none fashion.
Fig. 1.
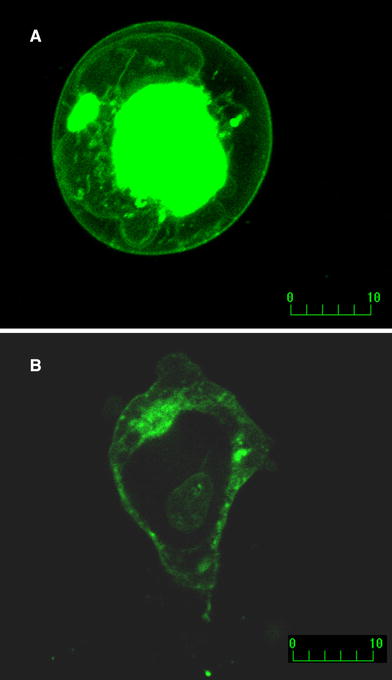
Confocal microscopy of the live tsA201 cells transfected with Nav1.5 + β1 (a) or Nav1.5 + β2 (b). Fluorescence of GFP linked to β subunit C-terminus at the cell membrane is evident for both β subunits. Optical slices were 0.5 μm (Zeiss Axiovert 100, Bio-Rad MRC 1024, excitation/emission wavelength 488/522 nm, laser power 10%)
To carefully evaluate I NaL, we averaged 20–50 original current traces, and the “zero” current (obtained after TTX 25 μM application) was subtracted from the current traces (typical examples are shown in Fig. 2). We determined effects of β subunits on I NaL kinetics and I NaL relative amplitude compared with that of I NaT (Fig. 3). We found that co-expression of α with the β1 subunit but not the β2 subunit significantly slows I NaL decay (Fig. 3a) and substantially increases the relative amplitude of I NaL (Fig. 3b). Accordingly α + β2 can be interpreted as a mock control for these studies. The effect of the β1 subunit on these I NaL values was independent of the reporter GFP gene. This is evident from comparison of the β1-GFP construct with β1 alone (Fig 3b, c). Furthermore, the β1 subunit caused a significant rightward shift of the SSI curves for both peak I NaT and I NaL (measured at 200 ms; not shown). Representative examples of I NaT are shown in Fig. 4, statistics for SSI data are given in Table 1. The current–voltage relationship and the steady-sate activation values remained unchanged (Fig. 5, Table 1).
Fig. 2.
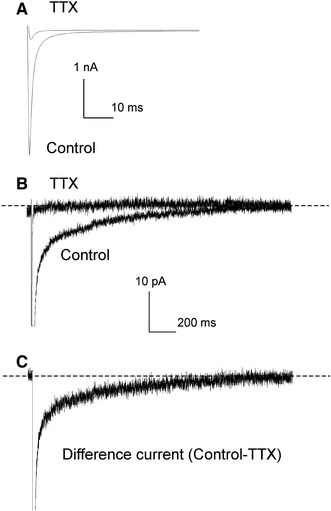
Experimental approach used to elucidate I NaL of heterologously expressed Nav1.5. a, b Typical examples of the I Na currents recorded in tsA201 cells transiently transfected by α + β1-GFP. Shown are averaged traces from 50 sweeps before and after TTX (25 μM) application to assess “zero” current. c Difference I NaL current obtained by subtraction of “zero” current. Note different currents and time scales in a, b, and c to demonstrate peak (I NaT) and I NaL. V h = −120 mV, V m = −30 mV, 23°C
Fig. 4.

Effect of β1 subunit on the steady-state inactivation (SSI) voltage-dependency of the heterologous expressed Nav1.5. a, b Representative raw current recordings at −30 mV in response to a different 2-s-long prepulse potential (voltage procedure is shown in c). c Data points of the relative (normalized to maximum) peak I NaT measured at −30 mV and plotted against prepulse voltage (V p). Solid lines represent the Bolzmann fit (Eq. 2 in “Methods”), and values are given on the plots. Statistics for values evaluated in numerous cells are given in Table 1. V h = −140 mV, [Na]0 = 140 mM, 24°C
Table 1.
