

Abstract
A new method for rapid proteolytic digestion of proteins under high pressure that uses pressure cycling technology in the range of 5 to 35 kpsi was demonstrated for proteomic analysis. Successful in-solution digestions of single proteins and complex protein mixtures were achieved in 60 s and then analyzed by reversed phase liquid chromatography-electrospray ionization ion trap-mass spectrometry. Method performance in terms of the number of Shewanella oneidensis peptides and proteins identified in a shotgun approach was evaluated relative to a traditional “overnight” sample preparation method. Advantages of the new method include greatly simplified sample processing, easy implementation, no cross contamination among samples, and cost effectiveness.
Introduction
“Omics” technologies, such as genomics, proteomics, metabolomics, etc., have not only provided information needed to understand relationships and interactions in biological systems but have also contributed greatly to the advancement of clinical and biotechnological sample processing1,2. Key in the advancement of these disciplines is a reduction in analysis time. An increase in the throughput of sample processing and analysis directly translates into increased experimental success in terms of depth of knowledge gained and decreased costs, which are important to both academic and industrial realms.
Mass spectrometry (MS)-based proteomics is a powerful technique for characterizing individual proteins or very complex protein mixtures, such as whole cell lysates. The integration of several key elements, such as improved mass spectrometry (MS) instrumentation3-5, multidimensional chromatographic separations6, computational biology, and signal processing applied to MS data7 has resulted in increasingly fast proteomics analyses and data processing; however, sample preparation times can still remain a significant bottleneck in the analysis pipeline. While several sample preparation schemes have been used in an attempt to increase throughput8, none have been adopted with universal appeal. Traditional strategies include the enzymatic digestion of proteins either in-solution or from gel spots after polyacrylamide gel electrophoresis (PAGE). In both strategies, the enzymatic digestion of the proteins is a critical and often times a time consuming step.
Protein digestion has traditionally been performed using serine protease trypsin in a buffered medium over a defined length of time, generally overnight (~12 h), which makes protein digestion one of the most time consuming steps in the proteome analysis workflow. The success of a trypsin digestion is primarily defined by the time it takes to obtain a complete and accurate proteolysis and the selectivity of the enzyme to access the reactive amino acid cleavage sites. Enzymatic reactions rates strongly depend on many environmental factors, including temperature, solvents used, pH range, and enzyme-to-substrate ratio. With regard to temperature, higher temperatures usually facilitate faster reaction rates; that is until the temperature is so high that it denatures the active site of the enzyme. Attempts at creating a more thermo-stable trypsin, in particular, are continuously under study in hopes of accelerating its digestion efficiency9 in proteomics.
For in-solution digestions, an increase in temperature may not be ideal since the heat can cause some proteins to aggregate and precipitate. Even if higher temperatures are avoided, a means of denaturing the proteins in order for trypsin to have access to the active cleavage sites on the proteins is still necessary, and this has been traditionally achieved by the use of chaotropes and/or surfactants. However, the use of these chemical denaturants can present their own challenges, including the inactivation of enzyme activity or incompatibility with downstream MS analysis. For example, when urea is used as chaotrope, the formation of ammonium cyanate can result in the carbamylation of free amine groups on the proteins and peptides10.
Several works have been reported in which chemical denaturants were replaced by solvent assisted digestions11. The use of mixed-solvent buffers containing various concentrations of organic solvent (i.e., methanol, acetonitrile or isopropanol) not only resulted in protein denaturation, but also in better solubilization of hydrophobic proteins12. Because these mixed aqueous-organic buffer systems can affect enzymatic properties (due to the distortion of the protein solvation shell by the organic solvent which can positively affect protein stability), optimizing these buffers has resulted in the achievement of higher efficiencies in the proteolytic process in terms of more peptide identifications and shorter reaction times11,13,14. Another important factor that dictates enzyme reaction rates is the protease:substrate ratio. Higher enzyme concentrations produce faster reaction times, but at the expenses of increasing the concentration of unwanted peptides produced by autolysis, which ultimately results in a loss of sensitivity.
Combined optimization of the aforementioned parameters has proven to decrease digestion times9. Havlis et al. developed a procedure to reduce the trypsin digestion time to only 30 min for proteins isolated by gel electrophoresis. The procedure made use of a modified trypsin and was based on: 1) a higher trypsin concentration in the digestion buffer, 2) an increase in the digestion temperature, which was performed at 57 °C (due to the use of a thermo-stable trypsin) instead of the more typical 30-37°C, and 3) the addition of organic solvents, which helped the enzyme gain access to the substrate cleavage sites.
