

Abstract
Amyotrophic Lateral Sclerosis (ALS) is a fatal neurodegenerative disorder characterized by progressive paralysis due to motor neuron degeneration. Despite the fact that many different therapeutic strategies have been applied to prevent disease progression, no cure or effective therapy is currently available for ALS. We found that L-arginine protects cultured motor neurons from excitotoxic injury. We also found that L-arginine supplementation both prior to and after the onset of motor neuron degeneration in mtSOD1 (G93A) transgenic ALS mice significantly slowed the progression of neuropathology in lumbar spinal cord, delayed onset of motor dysfunction, and prolonged lifespan. Moreover, L-arginine treatment was associated with preservation of arginase I activity and neuroprotective polyamines in spinal cord motor neurons. Our findings show that L-arginine has potent in vitro and in vivo neuroprotective properties and may be a candidate for therapeutic trials in ALS.
Keywords: amyotrophic lateral sclerosis (ALS), L-arginine, motor neuron, polyamines, superoxide dismutase 1 (SOD1)
Introduction
ALS is a devastating fatal neurodegenerative disorder primarily targeting upper and lower motor neurons resulting in progressive muscle wasting and paralysis (1). Most ALS cases are sporadic but about 5-10% of cases are familial. Missense mutations of copper/zinc SOD1 cause about 25% of familial ALS cases (2), whose clinical and pathological features are indistinguishable from those in sporadic ALS. The mechanism of motor neuron degeneration in ALS is under intense investigation. There is increasing evidence that oxidative stress, neurofilament disorganization, protein misfolding, and glutamate toxicity are involved in disease pathogenesis (3). Therapies based on those mechanisms have been tested but results have been disappointing and no clinically effective therapy is currently available. Transgenic mice expressing the G93A or G37R human SOD1 mutations with elevated levels of SOD1 activity and mice expressing G85R mutant SOD1 with protein and activity levels essentially equal to endogenous levels, develop progressive hind limb weakness, muscle wasting, and neuropathological sequelae similar to that observed in ALS patients (3-5). Disease modifying therapies that have been efficacious in ALS transgenic mice to date have been minimally effective in patients with ALS (3-7).
L-arginine is a semi-essential amino acid that serves as sole substrate for enzymes involved in diverse cell processes. It is noteworthy that L-arginine concentrations have been reported to be low in the plasma of ALS patients, potentially due to malnutrition associated with advanced ALS (8). The relationship of L-arginine to motor neuron death in ALS has not been previously investigated. The goal of our study is to evaluate the neuroprotective effects of L-arginine on motor neuron cultures and in mtSOD1 (G93A) transgenic mouse model of ALS. Here we report that L-arginine supplementation slows motor neuronal loss and prolongs the survival of mtSOD1 (G93A) ALS transgenic mice.
Materials and Methods
Motor neuron culture
The procedure for motor neuron culture was based on previously described methods with several modifications (9, 10). Spinal motor neurons were prepared from mouse embryo (embryonic day 12.5 [E12.5]) spinal cord by a combination of BSA cushion and Opti-prep (Axis Shield PoC, Oslo, Norway) gradient centrifugation. Briefly, ventral spinal cords were dissected in modified F10 medium (without calcium and magnesium, and with sodium pantothenate), treated with 0.05% trypsin for 15 min at 37°C, and dissociated by gentle trituration in 0.1% bovine serum albumin (BSA) and 0.1 mg/ml DNase I in basal culture medium. The single-cell suspension was centrifuged on a 5.7% Optiprep density gradient for 15 min at 500g. Motor neurons were removed from the interface. Motor neurons were plated at a density of 280 cells/cm2 on 35 mm dishes or at a density of 103 cells/cm2 on glass coverslips precoated with polyornithine-laminin in basal culture medium. The neurons were fed with media made fresh each week. Neurobasal culture media contains: apotransferrin (0.1 mg/ml), chick albumin (1 mg/ml), 2% heat inactivated horse serum, penicillin/ streptomycin, B27, GDNF (10 ng/ml, R&D Systems, Minneapolis, MN), CNTF (10 ng/ml, R&D Systems, Minneapolis, MN) and BDNF (1 ng/ml, R&D Systems, Minneapolis, MN). The purity of motor neuron was determined by using antibodies against Islet-1 (4D5 monoclonal antibody from the Developmental Studies Hybridoma Bank, Iowa City, IA), neurofilament 200 (SMI32 monoclonal antibody, Sternberger Monoclonals, Baltimore, MD) and choline acetyltransferse (ChAT, Abcam Inc., Cambridge, MA). Cultures were maintained at 37°C in an atmosphere 5% CO2/95% humidified air.