Steady-state activation (SSA) and inactivation (SSI) data for I Na in the heterologously expressed cardiac Nav1.5 (α) without or with its auxiliary β subunits
| Conditions | SSI data | SSA data | ||||
|---|---|---|---|---|---|---|
| V 1/2A (mV) | K A (mV) | n | V 1/2G (mV) | K G (mV) | n | |
| α alone | −88.2 ± 0.9 | −6.2 ± 0.2 | 11 | −35.4 ± 1.2 | 5.5 ± 0.2 | 11 |
| α + β1 + GFP | −85.1 ± 1.1* | −5.7 ± 0.4 | 13 | −36.1 ± 2.1 | 6.4 ± 0.4 | 11 |
| α + β1 (1:5) | −81.8 ± 0.9* | −5.8 ± 0.4 | 12 | −34.7 ± 1.2 | 5.9 ± 0.2 | 7 |
| α + β2 + GFP | −88.3 ± 1.8 | −6.1 ± 0.4 | 9 | −36.8 ± 1.2 | 5.9 ± 0.3 | 5 |
SSA and SSI data were obtained from I Na data fit to Eqs. 1 and 2 (“Methods”), respectively. Data represent means ± SE, n stands for the cell number
*P < 0.05 versus α alone. Statistically significant differences between results within the experimental groups were evaluated by ANOVA followed by Bonferroni’s post hoc test and were considered significant at P < 0.05
Fig 5.
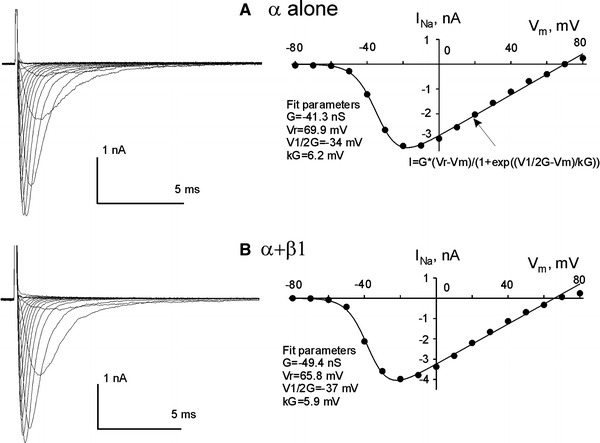
Voltage–current relationship for heterologous expressed Nav1.5 (α alone a, or with the β1 subunit b). a, b Left panels raw current recordings at different membrane potentials, right panels data point plots, with the fits (solid lines) to Eq. 1 (“Methods”). The equation is given in a, and fit values for the steady-state activation are indicated on the plots in a and b. The statistical data for the steady-state activation are given in Table 1
Discussion
For the first time we demonstrate that the β1 subunit can significantly modulate I NaL produced by the heterologously expressed Nav1.5. The modulation includes slowed inactivation, augmented amplitude relative to I NaT, and rightward SSI shift.
Although β subunits do not form ion-conducting pores, they are important modulators of Nav function, expression levels at the plasma membrane (trafficking), and cell adhesion [23, 24]. Recent studies support the emerging significance of the β1 auxiliary subunit in modulation of Nav1.5 function. It has been shown that the β1-subunit:
is involved in abnormal NaCh activity associated with the LQT3 mutation [30];
aggravates NaCh dysfunction in Brugada syndrome [31];
modifies block of NaCh by fatty acids [32] and lidocaine [33]; and
modulates trafficking of Nav1.5 [34].
As to modulation of the late Na+ channel activity by the β subunits, there are only a few controversial reports. Both β1 and β3 subunits exhibit dual and opposite effects on the decay time course of the Na+ current produced by heterologously expressed rat brain αIIA (Nav1.2) [35]. The current decay was assessed by use of two exponential models and the fast time constant, τ 1, was accelerated whereas the slow time constant, τ 2, was increased more than twofold (10.9 vs. 25 ms) by β1 and β3, respectively. In line with this finding it has been shown that expression of the β1 subunit increased slowly, inactivating current (called persistent current by the authors) produced by the new epilepsy-related Nav1.1 mutant D1866Y [36]. These data are in line with our findings reported here.