More recently, alternative energy inputs have been applied to digestions to further increase enzyme reaction rates. One alternative method involves the use of microwave energy, which hastens enzymatic digestions to 3-5 min. This approach, referred to as microwave-assisted protein enzymatic digestion (MAPED) was useful for either in-solution15 or in-gel16 digestions and was shown to be applicable complex protein mixtures17. More recently, a new method18 applies high intensity focused ultrasound (HIFU) to small sample volumes (typically 20-50 μL) of protein in the presence of trypsin for ultra fast protein digestion of either in-solution or in-gel protein samples. HIFU treatment was believed to act by boosting the enzyme-substrate kinetics due to the cavitation effects produced during the ultrasound irradiation19. In-gel and in-solution protein digestions, using HIFU were achieved in 15 s to 30 s.
Considering the recent advances in sample preparation, particularly MAPED and HIFU, one optimization parameter that has yet to be fully studied is the effect of pressure on enzymatic activity. High pressure processing (HPP) has been a well known and established technology for many applications in the food and biotechnology industries. The majority of the work published in this area has been related to pressure food processing to inactivate food pathogens and inhibit those enzymes that degrade food20. In addition, substantial evidence exists to suggest that low to moderate pressures (e.g., 100 to 400 MPa) can activate enzyme activity21, whereas higher pressures inactivate the same enzymes.
In the study reported herein, we used these premises as the basis for applying pressure cycling technology (PCT) to prepare samples for proteomics analysis. PCT has been used in sample preparation for many different analytical tasks and has been marketed as an efficient method for cell lysis22. In our study, we evaluated the performance of PCT as a means of enhancing enzyme activity. Using different organic solvent compositions, the samples were subjected to PCT at different pressures and cycle counts for 60 s. The pressurized trypsin digestion procedure was compared to a common overnight digestion to demonstrate proof of concept in a shotgun proteomics analysis.
Materials and Methods
Materials and reagents
Sequencing grade trypsin was obtained from Promega (Madison,WI). Bovine serum albumin (BSA), myoglobin, urea, dithiothretiol (DTT), iodoacetamide (IAA), ammonium bicarbonate (Ambic), formic acid, and HPLC grade solvents were purchased from Sigma-Aldrich (St. Louis, MO). 0.1 mm zirconia/silica beads were ordered from Biospec (Bartlesville, OK).
In-solution digestions
Bovine serum albumin was used as a standard protein to evaluate the method under different conditions. First, 6 mg of BSA was denatured in 8 M urea and reduced with 10 mM DTT in 25 mM ammonium bicarbonate (pH 8.25) at 37 °C for 1 h. Iodoacetamide was added to a final concentration of 50 mM, and the resulting mixture was incubated at room temperature in the dark for 45 min. Twelve 50-μg aliquots were diluted 4 fold to reduce the urea concentration, using either 25 mM ammonium bicarbonate, 20% MeOH, or 80% MeOH. Trypsin was added (1:50 protease-to-protein ratio), to a final volume of 1.4 mL and the solutions were placed in pulse tubes. The Barocycler™ NEP-3229 instrument and disposable polypropylene PULSE tubes FT-500 were obtained from Pressure BioSciences (West Bridgewater, MA, USA) and were used for all experiments. The pulse tubes were subjected to the Barocycler™ program, using 4 or 8 pressure pulses for a total of 1 minute per run. Finally, the enzymatic digests were transferred to new centrifuge tubes, acidified, and frozen with liquid N2 to stop the reaction. The samples were then dried down by centrifugal evaporation and stored at -20 °C.
The Shewanella oneidensis, strain MR-1, whole cell protein tryptic digest was prepared as described elsewhere23 with minor modifications. Briefly, cells were lysed by bead beating, using 0.1 mm zirconia/silica beads in a mini-bead beater (Biospec, Bartlesville, OK) for 180 s at 4500 rpm. The lysate was collected and placed immediately on ice to inhibit proteolysis, then denatured with 8 M urea, 25 mM ammonium bicarbonate, 10 mM DTT, (pH 8), and incubated for 1 h at 37 °C. Iodoacetamide was added to a final concentration of 50 mM, and the resultant mixture was incubated for 45 min at room temperature in the dark. The mixture was diluted 4 fold, and following the addition of trypsin (1:50 protease-to-protein ratio), was incubated either overnight at 37 °C overnight or for 1 min using PCT at 35 kpsi.