Glutamate cytotoxicity and determination of motor neuron survival
Motor neurons were cultured in vitro for 2 weeks and then exposed to glutamate (100 μM) in a HEPES-buffered salt solution (HBSS) containing 120 mM NaCl, 5.4 mM KCl, 0.8 mM MgCl2, 1.8 mM CaCl2, 15 mM glucose, and 20 mM HEPES, pH 7.4. After 2 hr at room temperature the HBSS was aspirated, and the original neurobasal media was returned to the cells. Motor neuronal survival was determined by the MTT [3-(4,5-dimethyldiazol-2-yl)-2,5-diphenyl tetrazolium bromide] assay as described (7), which correlates with cell death as determined by trypan blue exclusion and a colony-forming assay. At 24 hr after the addition of glutamate, MTT solution (2.5 mg/ml) is added to each well and the cells are incubated for 3 hr at 37°C. Then 100 μl of solubilization solution (50% dimethylformamide and 20% SDS, pH 4.8) is added to the wells, and the next day the absorption values at 570 nm are measured. The results are expressed relative to the controls specified in each experiment. They are expressed as the mean of triplet determinations within the same experiment ± SEM.
Immunofluorescence staining and confocal microscopy
Indirect labeling methods were used to determine arginase I, SMI32, ChAT, spermidine, and spermine as previously described (7). Images were taken by a spinning confocal microscopy (Olympus DSU, Tokyo, Japan). Size of motor neuronal cell body was analyzed by AQI-X-COMBO-CWF program (Media cybernetics Inc., Bethesda, MD).
Determination of arginase activity
Arginase activity was determined from the lysate of spinal cord. Spinal cord tissue was homogenized in 0.1% Triton X-100 containing 2 mM Pefabloc, 2 μg/ml pepstatin A, and 10 μg/ml leupeptin. Lysates were centrifuged at 12,000 × g for 10 min, and supernatants were used for assays. Protein concentration was determined by the BCA assay (Pierce Chemical). Arginase activity was measured by the conversion of [guanido-14C] L-arginine to [14C] urea, using a derivation of the method of Russell and Ruegg (11). After a 30-min incubation at 37°C, the reaction was terminated by heating at 100°C for 3 min, and the reaction mixture was incubated for an additional 45 min at 37°C with jack bean urease as described previously (11). The urease reaction was terminated by addition of 2 N HCl, and incubation was continued at 37°C for 30 min. Liberated 14CO2, trapped as Na214CO3, was quantified by scintillation counting.
Western blot analysis
The Western blot analysis was performed as previously described (7).
Animals
Male transgenic ALS mice of the mtSOD1 (G93A) H1 high-expressor strain (Jackson Laboratories, Bar Harbor, Maine) are bred with females with a similar background (B6/SJLF1). Mice were randomized from 24 litters all within 4 days of age from the same ‘f’ generation removed from the founding mice in our colony. Disease onset may be delayed in female G93A mice due to the neuroprotective effects of female sex hormones (12). For this reason we only used male G93A transgenic ALS mice as in our previous studies (7). These experiments were carried out in accordance with the NIH Guide for the Care and Use of Laboratory Animals.