Transient expression of Nav1.5 into the HEK293 cell line stably expressing β1 subunit reduced a non-inactivating current component measured 750 ms after depolarization onset [37]. A similar non-inactivating Na+ current has been also reported in rat cardiomyocytes; it was highly TTX-sensitive and could be augmented by hypoxia or cyanides [38, 39]. In both cases, when the non-inactivated current was measured, the authors used intracellular solutions deprived of ATP and containing artificial (non-physiological) anion fluoride. It has long been known that fluoride can retard Na+ channel inactivation significantly, as was shown in internally perfused axons [40], and in cardiac cells [41]. Metabolic regulation of Na+ channel inactivation and its rundown was demonstrated in neonatal cultured rat cardiomyocytes [42]. Therefore, this non-inactivating I Na component is likely to be related to these non-physiological experimental conditions. In contrast, our current recordings were performed in close-to-physiological conditions (physiological [Na+] and with no fluoride) and they did not reveal any non-inactivating current either in heterologously expressed Nav1.5 clone (see Fig. 2, and Ref. [19]) or in cardiomyocytes from human hearts [3, 8]. In addition to the non-inactivating current discussed above, a background Na+ current that could be recorded at very negative membrane potentials (−120 mV) has been reported in rabbit cardiac Purkinje cells and ventricular cardiomyocytes [43]. In contrast, recent studies [11, 12] show that canine cardiac Purkinje cells exhibit slowly inactivating I NaL, similar to that described in ventricular cardiomyocytes of humans and dogs [3, 8]. Unlike background Na+ currents, this I NaL in Purkinje cells possesses steady state inactivation and is not activated at resting membrane potentials. Furthermore, non-inactivating Na+ current was not present in human cardiomyocytes [3, 8]. Our single-channel data, in fact, excluded the presence of the non-inactivating component under close-to-physiological conditions for both Nav1.5 clone and for human cardiomyocytes [2, 19]. Thus, the non-inactivating or background Na+ currents are more likely to be species-dependent and/or were recorded in the presence of artificial anion fluoride and absence of ATP in the intracellular milieu. The molecular and genetic origins of background currents in cardiac cells remain unknown, but Denis Noble in his recent review [6] suggested that they could result from a leak form of Na+–K+ ATPase [44] or from NCX [45]. Accordingly, it is important to emphasize that we report here for the first time the modulatory effect of the β1 subunit on cardiac-type late Na+ current I NaL (also known as persistent Na+ current I pNa) rather than on background non-inactivated currents of yet unknown nature reported in some previous studies.
The potency of the β1 subunit to modulate I NaL shown herein has been confirmed in the native cell environment by our preliminary study in normal dog cardiomyocytes in which antisense inhibition of SCN1B significantly accelerated I NaL decay ([46], see Fig. 8A in Ref. [7]). It has been also shown that at the protein level Nav1.5 is downregulated whereas β1 remained unchanged in HF, pointing toward relative higher membrane content of β1 [27], but the SSI shift was not found in HF [2, 3, 29]. The SSI shift is dependent on variety of factors, including intracellular [Ca2+], cytoskeleton, and membrane lipid content, that may affect SSI in different ways negating the β1-related effect ([22], see review [7]). These data together with the findings of our study suggest a potential mechanism for the contribution of β1 to HF-related I NaL alterations [1, 3]. Furthermore, in addition to the I NaL decay slowing, β1 can also change I NaL via its well-known effect of SSI shift. This β1-induced SSI shift under physiological conditions (i.e. at a resting potential of ~−80 mV) may have profound effect on I NaL enhancement during action potentials, as more Na+ channels operating in late modes become available.
The next important question is how, specifically, the β1 subunit interacts with Na+ channel to produce the observed I NaL changes. One possibility, however, could be related to the C-terminus (CT). The role of the CT in regulating Nav1.5 inactivation via the Ca2+-calmodulin-dependent interaction with the III–IV linker, responsible for the initial fast inactivation, has recently been suggested [47, 48]. Direct interaction between the cytoplasmic CT domain of Nav1.1 with β1 and β3 has recently been demonstrated [36] and thus provides a possible molecular mechanism for the I NaL modulation found here.
Single-channel studies in heterologously expressed Nav1.5 and human cardiomyocytes show that late NaCh activity is arranged in two major gating modes—late scattered mode (LSM) and ‘burst” mode (BM) [19]. Numerical evaluation based on the Markovian chain model revealed that BM + LSM is responsible for the intermediate phase (40–300 ms) whereas LSM is responsible for the ultra-late (>300 ms) phase on I NaL inactivation [2]. In this study we analyzed the amplitude and decay of I NaL after 200 ms, thus presumably the function of LSM mode. Therefore, the β1 subunit may increase the probability of occurrence of these modes (i.e. make modal switch more probable), thus increasing relative I NaL/I NaT. Also β1 affects the gating kinetics (inactivation) of LSM. The idea of β1 subunit involvement in the modal switch has previously been suggested for muscular isoform Nav1.4 [49].