A solution with a final concentration of 1 μM protein in 12.5 mM ammonium bicarbonate was prepared for the myoglobin experiments. Trypsin was added and the samples were digested (1:50 protease-to-protein ratio) during the pressure cycles in the Barocycler™.
MS analysis
For BSA analyses, 500 fmol of the protein digest was analyzed by LC-MS/MS. An Agilent HPLC-Chip system was coupled to a MSD Trap XCT Ultra ion trap (Agilent Technologies, Santa Clara, CA). The Agilent auto sampler was used to load the samples at 4 °C. Separations were performed using a 40-nL enrichment column and 43 mm × 75 μm analytical column packed with 5 μm ZORBAX 300SB C18 particles. A flow rate of 1 μL/min was employed for enrichment and 600 nL/min afterwards. Peptides were eluted using a 5 min gradient from 5% to 90% Solvent B (0.5% formic acid, 90% acetonitrile; Solvent A: 0.5% formic acid in water:acetonitrile 97:3), with a separation window of ~2 min. The total analysis time was 12 min. Each sample was analyzed in triplicate. To prevent cross contamination among different samples, a blank was run between each set of replicates.
The data were acquired in survey scans from 500 to 1600 amu (3 microscans) followed by five data dependent MS/MS scans, using an isolation width of 3 amu, a normalized collision energy of 35%, and a dynamic exclusion period of 2 min. MS/MS data were analyzed using Spectrum Mill software against an in-house FASTA database that contained S. oneidensis MR-1 and BSA proteins. Spectra that matched to BSA were manually verified.
For the complex protein mixture analysis, 2 μg of the S. oneidensis digest were analyzed using a custom-built capillary LC system coupled online to a linear ion trap mass spectrometer (LTQ; Thermo-Fisher, San Jose, A) with an in-house developed ESI source 23, 24. The LTQ mass spectrometer was operated in a data-dependent MS/MS mode (m/z 400-2,000), in which a full MS scan was followed by ten MS/MS scans, using a normalized collision energy of 35% with a dynamic exclusion of 1 min. Protein identification was carried out using SEQUEST to deduce protein sequences from the S. oneidensis MR-1 genome sequence 25. Database search parameters included a dynamic modification search (i.e., the presence and absence of the modification was searched) for Met oxidation and a static search (i.e., presence of the modification was searched only) for carbamidomethylation on Cys. Error rates for peptide identifications were calculated as reported previously26.
To study myoglobin folding, the protein was directly infused by a syringe pump (KD Scientific, Holliston, MA) at 1 μL/min either with or without previous pressure treatment, into an Agilent TOF MS through an ESI interface developed in house 27. MS data were recorded over an m/z range of 500-2500 at a scan rate of 1 scan/sec.
Results and Discussion
Effect of pressure treatment on in-solution enzymatic hydrolysis
PCT studies were performed using a Barocycler™ instrument, which utilizes changes in hydraulic pressure to manipulate the samples under analysis within a closed system (i.e., the Pulse Tube™). These tubes allow the volume of the sample to be altered during the pressure cycles by the movement of a small piston in each tube, which reacts to the pressure of hydraulic fluid (30% ethylene glycol) surrounding the Pulse Tube™. Initial studies focused on investigating the effect of pressure on enzymatic digestion. The behavior of enzymes under pressure is complicated by a wide range of experimental conditions that can affect the reaction. For this experiment we used BSA as a standard protein and trypsin as the proteolytic enzyme. BSA is a globular protein with a large number of disulfide bridges that provide a high level of stability to its tertiary structure, making it very resistant to unfolding and thus to protein digestion. To combat this resistance to unfolding, the BSA samples were reduced and alkylated. In addition all sample analyses were performed in a buffered media of ammonium bicarbonate at pH 8.1, which is optimal for trypsin activity in traditional (i.e., ambient pressure) protocols. Because several studies have shown at least a three-fold increase in enzyme activity, which corresponds to an increase in pressure of at least 30 fold, we first analyzed the effect of enzyme activity in terms of protein proteolytic products at 5, 10, 20, and 35 kpsi for 60 s at each pressure (Figure 1). Note that this range was chosen based on the operating pressures of the Barocycler™. Additionally, pressures above 40 kpsi tend to destabilize protein structures with a consequential loss of activity28.