L-Arginine supplementation
6% L-arginine was dissolved in water made freshly twice a week and administered to groups (n=20) of wild type and G93A according to the method described by Ignarro and colleagues (13, 14). L-arginine supplementation was started at either 30 or 70 days of age, that is, either prior to or after the onset of motor neuron degeneration in G93A mice until mice reached criteria for euthenasia. Control groups of G93A mice were supplemented with water only.
Histopathological evaluation and densitometry
Groups of 10 animals from each treatment paradigm were deeply anesthetized and transcardially perfused with 4% PFA at 120 days of age. Serial-cut lumbar spinal cord tissue-sections (n=10), from L3-L5 spinal cord segments were used for neuronal analysis. Spinal cord tissue sections were stained for Nissl substance and immunostained for glial fibrillary antigen protein (GFAP), arginase I, ChAT, and etc as previously described (7). The intensity (pixels) of immunoreactivity in motor neurons was analyzed using NIH ImageJ image analysis software.
Clinical assessment and survival
Both body weight and survival were measured throughout the study. Body weights were recorded twice weekly at the same time of day. G93A mice were assessed twice daily (mid-morning and late afternoon) for morbidity and mortality.
Statistics
The data was expressed as the mean + standard error of the mean. Survival data was analyzed by the Kaplan-Meier survival curves. Values were analyzed by oneway ANOVA followed by Dunn's multiple-comparison test. All statistics and graphs were performed using Prism 4.0c (GraphPad Software, San Diego, CA).
Results
L-Arginine prevents glutamate-induced motor neuron death
To investigate whether L-arginine is a critical factor for motor neuron survival, we established an in vitro primary culture system for motor neurons. We used SMI32, a motor neuron marker, in conjunction with immunofluorescence staining to confirm the purity of our motor neuron cultures (Fig. 1A). Then, we evaluated the effects of L-arginine depletion on motor neuron morphology and viability by depleting it from the culture medium. L-arginine deficiency resulted in significant cell death and dramatic shrinkage of cell body and neurite morphology as monitored by phase contrast microscopy (Fig. 1B).
Figure 1. L-Arginine prevents glutamate-induced motor neuron death.
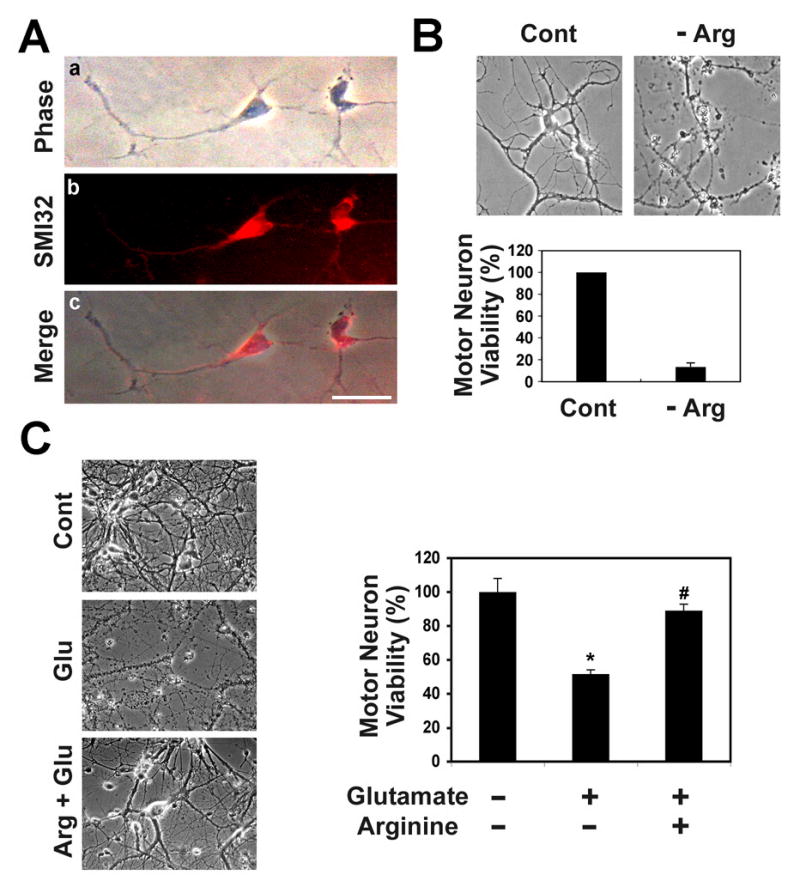