We did not find β2-related effects on I NaL in our experimental setting. The role of this subunit in physiological function of NaCh is not clear. Initially the β2 subunit was implicated in intercellular adhesion and recruitment of the cytoskeletal protein ankyrin to the plasma membrane at sites of cell-to-cell contact [50]. Accordingly the direct role of the β2 subunit on NaCh gating was not suggested, because NaCh protein has direct attachment to the sub-membrane cytoskeleton via ankyrin (for a review see Ref. [48]) and may directly be related to the cytoskeleton-dependent effects on Na+ channel gating [51, 52]. It has been recently demonstrated that β1/β2 chimeras may cause the additional closed state for Nav1.5 that is accessible at hyperpolarized potentials, although this effect was not produced by the wild type β2 subunit [53]. Given the multi-modal origin of Nav1.5-related I NaL it is not obvious how this subunit can play a role in the kinetic transition constants assuming the new closed state [2]. Although our experiments do not reveal apparent effects on the I NaL properties studied, we cannot exclude the possibility that the β2 subunit can still affect late Na+ channel openings taking into account a possible role of neuronal isoforms (for a review see Ref. [7]).
We conclude that the auxiliary subunit β1 modulates I NaL, produced by the human cardiac Na+ channel Nav1.5 by slowing its decay and increasing I NaL amplitude relative to I NaT. Because expression of Nav1.5 reportedly decreases but β1 remains unchanged in chronic HF, the relatively higher expression of β1 may contribute to known I NaL increase in HF via the modulation mechanism found in this study.
Electronic supplementary material
Acknowledgments
The study was supported by grants from the National Heart, Lung, and Blood Institute HL074328 (A. Undrovinas), HL-65661 (J. W. Kyle), and by American Heart Association grant 0350472Z (A. Undrovinas).
References
- 1.Undrovinas AI, Maltsev VA, Sabbah HN. Repolarization abnormalities in cardiomyocytes of dogs with chronic heart failure: role of sustained inward current. Cell Mol Life Sci. 1999;55:494–505. doi: 10.1007/s000180050306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Maltsev VA, Undrovinas AI. A multi-modal composition of the late Na+ current in human ventricular cardiomyocytes. Cardiovasc Res. 2006;69:116–127. doi: 10.1016/j.cardiores.2005.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Maltsev VA, Silverman N, Sabbah HN, Undrovinas AI. Chronic heart failure slows late sodium current in human and canine ventricular myocytes: implications for repolarization variability. Eur J Heart Fail. 2007;9:219–227. doi: 10.1016/j.ejheart.2006.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Maltsev VA, Sabbah HN, Tanimura M, Lesch M, Goldstein S, Undrovinas AI. Relationship between action potential, contraction-relaxation pattern, and intracellular Ca2+ transient in cardiomyocytes of dogs with chronic heart failure. Cell Mol Life Sci. 1998;54:597–605. doi: 10.1007/s000180050187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Undrovinas AI, Belardinelli L, Undrovinas NA, Sabbah HN. Ranolazine improves abnormal repolarization and contraction in left ventricular myocytes of dogs with heart failure by inhibiting late sodium current. J Cardiovasc Electrophysiol. 2006;17:S169–S177. doi: 10.1111/j.1540-8167.2006.00401.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Noble D, Noble PJ. Late sodium current in the pathophysiology of cardiovascular disease: consequences of sodium-calcium overload. Heart. 2006;92:iv1–iv5. doi: 10.1136/hrt.2005.078782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Maltsev VA, Undrovinas A. Late sodium current in failing heart: friend or foe? Prog Biophys Mol Biol. 2008;96:421–451. doi: 10.1016/j.pbiomolbio.2007.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maltsev VA, Sabbah HN, Higgins RSD, Silverman N, Lesch M, Undrovinas AI. Novel, ultraslow inactivating sodium current in human ventricular cardiomyocytes. Circulation. 1998;98:2545–2552. doi: 10.1161/01.cir.98.23.2545. [DOI] [PubMed] [Google Scholar]