Figure 1.

a) LC-MS/MS chromatograms of high pressure assisted digestions as a function of digestion pressure. High pressure in-solution digestions of BSA were performed at 5, 10, 20, and 35 kpsi . b) Histograms depict the number of unique identified peptides for each method.
The chromatograms in Figure 1a indicate that trypsin activity was not compromised at any of the pressures. However, on the basis of the number of identified peptides (Figure 1b), digestion was not as complete at 5 kpsi as those achieved at higher pressures, even though the chromatograms at 5, 10, and 20 kpsi are similar. Although chromatograms belonging to the 35 kpsi samples look significantly different as compare with the others. Manual inspection of the MS spectra showed the same peptides between chromatograms. When pressure was applied to solutions that contained BSA in the absence of trypsin, no protein degradation products were observed, which indicates that the pressure treatment itself did not cause protein fragmentation (data not shown).
Effect of pressure, static time, number of cycles and buffer composition using pressure cycling technology
PCT can disrupt tissues, cells, and cellular structures in buffers or other solutions22, as a result, it is frequently used for extracting proteins and nucleic acids in reaction tubes. Our goal was to determine if multiple pressure cycles would influence trypsin activity. To evaluate the influence of rapid cycling between high and low pressures, BSA was digested under pressure, using either 4 or 8 differential pressure cycles for a total of 60 s. To further analyze the combined effect of pressure in the presence of an organic solvent for a trypsin digestion, identical BSA protein aliquots were subjected to pressure-digestion at 35 kpsi in the presence of 1) ammonium bicarbonate, 2) an 80:20 (v/v) mixture of ammonium bicarbonate:methanol, and 3) a 20:80 (v/v) mixture of ammonium bicarbonate:methanol. Organic media have been widely used in proteomics applications involving enzymatic reactions11, 14, especially those applied to membrane proteins. The properties of enzymes in mixed organic-aqueous solvent systems are influenced by factors such as protein structure, presence of phase interfaces, dielectric constants, etc.; all of which contribute to the performance of an enzyme in its biocatalytic system.
The histogram in Figure 2a shows that nearly identical results in terms of the number of unique peptide identifications were obtained for samples digested in comparable digestion buffers, regardless of the number of cycles. The chromatographic profiles in Figure 2b are also very similar for comparable buffers. A comparison of these results obtained at cyclic pressures to those obtained at constant pressure revealed no significant differences, which suggests that at least for trypsin, there is no significant effect in activity due to the number of differential pressure cycles used with PCT during digestion. The number of identified peptides was not significantly different between aqueous (i.e., ammonium bicarbonate) and 20% methanol digestions for either the 4- and 8-cycle protocols (Figure 2a), although proteolysis in mixed organic-aqueous solvent systems is reportedly favored14. However, fewer peptides were identified for digestion in the 80% methanol. The chromatograms for this solvent condition exhibited profiles characteristic of an incomplete protein digestion, i.e., the whole protein or large peptide fragments elute late in the gradient.
Figure 2.

a) Unique peptides identified as a function of the number of PCT cycles performed at 35 kpsi and with different organic solvent compositions. Ammonium bicarbonate buffer digestions correspond to the diagonal lines; 20% methanol (v/v) digestions, to the vertical lines; and 80% methanol (v/v) digestions, to the solid white bars. b) Chromatograms corresponding to the LC-MS/MS of PCT assisted digestion performance as a function of the number of cycles and the organic content in the digestion buffer. 4- and 8-cycle high pressure in-solution digestions of BSA were performed, using ammonium bicarbonate buffer, 20% methanol, or 80% methanol.
As a general note, concentrated organic solvents typically enhance enzymatic digestions of proteins by changing and denaturing the sample proteins, which allows the enzyme to access protein cleavage sites. In some cases the thermo-stability of certain enzymes even increases when they are suspended in micro-aqueous organic solvents28.
Although trypsin digestions can be performed with high solvent concentrations, such as 60% methanol and 80% acetonitrile 14, our results obtained for a highly concentrated organic solvent (i.e., 80% methanol) at 35 kpsi showed a loss of enzymatic activity, most likely due to the denaturation of trypsin itself into a non-active conformation. Thus digestions in buffers with a high organic concentration at increased pressures should be avoided.