A, Mouse primary cultured spinal cord motor neurons at 3 days old: phase contrast microscopic view (a); SMI-32, a motor neuron marker, immunofluorescence staining (b); a merged image of motor neurons with SMI-32 (c). Scale bar: 50 μm. B, L-Arginine (Arg) deficiency leads to primary motor neuron (2 weeks in vitro culture) death. Upper panel, phase contrast microscopic views of motor neurons. Lower panel, motor neuron viability by MTT assay. C, L-Arginine prevents motor neuron death in the presence of glutamate (Glu)-induced excitotoxicity. Left panel, phase contrast microscopic view of motor neurons. Motor neurons were cultured in vitro for 2 weeks. Right panel, motor neuron viability by MTT assay. *, Significantly different from untreated motor neurons at p<0.05. #, L-arginine supplementation results in motor neuronal protection in comparison to glutamate-treated motor neurons at p<0.05.
Because previous evidence has established that arginase can prevent excitotoxic necrosis in cortical cultures by inhibiting nitric oxide generation (15, 16), we hypothesized that L-arginine, the substrate of arginase, prevent motor neuron death by a similar mechanism. Pre- or co-treatment of motor neurons with glutamate (100 μM) induced marked motor neuron death (Fig. 1C). However, treatment of motor neurons with L-arginine (10 mM) prevented cell death in the presence of glutamate. This suggests that L-arginine and/or its metabolites protect motor neurons from glutamate-induced neurotoxicity.
L-Arginine supplementation protects motor neurons in the lumbar spinal cord of G93A ALS mice
In order to examine the potential protective role of L-arginine in G93A ALS mice, we supplemented L-arginine (6% drinking water) from 70 days to 120 days of age followed by neuropathological evaluation. Control G93A mice showed marked motor neuron loss, gross atrophy and increased astrogliosis in the lumbar spinal cord (Fig. 2A). G93A ALS mice receiving L-arginine treatment showed much less neuronal loss than their control counterparts (Fig. 2B). To measure the size change of motor neurons, we analyzed the cell body volume using spinning confocal microscopy with AQI-X-COMBO-CWF image analysis program (Media cybernetics Inc. Bethesda, MD). Choline acetyltransferase (ChAT)-positive motor neurons were significantly reduced in both number and size in the lumbar spinal cord of the G93A ALS mice compared to littermate control mice (Fig. 2C). L-arginine treatment prevented both neuronal loss and shrinkage in G93A ALS mice (Fig. 2D).
Figure 2. L-Arginine supplementation prevents motor neuron death in mutant SOD1 (G93A) ALS mice.
