

Abstract
Background
We sought to identify two sets of familial/genetic risk factors for major depression (MD): 1) high familial loading for MD, which we predicted would be most prominent in cases of MD with an early age at onset (AAO), and 2) high familial loading for vascular disease (VD), which should be strongest in MD cases with a late AAO.
Methods
We examined 4785 twin pairs from the Swedish Twin Registry, assessed at a mean age of 54.0 (SD = 7.4), where both members of the pair were evaluated by interview and at least one member reported a lifetime history of modified DSM-IV MD. Risk for VD was assessed from hospital discharge information and death certificates.
Results
Using Cox proportional hazard models and controlling for zygosity, age, and sex, early AAO in depressed twins predicted risk for MD in their cotwins, whereas late AAO predicted cotwin risk for VD. Using piecewise regression, the hazard ratio (HR) relating AAO per decade to risk for MD in cotwin was much stronger for AAO from 13–23 years (HR = .62) than for AAO 24 – 65 years (HR = 0.94). The HR relating AAO of MD in twin and risk for VD in cotwin was twice as strong for AAO from 47– 65 years (HR = 1.17) as for AAO 13– 46 years (1.08).
Conclusions
From a familial/genetic perspective, MD is etiologically heterogeneous. Early and late onset MD are indexed, respectively, by the risk for MD and VD in relatives.
Keywords: Age at onset, depression, genetics, heterogeneity, twin study, vascular disease
Major depression is a familial and moderately heritable multifactorial disorder generally believed to result from a wide range of genetic and environmental risk factors (1–5). Efforts are actively underway to detect variants in specific genes that alter risk for MD (6,7). These genes are likely to reflect diverse biological functions (8).
This report addresses the question of whether it is possible, using a genetically informative twin design, to detect two distinct sets of familial risk factors for MD. We know that the risk for MD is substantially increased in those with relatives also affected with MD and that most, or all, of this risk is genetically transmitted (1). There is also good evidence that individuals with an early age at onset (AAO) of MD are particularly likely to have a strong family history for depression (1,9,10).
Another etiologic pathway to MD is through vascular disease (VD). A range of studies have suggested a possible relationship between risk for MD and risk for various forms of VD (11–13). Of particular interest has been the concept of “vascular depression,” which tends to have a late AAO and is associated with ischemic brain lesions (14–18). The risk for VD is itself strongly influenced by genetic factors (19–21).
We evaluated the hypothesis that two sets of familial risk factors for MD can be identified: 1) high familial loading for MD, which will be most prominent in cases of MD with an early AAO, and 2) high familial loading for VD, which will be pronounced in cases of MD with a late AAO. We examined this question in 4785 twin pairs from the Swedish Twin Registry, assessed at a mean age of 54.0 (SD = 7.4). In these pairs, at least one member reported a lifetime history of MD. Their cotwin was evaluated at personal interview for a history of MD, and their risk for VD was assessed from hospital discharge information and death certificates.
Methods and Materials
Sample
The sample for this study came from the Swedish Twin Registry (22), which includes all like-sex twins born in Sweden between 1886 and 1904, and all twin pairs from 1904 to 1958. The Screening Across the Lifespan Twin (SALT) study completed phone interviews between March 1998 and January 2003 with all cooperative, living members of the Registry born in or before 1958. According to standard Swedish practice, a letter about the study was sent before the telephone call, and informed verbal consent was obtained prior to interview. The project was approved by the Swedish Data Inspection Authority, the Ethics Committee of the Karolinska Institute, and the Institutional Review Board of the University of Southern California. The participation rate was 65% for twins born 1886–1925 and 74% for twins born 1926–1958.
The sample contained 32,168 individuals who were assessed, of whom 6368 were positive for a lifetime diagnosis of MD. In 6093 of these individuals, we had valid information about AAO. To avoid influential extreme values, we restricted our analyses to the 95.6% of MD cases in our sample with onsets from the ages of 13–65. Our Cox models, which required complete information from both members of the pair, were fitted to 5604 MD cases and their cotwins. This sample, made up of 4785 twin pairs, 819 of whom were concordant for lifetime MD, was 57.2% female, and 26% came from monozygotic pairs.
In the first phase of this study involving 19% of the sample, age at worst episode was recorded instead of age at first episode. Of that subgroup, 39% who experienced a lifetime history of MD reported multiple episodes. Thus, in 7% (.39 × .19) of this sample, we may have overestimated AAO by assuming that the worst episode was the first. We repeated our key analyses eliminating these individuals and the pattern of findings was similar.
Sources of Data
Information for the current study was obtained from the SALT interview and linkage to two national registers: the Inpatient Discharge Register (IDR) and the Cause of Death Register (COD). The IDR, established in 1964 on a regional basis and expanded until complete coverage was attained by 1987, contains primary and secondary hospital discharge diagnoses for all public hospitals in Sweden. The IDR uses the International Classification of Diseases (23) coding system in use at the time of the diagnosis. The IDR also contains surgical procedure codes from the Classification of Operations (versions 1–7; 1964–1996) and the Swedish Version of the Nordic Medico-Statistical Committee (NOMESCO) Classification of Surgical Procedures (Version 1.9; in use since 1997, revised 2004) (24). The COD, in place since 1749, is a nationwide register containing underlying and contributing causes of death classified by International Classification of Diseases (ICD).
As detailed elsewhere (25), a modified version of MD as defined by DSM-IV (26), was assessed in the SALT telephone interview using the Composite International Diagnostic Interview—Short Form (CIDI-SF) (26,27). We used the recommended cutoff of four or more of the eight assessed criteria, as well as a small number of subjects who skipped out of the section because they volunteered for antidepressant usage, shown in this sample to be a valid substitute for an MD diagnosis.