Application to complex protein mixtures using shotgun proteomics
We evaluated this novel method for ultra fast (1 min) protein digestion by applying PCT to a proteomics sample and then analyzing it using a shotgun proteomics approach. A total proteome extract from a preparation of S. oneidensis cells was separated into two identical aliquots, one of which was subjected to a PCT-assisted digestion at 35 kpsi for 8 cycles during a 60 s time frame and the other, to a trypsin digestion following the conventional overnight approach for comparative purposes. PCT conditions reflected the highest number of cycles and the highest pressure that the Barocycler™ is capable of operating that was shown previously to achieve a good trypsin activity. The digested peptide mixtures were analyzed by reversed phase HPLC using a 100 min gradient.
The total ion current chromatograms from the LC-MS/MS analyses of the trypsin digestion using the traditional method at typical ambient pressure and the PCT assisted digestion are provided in Figure 3 (a and b) for comparison. Note that the chromatograms display very similar intensity profiles in spite of different digestion reactions; however, the total number of identified peptides obtained using the pressure protocol is slightly higher (~10%) than that obtained by the conventional method (Figure 3c). The results from a more constrained study of the identified peptides, i.e., false discover rate (FDR) <1% showed that the number of peptides with more than one missed cleavage was much lower for the traditional protocol (Figure 3d), while approximately more than 95% of the PCT-assisted digested peptides had less than two missed cleavages. Nevertheless, both experiments had a wide overlap in terms of identified proteins, with the PCT-assisted digestion protocol producing more protein identifications (Figure 3e). The number of non-tryptic peptides was insignificant in both samples (Figure 3f) and within the error limits we set for identifying peptides (i.e., <1% FDR). The population of semi-tryptic peptides, defined as a peptide with one end that is not tryptic, is very similar for both digestions. This population may originate from protein degradation during sample processing, as no protease inhibitor cocktail was added. Additionally, it could be expected that natural protein degradation processes were occurring at the time of cell harvest or post-digestion trimming29. Our results confirm that the protease seems to cleave exclusively at the C-terminals of arginine and lysine residues, which is in agreement with Olsen et al.30
Figure 3.

Comparison of a traditional digestion protocol versus a PCT- assisted digestion. Panels a) and b) show chromatograms obtained from the LC-MS/MS analysis from traditional and PCT digestions, respectively. Panel c) compares the number of identified peptides in both samples at a given FDR following SEQUEST analysis. Filled circles correspond to the regular digestion protocol and empty circles to the PCT-assisted digestion protocol. d) Histogram showing the number of identified peptides with missed cleavages. Grey bars correspond to the traditional digestion protocol and black bars to the PCT-assisted digestion protocol. e) Overlap between the identified proteins. PCT-assisted (I) digestion protocol and the traditional (II) digestion protocol. f) Comparison of the different identified peptides in terms of trypsin specificity.
Finally, to test the hypothesis that a better digestion yield is due to the unfolding effect caused by pressure, myoglobin was dissolved in 12 mM ammonium bicarbonate and subjected to 10 kpsi for 60 s prior to direct infusion into the mass spectrometer. Myoglobin is an oxygen-binding protein in muscle cells that works in the storage and transport of oxygen. It is composed of a single chain of 123 amino acids and a heme group that is non-covalently attached. This protein natively displays a compact tertiary structure with the heme group embedded in the hydrophobic cavity of the folded amino acid chain. For comparative purposes, a native myoglobin protein sample not subjected to pressure treatment was also analyzed by TOF-MS. As shown in Figure 4, the spectra for pressure-treated and non-pressure-treated proteins taken at a neutral pH are completely different. Remarkably, the charge states corresponding to the native protein are much lower than those corresponding to the pressure treated protein. The native protein yielded the maximum signal at a MW of 17,568 Da, whereas the distribution of the charge states for the pressure-treated protein shifted to higher charge states, and the MW corresponded to 16,952 Da. This finding indicates a loss of the heme group (-616 Da) and complete denaturation of the protein due only to the pressure treatment; unfolding of the native protein permitted a reduction of charge-charge repulsion forces, which in turn allowed a larger degree of protonation.
Figure 4.