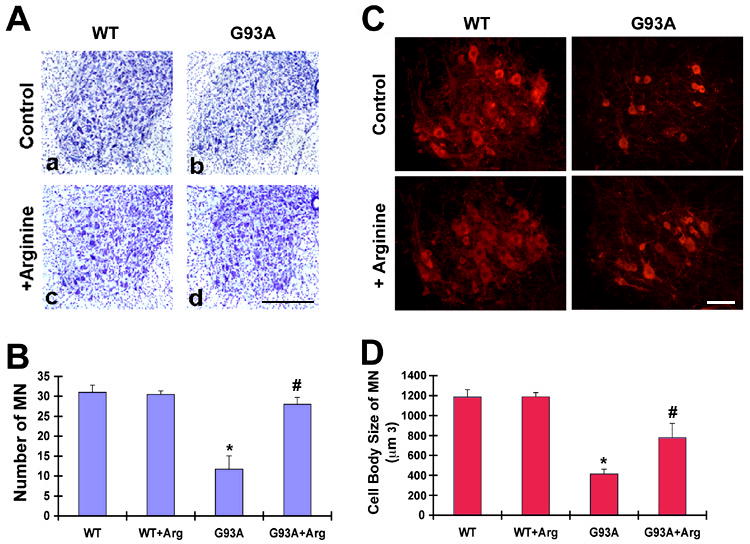
A, Nissl-stained tissue sections from the lumbar spinal cord showed marked reduction of ventral neuronal loss and astrogliosis in G93A mice (b and d) by L-arginine. Scale bar (black): 250 μm. B, L-Arginine (Arg) prevents loss of motor neurons in the lumbar spinal cord in G93A mice. Scale bar (white): 50 μm. C, L-Arginine prevents loss of choline acetyltransferase (ChAT)-positive motor neurons in the ventral horn of G93A mice. D, L-Arginine improves the size of motor neuron cell body. The size of ChAT-positive motor neurons was analyzed by AQI-X-COMBO-CWF program (Media cybernetics Inc. Bethesda, MD). *, Significantly different from WT mice at p<0.01. #, Significantly different from untreated G93A mice at p<0.05.
L-Arginine supplementation delays onset of disease and prolong the life span of G93A ALS mice
Consistent with the in vitro results L-arginine-treated G93A mice had a delay in disease onset, extended survival, and the ability to maintain their body weight all consistent with neuropathological sequelae (Fig. 3). Supplementation of drinking water with 6% L-arginine significantly extended the lifespan G93A mice both prior to and after the onset of motor neuron degeneration by 20% and 9% respectively compared to untreated G93A mice (Fig. 3A and C). Kaplan-Meier probability of survival analysis showed that supplementation with L-arginine prior to the onset of motor neuron degeneration was significantly more effective. In addition, L-arginine supplementation prevented weight loss both prior to and after the onset of motor neuron degeneration in mtSOD1 (G93A) mice in comparison with untreated controls (Fig. 3 B and D). The photograph in the Figure 3 E shows that L-arginine supplementation also delayed the onset of hindlimb muscle wasting in G93A mice.
Figure 3. L-Arginine prolongs the life-span of mutant SOD1 (G93A) ALS mice.

A, Supplementation with L-arginine prior to the onset of motor neuron degeneration prolongs the lifespan of G93A ALS mice as shown by Kaplan-Meier probability of survival analysis (n=20). B, Supplementation with L-arginine prior to the onset of motor neuron degeneration prevented weight loss in G93A mice after 14 weeks (n=20). C, Supplementation with L-arginine after the onset of motor neuron degeneration extends the survival of G93A mice (n=20). D, Supplementation with L-arginine after the onset of motor neuron degeneration improved the bodyweight of G93A mice after 14 weeks (n=20). E, Photographs of mice showed that L-arginine markedly reduces hindlimb muscle wasting of G93A mice (c) in comparison to non-treated G93A mice (b). a, wild type mice.
L-Arginine treatment restores depleted Arginase I in G93A ALS mice
We immunostained lumbar spinal cord sections from control and G93A ALS mice at 30, 70, 90, and 120 days of age. In control mice arginase immunoreactivity was primarily localized within motor neurons that were readily identifiable at all time points. In contrast arginase I immunoreactivity which was clearly present at 30 days of age in G93A mice became progressively depleted over time (Fig. 4A). At 30 days of age Arginase 1 enzyme activity was identical in both wildtype and G93A mice. Between 30 and 120 days, arginase 1 activity increased over time in control mice in contrast to G93A mice where levels fell about 15% (Fig. 4). Arginase I immunoreactivity within motor neurons was preserved in L-arginine-treated G93A mice compared to vehicle-treated controls (Fig. 4C). Western blots confirmed that arginase 1 protein levels were significantly greater in G93A mice treated with L-arginine (Fig. 4D). Moreover, spinning disk confocal microscopic data showed that L-arginine supplementation preserved arginase I immunoreactivity in the cell body and axonal arbors of ChAT-positive motor neurons in G93A mice (Fig. 4E).