- 9.Maltsev VA, Sabbah HN, Undrovinas AI. Late sodium current is a novel target for amiodarone: studies in failing human myocardium. J Mol Cell Cardiol. 2001;33:923–932. doi: 10.1006/jmcc.2001.1355. [DOI] [PubMed] [Google Scholar]
- 10.Zygmunt AC, Eddlestone GT, Thomas GP, Nesterenko VV, Antzelevitch C. Larger late sodium conductance in M cells contributes to electrical heterogeneity in canine ventricle. Am J Physiol. 2001;281:H689–H697. doi: 10.1152/ajpheart.2001.281.2.H689. [DOI] [PubMed] [Google Scholar]
- 11.Vassalle M, Bocchi L, Du F. A slowly inactivating sodium current (INa2) in the plateau range in canine cardiac Purkinje single cells. Exp Physiol. 2007;92:161–173. doi: 10.1113/expphysiol.2006.035279. [DOI] [PubMed] [Google Scholar]
- 12.Bocchi L, Vassalle M. Characterization of the slowly inactivating sodium current INa2 in canine cardiac single Purkinje cells. Exp Physiol. 2008;93:347–361. doi: 10.1113/expphysiol.2007.040881. [DOI] [PubMed] [Google Scholar]
- 13.Sakmann BF, Spindler AJ, Bryant SM, Linz KW, Noble D. Distribution of a persistent sodium current across the ventricular wall in guinea pigs. Circ Res. 2000;87:910–914. doi: 10.1161/01.res.87.10.910. [DOI] [PubMed] [Google Scholar]
- 14.Belardinelli L, Shryock JC, Fraser H. Inhibition of the late sodium current as a potential cardioprotective principle: effects of the late sodium current inhibitor ranolazine. Heart. 2006;92:iv6–iv14. doi: 10.1136/hrt.2005.078790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.La C, You Y, Zhabyeyev P, Pelzer DJ, McDonald TF. Ultraviolet photoalteration of late Na+ current in guinea-pig ventricular myocytes. J Membr Biol. 2006;210:43–50. doi: 10.1007/s00232-005-0844-6. [DOI] [PubMed] [Google Scholar]
- 16.Wu L, Shryock JC, Song Y, Belardinelli L. An increase in late sodium current potentiates the proarrhythmic activities of low-risk QT-prolonging drugs in female rabbit hearts. J Pharmacol Exp Ther. 2006;316:718–726. doi: 10.1124/jpet.105.094862. [DOI] [PubMed] [Google Scholar]
- 17.Chattou S, Coulombe A, Diacono J, Le Grand B, John G, Feuvray D. Slowly inactivating component of sodium current in ventricular myocytes is decreased by diabetes and partially inhibited by known Na+-H+ exchange blockers. J Mol Cell Cardiol. 2000;32:1181–1192. doi: 10.1006/jmcc.2000.1151. [DOI] [PubMed] [Google Scholar]
- 18.Wagner S, Dybkova N, Rasenack EC, Jacobshagen C, Fabritz L, Kirchhof P, et al. Ca/calmodulin-dependent protein kinase II regulates cardiac Na channels. J Clin Investig. 2006;116:3127–3138. doi: 10.1172/JCI26620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Undrovinas AI, Maltsev VA, Kyle JW, Silverman NA, Sabbah HN. Gating of the late Na+ channel in normal and failing human myocardium. J Mol Cell Cardiol. 2002;34:1477–1489. doi: 10.1006/jmcc.2002.2100. [DOI] [PubMed] [Google Scholar]
- 20.Josephson ME, Rosen MR, Tomaselli GF. The year in arrhythmias—2007: part I. Heart Rhythm. 2008;5:742–748. doi: 10.1016/j.hrthm.2008.03.012. [DOI] [PubMed] [Google Scholar]
- 21.Maltsev VA, Kyle JW, Mishra S, Undrovinas A. Molecular identity of the late sodium current in adult dog cardiomyocytes identified by Nav1.5-antisense inhibition. Am J Physiol Heart Circ Physiol. 2008;295:H667–H676. doi: 10.1152/ajpheart.00111.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maltsev VA, Reznikov V, Undrovinas NA, Sabbah HN, Undrovinas A. Modulation of the late sodium current by Ca2+, calmodulin, and CaMKI I in normal and failing dog cardiomyocytes: similarities and differences. Am J Physiol Heart Circ Physiol. 2008;294:H1597–H1608. doi: 10.1152/ajpheart.00484.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nerbonne JM, Kass RS. Molecular physiology of cardiac repolarization. Physiol Rev. 2005;85:1205–1253. doi: 10.1152/physrev.00002.2005. [DOI] [PubMed] [Google Scholar]