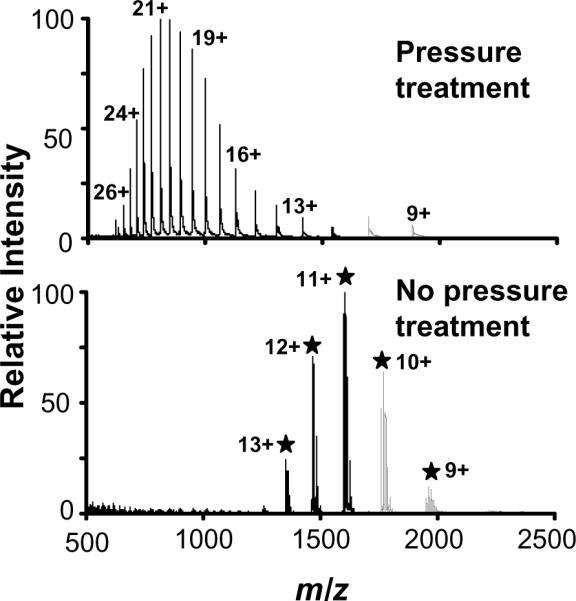
ESI mass spectra of horse myoglobin dissolved in 12.5 mM ammonium bicarbonate. The upper panel shows the pressure treated protein yielding denaturated horse myoglobin and elimination of the heme group. Bottom panel shows the native myoglobin without pressure treatment and the heme group attached.
Conclusions
The results shown in this work demonstrate how an increase in pressure can dramatically increase the rate of the enzymatic digestion of proteins in proteomic samples. PCT simplified sample preparation compared with other newer rapid digestion methods, such as MAPED and HIFU technologies. Among the advantages afforded by PCT are automated sample preparation, high sample throughput (up to three samples per minute in our setup) without compromising the digestion yields, high reproducibility, no aerosolization (a common effect that occurs when HIFU is applied), and the acquisition of results comparable to those obtained using regular digestion protocols but in a much shorter time-frame (i.e., 1 min). Since digestions can be completed at 20 °C, undesired protein modification can be avoided. PCT technology is reportedly effective for protein extraction from lipid-rich samples and cell membranes 31; as such, our laboratory is exploring new strategies for membrane proteomics using PCT without detergents. In spite of challenges that must be overcome, e.g., working with smaller volumes and processing samples more rapidly, this technology opens up new possibilities for sample preparation in proteomics applications in areas such as biotechnology and food and pharmaceutical science, to name a few, where high sample throughput is important.
Acknowledgements
The authors thank Dr. Natacha Lourette, David Prior, Karl Weitz, and Penny Colton for their helpful assistance and suggestions. Portions of this work were supported by the NIH National Center for Research Resources (RR018522), NIH National Cancer Institute (R21 CA12619-01), and the Pacific Northwest National Laboratory's (PNNL) Laboratory Directed Research and Development Program. PNNL is operated by Battelle for the U.S. Department of Energy through contract DE-AC05-76RLO1830.
Abbreviations
- ESI
Electrospray Ionization
- FDR
False Discovery Rate
- HIFU
High Intensity Focused Ultrasound
- HPP
High Pressure Processing
- IT
Ion trap
- MAPED
Microwave-assisted Protein Enzymatic Digestion
- MS
Mass Spectrometry
- MS/MS
Tandem Mass Spectrometry
- MW
Molecular Weight
- PCT
Pressure Cycling Technology
- RP
Reversed Phase
- TOF
Time-of-Flight
References
- 1.Fischer HP. Towards quantitative biology: integration of biological information to elucidate disease pathways and to guide drug discovery. Biotechnol. Annu. Rev. 2005;11:1–68. doi: 10.1016/S1387-2656(05)11001-1. [DOI] [PubMed] [Google Scholar]
- 2.Hood L, Perlmutter RM. The impact of systems approaches on biological problems in drug discovery. Nat. Biotechnol. 2004;22(10):1215–7. doi: 10.1038/nbt1004-1215. [DOI] [PubMed] [Google Scholar]
- 3.Blackler AR, Klammer AA, MacCoss MJ, Wu CC. Quantitative comparison of proteomic data quality between a 2D and 3D quadrupole ion trap. Anal. Chem. 2006;78(4):1337–44. doi: 10.1021/ac051486a. [DOI] [PubMed] [Google Scholar]
- 4.Denison C, Rudner AD, Gerber SA, Bakalarski CE, Moazed D, Gygi SP. A proteomic strategy for gaining insights into protein sumoylation in yeast. Mol. Cell. Proteomics. 2005;4(3):246–54. doi: 10.1074/mcp.M400154-MCP200. [DOI] [PubMed] [Google Scholar]