Figure 4. L-Arginine supplementation increases the level of arginase I in motor neurons of mtSOD1 (G93A) ALS mice.

A, Arginase I immunoreactivity is markedly reduced in the motor neurons in the ventral horn of G93A ALS mouse lumbar spinal cords at 70D, 90D and 120 D compared to age matched control mice. B, Arginase I activity is significantly decreased in mtSOD1 (G93A) ALS mice compared to WT at 120 days of age. *, Significantly different from WT mice at p<0.01. C, L-Arginine supplemented G93A mice show increased arginase I immunoreactivity in spinal motor neurons (d). D, Western blot analysis shows an increase in protein level of arginase I in the lumbar spinal cord of mtSOD1 (G93A) ALS mice as compared to age matched controls. AN, animal number. The numbers refer to individual animals. The lower panel shows the densitometric analysis of arginase I protein derived from the upper panel. **, Significantly different from WT mice at p<0.05. E, Spinning confocal microscopy shows that L-arginine supplementation increases arginase I immunoreactivity in cell body and axonal arborization in ChAT-positive motor neurons of ALS mice. Scale bars: black, 250 μm; white, 50 μm.
L-Arginine supplementation preserves neuronal spermidine and spermine immunoreactivity in G93A ALS mice
In order to determine whether L-arginine mediated neuroprotection is through the generation of polyamines, we further examined the level of polyamines by immunohistochemistry and spinning disk confocal microscopy in both groups. In control mice, intense spermine and spermidine immunoreactivity was largely confined to large motor neurons (Supplementary Figure 1). This pattern was not changed by L-arginine supplementation. As expected, G93A mice euthanized at 120 days showed extensive neuronal loss and reduced polyamine immunoreactivity. Spinning confocal microscopy showed that polyamine immunoreactivity within surviving motor neurons was clearly reduced in untreated G93A mice whereas in L-arginine supplemented G93A mice polyamine immunoreactivity was preserved.
Discussion
Several potential pathways can be invoked to explain the neuroprotective role of L-arginine. Abnormalities of glutamate regulation have been identified in ALS suggesting that excessive synaptic glutamate may initiate or propagate motor neuron loss (17-20). We found that L-arginine protects motor neurons from glutamate induced toxicity in vitro so it is possible that this effect is also operative in vivo. It is noteworthy that L-arginine is among the handful of amino acids that are reduced in the plasma of ALS patients (8). It is possible that depleted levels of L-arginine could render neurons more sensitive to excitoxic injury in patients with ALS.
L-arginine is a substrate for both arginase I and NOS. L-arginine is also a major precursor for polyamine synthesis. Arginase I converts L-arginine to ornithine, which is ultimately converted to the polyamines spermine and spermidine. Spermine and spermidine are ubiquitous organic low molecular weight cations involved in a number of cellular functions (21, 22). Neurite outgrowth depends on polyamines and we found that polyamines, either synthesized endogenously or supplied exogenously, are essential for neuronal survival and regeneration, as reported by others (23). We also found that spermidine and spermine levels are decreased in motor neurons in the lumbar spinal cord of G93A mice suggesting that L-arginine metabolism and/or polyamine synthesis may be dysfunctional. Polyamine deficiency could contribute to neuronal degeneration in ALS pathogenesis by inhibiting neurite outgrowth and triggering neurodegeneration.
In conclusion, our study shows that L-arginine has potent in vitro and in vivo neuroprotective effects and prolongs the lifespan of ALS (G93A) mice. The mechanism underlying these effects is not known but may be related to the synthesis of neuroprotective polyamines. In any case, our results suggest that L-arginine may be a candidate for therapeutic trials in ALS.