- 24.Meadows LS, Isom LL. Sodium channels as macromolecular complexes: implications for inherited arrhythmia syndromes. Cardiovasc Res. 2005;67:448–458. doi: 10.1016/j.cardiores.2005.04.003. [DOI] [PubMed] [Google Scholar]
- 25.Kazen-Gillespie KA, Ragsdale DS, D’Andrea MR, Mattei LN, Rogers KE, Isom LL. Cloning, localization and functional expression of sodium channel β1A subunits. J Biol Chem. 2000;275:1079–1088. doi: 10.1074/jbc.275.2.1079. [DOI] [PubMed] [Google Scholar]
- 26.Messner DJ, Catterall WA. The sodium channel from rat brain. Role of the beta 1 and beta 2 subunits in saxitoxin binding. J Biol Chem. 1986;261:211–215. [PubMed] [Google Scholar]
- 27.Zicha S, Maltsev VA, Nattel S, Sabbah HN, Undrovinas AI. Post-transcriptional alterations in the expression of cardiac Na+ channel subunits in chronic heart failure. J Mol Cell Cardiol. 2004;37:91–100. doi: 10.1016/j.yjmcc.2004.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Qu Y, Rogers JC, Chen SF, McCormick KA, Scheuer T, Catterall WA. Functional roles of the extracellular segments of the sodium channel alpha subunit in voltage-dependent gating and modulation by beta1 subunits. J Biol Chem. 1999;274:32647–32654. doi: 10.1074/jbc.274.46.32647. [DOI] [PubMed] [Google Scholar]
- 29.Maltsev VA, Sabbah HN, Undrovinas AI. Down-regulation of sodium current in chronic heart failure: effects of long-term therapy with carvedilol. Cell Mol Life Sci. 2002;59:1561–1568. doi: 10.1007/s00018-002-8529-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.An RH, Wang XL, Kerem B, Benhorin J, Medina A, Goldmit M, et al. Novel LQT-3 mutation affects Na+ channel activity through interactions between alpha- and beta1-subunits. Circ Res. 1998;83:141–146. doi: 10.1161/01.res.83.2.141. [DOI] [PubMed] [Google Scholar]
- 31.Makita N, Shirai N, Wandg DW, Sasaki K, George ALJ, Kanno M, et al. Cardiac Na+ channel dysfunction in Brugada syndrome is aggravated by β1-subunit. Circulation. 2000;101:54–60. doi: 10.1161/01.cir.101.1.54. [DOI] [PubMed] [Google Scholar]
- 32.Xiao YF, Wright SN, Wang GK, Morgan JP, Leaf A. Coexpression with β1-subunit modifies the kinetics and fatty acid block of hH1α Na+ channels. Am J Physiol. 2000;279:H35–H46. doi: 10.1152/ajpheart.2000.279.1.H35. [DOI] [PubMed] [Google Scholar]
- 33.Makielski JC, Limberis J, Fan Z, Kyle JW. Intrinsic lidocaine affinity for Na channels expressed in Xenopus oocytes depends on α (hH1 vs. rSkM1) and β1 subunits. Cardiovasc Res. 1999;42:503–509. doi: 10.1016/S0008-6363(99)00024-3. [DOI] [PubMed] [Google Scholar]
- 34.Zhou J, Yui J, Hu NN, George ALJ, Murray KT. Activation of protein kinase A modulates trafficking of the human cardiac sodium channel in Xenopus oocytes. Circ Res. 2000;87:33–38. doi: 10.1161/01.res.87.1.33. [DOI] [PubMed] [Google Scholar]
- 35.Morgan K, Stevens EB, Shah B, Cox PJ, Dixon AK, Lee K, et al. beta 3: An additional auxiliary subunit of the voltage-sensitive sodium channel that modulates channel gating with distinct kinetics. Proc Natl Acad Sci USA. 2000;97:2308–2313. doi: 10.1073/pnas.030362197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Spampanato J, Kearney JA, de Haan G, McEwen DP, Escayg A, Aradi I, et al. A novel epilepsy mutation in the sodium channel SCN1A identifies a cytoplasmic domain for beta subunit interaction. J Neurosci. 2004;24:10022–10034. doi: 10.1523/JNEUROSCI.2034-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Valdivia C, Nagatomo T, Makielski J. Late Na currents affected by alpha subunit Isoform and beta1 subunit co-expression in HEK293 cells. J Mol Cell Cardiol. 2002;34:1029. doi: 10.1006/jmcc.2002.2040. [DOI] [PubMed] [Google Scholar]