- 5.Hu Q, Noll RJ, Li H, Makarov A, Hardman M, Graham Cooks R. The Orbitrap: a new mass spectrometer. J. Mass. Spectrom. 2005;40(4):430–43. doi: 10.1002/jms.856. [DOI] [PubMed] [Google Scholar]
- 6.Washburn MP, Wolters D, Yates JR., 3rd Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nat. Biotechnol. 2001;19(3):242–7. doi: 10.1038/85686. [DOI] [PubMed] [Google Scholar]
- 7.Hernandez P, Muller M, Appel RD. Automated protein identification by tandem mass spectrometry: issues and strategies. Mass. Spectrom. Rev. 2006;25(2):235–54. doi: 10.1002/mas.20068. [DOI] [PubMed] [Google Scholar]
- 8.López-Ferrer D, Cañas B, Vázquez J, Lodeiro C, Rial-Otero R, Moura I, Capelo JL. Sample Treatment for Protein Identification by Mass Spectrometry-Based Techniques. Trends in Analyt. Chem. 2006;25:996–1005. [Google Scholar]
- 9.Havlis J, Thomas H, Sebela M, Shevchenko A. Fast-response proteomics by accelerated in-gel digestion of proteins. Anal. Chem. 2003;75(6):1300–6. doi: 10.1021/ac026136s. [DOI] [PubMed] [Google Scholar]
- 10.Stark GR, Stein WH, Moore S. Reactions of the Cyanate Present in Aqueous Urea with Amino Acids and Proteins. J. Biol. Chem. 1960;235(11):3177–3181. [Google Scholar]
- 11.Hervey W. J. t., Strader MB, Hurst GB. Comparison of Digestion Protocols for Microgram Quantities of Enriched Protein Samples. J. Proteome. Res. 2007;6(8):3054–3061. doi: 10.1021/pr070159b. [DOI] [PubMed] [Google Scholar]
- 12.Blonder J, Chan KC, Issaq HJ, Veenstra TD. Identification of membrane proteins from mammalian cell/tissue using methanol-facilitated solubilization and tryptic digestion coupled with 2D-LC-MS/MS. Nat. Protoc. 2006;1(6):2784–90. doi: 10.1038/nprot.2006.359. [DOI] [PubMed] [Google Scholar]
- 13.Chen EI, Cociorva D, Norris JL, Yates JR., 3rd Optimization of mass spectrometry-compatible surfactants for shotgun proteomics. J. Proteome. Res. 2007;6(7):2529–38. doi: 10.1021/pr060682a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Russell WK, Park ZY, Russell DH. Proteolysis in Mixed Organic-aqueous Solvent Systems: Applications for Peptide Mass Mapping Using Mass Spectrometry. Anal. Chem. 2001;73(11):2682–2685. doi: 10.1021/ac001332p. [DOI] [PubMed] [Google Scholar]
- 15.Pramanik BN, Mirza UA, Ing YH, Liu YH, Bartner PL, Weber PC, Bose AK. Microwave-enhanced enzyme reaction for protein mapping by mass spectrometry: a new approach to protein digestion in minutes. Protein Sci. 2002;11(11):2676–87. doi: 10.1110/ps.0213702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sun W, Gao S, Wang L, Chen Y, Wu S, Wang X, Zheng D, Gao Y. Microwave-assisted protein preparation and enzymatic digestion in proteomics. Mol. Cell. Proteomics. 2006;5(4):769–76. doi: 10.1074/mcp.T500022-MCP200. [DOI] [PubMed] [Google Scholar]
- 17.Zhong H, Zhang Y, Wen Z, Li L. Protein sequencing by mass analysis of polypeptide ladders after controlled protein hydrolysis. Nat. Biotechnol. 2004;22(10):1291–6. doi: 10.1038/nbt1011. [DOI] [PubMed] [Google Scholar]
- 18.Lopez-Ferrer D, Capelo JL, Vazquez J. Ultra fast trypsin digestion of proteins by high intensity focused ultrasound. J. Proteome. Res. 2005;4(5):1569–74. doi: 10.1021/pr050112v. [DOI] [PubMed] [Google Scholar]