Supplementary Material
Acknowledgments
We thank Dr. Alvaro Estévez for his technical help and advice on the motor neuron culture. J.L. is an awardee of Les Turner ALS Foundation Grant. This work was supported by NIH P30 AG13846 (J.L.), NIH NS52724 (H.R.), and the Merit Review Grant from the Department of Veterans Affairs (J.L., N.W.K.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Rowland L, Shneider N. Amyotrophic lateral sclerosis. N Engl J Med. 2001;344:1688–1700. doi: 10.1056/NEJM200105313442207. [DOI] [PubMed] [Google Scholar]
- 2.Rosen DR. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- 3.Weiss MD, Weydt P, Carter GT. Current pharmacological management of amyotrophic lateral sclerosis and a role for rational polypharmacy. Expert Opin Pharmacother. 2004;5:735–746. doi: 10.1517/14656566.5.4.735. [DOI] [PubMed] [Google Scholar]
- 4.McGeer EG, McGeer PL. Pharmacologic approaches to the treatment of amyotrophic lateral sclerosis. BioDrugs. 2005;19:31–37. doi: 10.2165/00063030-200519010-00004. [DOI] [PubMed] [Google Scholar]
- 5.Festoff BW, Suo Z, Citron BA. Prospects for the pharmacotherapy of amyotrophic lateral sclerosis : old strategies and new paradigms for the third millennium. CNS Drugs. 2003;17:699–717. doi: 10.2165/00023210-200317100-00002. [DOI] [PubMed] [Google Scholar]
- 6.Rothstein JD. Of mice and men: reconciling preclinical ALS mouse studies and human clinical trials. Ann Neurol. 2003;53:423–426. doi: 10.1002/ana.10561. [DOI] [PubMed] [Google Scholar]
- 7.Ryu H, Smith K, Camelo SI, Lee J, Iglesias AH, Dangond F, Cormier KA, Cudkowicz ME, Brown RH, Jr, Ferrante RJ. Sodium phenylbutyrate prolongs survival and regulates expression of anti-apoptotic genes in transgenic amyotrophic lateral sclerosis mice. J Neurochem. 2005;93:1087–1098. doi: 10.1111/j.1471-4159.2005.03077.x. [DOI] [PubMed] [Google Scholar]
- 8.Izecka J, Stelmasiak Z, Solski J, Wawrzycki S, Szpetnar M. Plasma amino acids concentration in amyotrophic lateral sclerosis patients. Amino Acids. 2003;25:69–73. doi: 10.1007/s00726-002-0352-2. [DOI] [PubMed] [Google Scholar]
- 9.Henderson CE, Camu W, Mettling C, Gouin A, Poulsen K, Karihaloo M, Rullamas J, Evans T, McMahon SB, Armanini MP. Neurotrophins promote motor neuron survival and are present in embryonic limb bud. Nature. 1993;363:266–270. doi: 10.1038/363266a0. [DOI] [PubMed] [Google Scholar]
- 10.Pennica D, Arce V, Swanson TA, Vejsada R, Pollock RA, Armanini M, Dudley K, Phillips HS, Rosenthal A, Kato AC, Henderson CE. Cardiotrophin-1, a cytokine present in embryonic muscle, supports long-term survival of spinal motoneurons. Neuron. 1996;17:63–74. doi: 10.1016/s0896-6273(00)80281-0. [DOI] [PubMed] [Google Scholar]
- 11.Russell AS, Ruegg UT. Arginase production by peritoneal macrophages: a new assay. J Immunol Methods. 1980;32:375–382. doi: 10.1016/0022-1759(80)90029-0. [DOI] [PubMed] [Google Scholar]
- 12.Veldink JH, Bar PR, Joosten EA, Otten M, Wokke JH, van den Berg LH. Sexual differences in onset of disease and response to exercise in a transgenic model of ALS. Neuromuscul Disord. 2003;13:737–743. doi: 10.1016/s0960-8966(03)00104-4. [DOI] [PubMed] [Google Scholar]