- 38.Saint DA, Ju YK, Gage PW. A persistent sodium current in rat ventricular myocytes. J Physiol. 1992;453:219–231. doi: 10.1113/jphysiol.1992.sp019225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ju YK, Saint DA, Gage PW. Hypoxia increases persistent sodium current in rat ventricular myocytes. J Physiol. 1996;497:337–347. doi: 10.1113/jphysiol.1996.sp021772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Meves H. Inactivation of the sodium permeability in squid giant nerve fibres. Prog Biophys Mol Biol. 1978;33:207–230. doi: 10.1016/0079-6107(79)90029-4. [DOI] [PubMed] [Google Scholar]
- 41.Kohlhardt M. Gating properties of cardiac Na+ channels in cell-free conditions. J Membr Biol. 1991;122:11–21. doi: 10.1007/BF01872735. [DOI] [PubMed] [Google Scholar]
- 42.Kohlhardt M, Fichtner H, Frobe U. Metabolites of the glycolytic pathway modulate the activity of single cardiac Na+ channels. FASEB J. 1989;3:1963–1967. doi: 10.1096/fasebj.3.8.2542113. [DOI] [PubMed] [Google Scholar]
- 43.Zilberter YI, Starmer CF, Starobin J, Grant AO. Late Na channels in cardiac cells: the physiological role of background Na channels. Biophys J. 1994;67:153–160. doi: 10.1016/S0006-3495(94)80464-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Artigas P, Gadsby DC. Large diameter of palytoxin-induced Na/K pump channels and modulation of palytoxin interaction by Na/K pump ligands. J Gen Physiol. 2004;123:357–376. doi: 10.1085/jgp.200308964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hilgemann DW. New insights into the molecular and cellular workings of the cardiac Na+/Ca2+ exchanger. Am J Physiol Cell Physiol. 2004;287:C1167–C1172. doi: 10.1152/ajpcell.00288.2004. [DOI] [PubMed] [Google Scholar]
- 46.Undrovinas AI, Maltsev VA. Molecular basis for late Na+ current. Knockdown of Na+ channel subunits in adult cardiomyocytes by antisense oligonucleotides. Biophys J. 2002;82:89a. [Google Scholar]
- 47.Motoike HK, Liu H, Glaaser IW, Yang AS, Tateyama M, Kass RS. The Na+ channel inactivation gate is a molecular complex: a novel role of the COOH-terminal domain. J Gen Physiol. 2004;123:155–165. doi: 10.1085/jgp.200308929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Abriel H, Kass RS. Regulation of the voltage-gated cardiac sodium channel Nav1.5 by interacting proteins. Trends Cardiovasc Med. 2005;15:35–40. doi: 10.1016/j.tcm.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 49.Chang SY, Satin J, Fozzard HA. Modal behavior of the μ1 Na+ channel and effects of coexpression of the beta 1-subunit. Biophys J. 1996;70:2581–2592. doi: 10.1016/S0006-3495(96)79829-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Malhotra JD, Kazen-Gillespie K, Hortsch M, Isom LL. Sodium channel beta subunits mediate homophilic cell adhesion and recruit ankyrin to points of cell-cell contact. J Biol Chem. 2000;275:11383–11388. doi: 10.1074/jbc.275.15.11383. [DOI] [PubMed] [Google Scholar]
- 51.Undrovinas AI, Shander GS, Makielski JC. Cytoskeleton modulates gating of voltage-dependent sodium channel in heart. Am J Physiol. 1995;269:H203–H214. doi: 10.1152/ajpheart.1995.269.1.H203. [DOI] [PubMed] [Google Scholar]
- 52.Chauhan VS, Tuvia S, Buhusi M, Bennett V, Grant AO. Abnormal cardiac Na+ channel properties and QT heart rate adaptation in neonatal ankyrin(B) knockout mice. Circ Res. 2000;86:441–447. doi: 10.1161/01.res.86.4.441. [DOI] [PubMed] [Google Scholar]
- 53.Zimmer T, Benndorf K. The intracellular domain of the beta 2 subunit modulates the gating of cardiac Na v 1.5 channels. Biophys J. 2007;92:3885–3892. doi: 10.1529/biophysj.106.098889. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.