- 19.Postema M, van Wamel A, Lancee CT, de Jong N. Ultrasound-induced encapsulated microbubble phenomena. Ultrasound Med. Biol. 2004;30(6):827–40. doi: 10.1016/j.ultrasmedbio.2004.02.010. [DOI] [PubMed] [Google Scholar]
- 20.Cheftel JC. Effects of High Hydrostatic Pressure on Food Constituents: An Overview. In: Balny C, Hayashi R, Heremans K, Masson P, editors. High Pressure and Biotechnology. John Libbey Eurotext, Ltd.; London, U.K.: 1992. pp. 195–209. [Google Scholar]
- 21.Cano MP, Hernandez A, DeAncos B. High Pressure and Temperature Effects on Enzyme Inactivation in Strawberry and Orange Products. J. Food Sci. 1997;62(1):85–88. [Google Scholar]
- 22.Smejkal GB, Witzmann FA, Ringham H, Small D, Chase SF, Behnke J, Ting E. Sample preparation for two-dimensional gel electrophoresis using pressure cycling technology. Anal. Biochem. 2007;363(2):309–11. doi: 10.1016/j.ab.2007.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shen Y, Zhao R, Belov ME, Conrads TP, Anderson GA, Tang K, Pasa-Tolic L, Veenstra TD, Lipton MS, Smith RD. Packed Capillary Reversed-Phase Liquid Chromotaography with High-Performance Electrospray Ionization Fourier Transform Ion Cyclotron Resonance Mass Spectrometry for Proteomics. Anal. Chem. 2001;73(8):1766–1775. doi: 10.1021/ac0011336. [DOI] [PubMed] [Google Scholar]
- 24.Wang H, Qian WJ, Chin MH, Petyuk VA, Barry RC, Liu T, Gritsenko MA, Mottaz HM, Moore RJ, Camp Ii DG, Khan AH, Smith DJ, Smith RD. Characterization of the mouse brain proteome using global proteomic analysis complemented with cysteinyl-peptide enrichment. J. Proteome. Res. 2006;5(2):361–9. doi: 10.1021/pr0503681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Elias DA, Monroe ME, Smith RD, Fredrickson JK, Lipton MS. Confirmation of the expression of a large set of conserved hypothetical proteins in Shewanella oneidensis MR-1. J. Microbiol. Methods. 2006;66(2):223–33. doi: 10.1016/j.mimet.2005.11.009. [DOI] [PubMed] [Google Scholar]
- 26.Lopez-Ferrer D, Martinez-Bartolome S, Villar M, Campillos M, Martin-Maroto F, Vazquez J. Statistical model for large-scale peptide identification in databases from tandem mass spectra using SEQUEST. Anal. Chem. 2004;76(23):6853–60. doi: 10.1021/ac049305c. [DOI] [PubMed] [Google Scholar]
- 27.Ding J, Sorensen CM, Zhang Q, Jiang H, Jaitly N, Livesay EA, Shen Y, Smith RD, Metz TO. Capillary LC Coupled with High-Mass Measurement Accuracy Mass Spectrometry for Metabolic Profiling. Anal. Chem. 2007;79(16):6081–93. doi: 10.1021/ac070080q. [DOI] [PubMed] [Google Scholar]
- 28.Adams MW, Kelly RM. Biocatalysis at Extreme Temperatures; Enzyme Systems Near and Above 100 °C. (Series 498).ACS Symposium. 1991:86–107. 1991. [Google Scholar]
- 29.Gupta N, Tanner S, Jaitly N, Adkins JN, Lipton M, Edwards R, Romine M, Osterman A, Bafna V, Smith RD, Pevzner PA. Whole proteome analysis of post-translational modifications: Applications of mass-spectrometry for proteogenomic annotation. Genome Res. 2007 doi: 10.1101/gr.6427907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Olsen JV, Ong SE, Mann M. Trypsin Cleaves Exclusively C-terminal to Arginine and Lysine Residues. Mol. Cell. Proteomics. 2004;3(6):608–614. doi: 10.1074/mcp.T400003-MCP200. [DOI] [PubMed] [Google Scholar]
- 31.Ringham H, Bell RL, Smejkal GB, Behnke J, Witzmann FA. Application of pressure cycling technology to tissue sample preparation for 2-DE. Electrophoresis. 2007;28(6):1022–4. doi: 10.1002/elps.200600434. [DOI] [PubMed] [Google Scholar]