- 13.Napoli C, Williams-Ignarro S, de Nigris F, Lerman LO, Rossi L, Guarino C, Mansueto G, Di Tuoro F, Pignalosa O, De Rosa G, Sica V, Ignarro LJ. Long-term combined beneficial effects of physical training and metabolic treatment on atherosclerosis in hypercholesterolemic mice. Proc Natl Acad Sci USA. 2004;101:8797–8802. doi: 10.1073/pnas.0402734101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Napoli C, Williams-Ignarro S, de Nigris F, Lerman LO, D'Armiento FP, Crimi E, Byrns RE, Casamassimi A, Lanza A, Gombos F, Sica V, Ignarro LJ. Physical training and metabolic supplementation reduce spontaneous atherosclerotic plaque rupture and prolong survival in hypercholesterolemic mice. Proc Natl Acad Sci USA. 2006;103:10479–10484. doi: 10.1073/pnas.0602774103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dawson VL, Dawson TM, London ED, Bredt DS, Snyder SH. Nitric oxide mediates glutamate neurotoxicity in primary cortical cultures. Proc Natl Acad Sci USA. 1991;88:6368–6371. doi: 10.1073/pnas.88.14.6368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Esch F, Lin KI, Hills A, Zaman K, Baraban JM, Chatterjee S, Rubin L, Ash DE, Ratan RR. Purification of a multipotent antideath activity from bovine liver and its identification as arginase: nitric oxide-independent inhibition of neuronal apoptosis. J Neurosci. 1998;18:4083–4095. doi: 10.1523/JNEUROSCI.18-11-04083.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Plaitakis A, Constantakakis E, Smith J. The neuroexcitotoxic amino acids glutamate and aspartate are altered in the spinal cord and brain in amyotrophic lateral sclerosis. Ann Neurol. 1988;24:446–449. doi: 10.1002/ana.410240314. [DOI] [PubMed] [Google Scholar]
- 18.Urushitani M, Kurisu J, Tsukita K, Takahashi R. Proteasomal inhibition by misfolded mutant superoxide dismutase 1 induces selective motor neuron death in familial amyotrophic lateral sclerosis. J Neurochem. 2002;83:1030–1042. doi: 10.1046/j.1471-4159.2002.01211.x. [DOI] [PubMed] [Google Scholar]
- 19.Rothstein JD, Van Kammen M, Levey AI, Martin LJ, Kuncl RW. Selective loss of glial glutamate transporter GLT-1 in amyotrophic lateral sclerosis. Ann Neurol. 1995;38:73–84. doi: 10.1002/ana.410380114. [DOI] [PubMed] [Google Scholar]
- 20.Gurney ME, Cutting FB, Zhai P. Pathogenic mechanisms in familial amyotrophic lateral sclerosis due to mutation of Cu, Zn superoxide dismutase. Pathol Biol. 1996;44:51–56. [PubMed] [Google Scholar]
- 21.McCormack SA, Johnson LJ. Role of polyamines in gastrointestinal mucosal growth. Am J Physiol Gastrointest Liver Physiol. 1991;260:G795–G806. doi: 10.1152/ajpgi.1991.260.6.G795. [DOI] [PubMed] [Google Scholar]
- 22.Tabor CW, Tabor H. Polyamines. Annu Rev Biochem. 1984;53:749–790. doi: 10.1146/annurev.bi.53.070184.003533. [DOI] [PubMed] [Google Scholar]
- 23.Cai D, Deng K, Mellado W, Lee J, Ratan RR, Filbin M. Arginase I and polyamines act downstream from cyclic AMP in overcoming inhibition of axonal growth MAG and myelin in vitro. Neuron. 2002;35:711–719. doi: 10.1016/s0896-6273(02)00826-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.