Information on VD was obtained from diagnoses and surgical codes in the IDR and cause of death codes in the COD register, as reflected in the occurrence of two common atherosclerotic diseases: coronary artery disease and nonhemorrhagic stroke. The following ICD-10 (23) diagnoses (and equivalent diagnoses from earlier ICD versions, except as noted) were considered indicative of coronary artery disease: myocardial infarct (acute or subsequent; including old MI in ICD-9) (28), angina pectoris, other acute ischaemic heart disease, chronic ischemic heart disease (including coronary atherosclerosis [ICD-9] and asymptomatic ischemic heart disease [ICD-8] (29) excluding complications following acute MI [ICD-10] or aneurism and dissection of heart [ICD-9]).
Surgical procedures that were viewed as indicative of coronary artery disease included coronary artery bypass graft, coronary thrombendarterectomy, and all types of expansion and recanalization of the coronary artery (dilatation, percutaneous transluminal coronary angioplasty with or without stent, embolectomy, removal of foreign body from coronary artery, expansion using patch, and other recanalization). Excluded were repair of the coronary artery, closure of coronary fistula, and other operations on coronary arteries. Twins were classified as having had a nonhemorrhagic stroke if they had one of the following diagnoses in either the IDR or the COD register: cerebral embolism or cerebral thrombosis (ICD-7 [30] and ICD-8), occlusion of cerebral arteries (ICD-9) or cerebral infarction (ICD-10).
Statistical Methods
The risk for MD or VD in cotwins of the twins selected for their lifetime history of MD was assessed using Cox proportional hazards models (31). The AAO of MD in the twin was the major variable of interest, with sex of the cotwin and the pair zygosity included as covariates. The analysis was stratified into 5-year blocks based on birth year to adjust for cohort effects. Thus, with respect to birth year, our analyses, based on a series of 5-year blocks, in effect compared “like with like.” Adjustments were made for the effect of clustered data within twin pairs on standard errors and hypothesis tests using the method of Binder (32). Piecewise models with a single inflection point were developed using a grid search to find the single inflection point that maximized the −2 log likelihood of the model. For purposes of plotting the hazard ratios for MD, AAO was calculated relative to onset at age 40, the approximate mean AAO in our sample.
Results
Using a Cox regression model, we first predicted the hazard ratio (HR) for MD in the cotwin from a linear effect of AAO of MD (per decade) in the twin (HR = .90, χ2 = 20.6, p < .0001) controlling for zygosity (HR = 1.50, χ2 = 39.7, p < .0001; risk higher in MZ cotwin) and sex (HR = 2.06, χ2 = 147.7, p < .0001; risk higher in female cotwin). We then fit a piecewise regression model in which the optimal fit divided the sample into those with an AAO of 13–23 and 24–65 years. The HR relating AAO of MD in twin and risk for MD in cotwin was much stronger for AAO from 13 to 23 years (HR = 0.62) than for AAO of 24 to 65 years (HR = .94; Figure 1).
Figure 1.
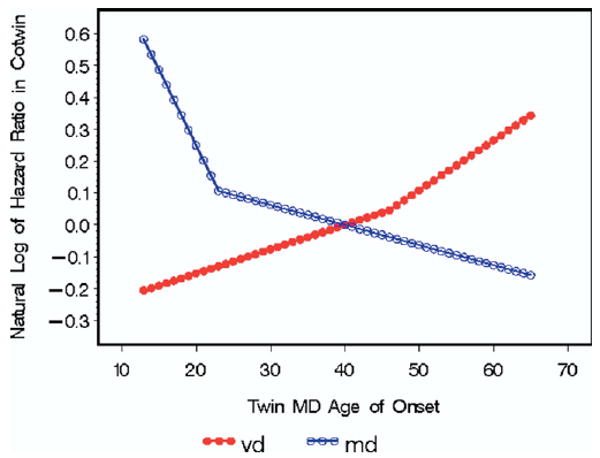
The relationship between the age at onset of major depression (MD) in an affected twin and the natural logarithm of the hazard ratio in the cotwin for MD (in open circles) and vascular disease (VD) (in filled-in circles). These results are obtained from a Cox proportional hazard model controlling for age, sex, and birth cohort. We fitted to these results piecewise models with a single inflection point using a grid search to find the single inflection point that maximized the model's −2 log likelihood.
Using the same statistical approach, we then predicted the HR for VD in the cotwin from a linear effect of AAO of MD in the twin (HR = 1.11, χ2 = 6.95, p < .008) controlling for zygosity (HR = .95, χ2 = .02, p = .69) and sex (HR = .55, χ2 = 37.1, p < .0001; risk higher in male cotwin). With a piecewise regression, the optimal fit divided the sample into those with an AAO of 13–46 and 47–65 years. The HR relating AAO of MD in twin and risk for VD in cotwin was approximately twice as strong for AAO from 47 to 65 years (HR = 1.17) than for AAO 13 to 46 years (1.08; Figure 1).
Discussion
This study was intended to detect evidence for two distinct familial pathways to MD. We hypothesized that the first pathway would be especially marked in those with an early AAO for MD and would be reflected in an increased risk for MD in relatives. The second hypothesized pathway would be prominent in those with a late AAO for MD and would be indexed by an increased risk for VD in relatives. Our results were supportive of these hypotheses. Consistent with prior reports in the literature (1,9) as well as prior analyses of the SALT data (10) (which differed from the current report in using a more restricted sample and different statistical methods), we found, in a large epidemiologic sample of Swedish twins in middle to late adult life who reported a lifetime history of MD, that risk for MD in the cotwin was significantly and inversely related to the AAO of MD in the twin. That is, the familial/genetic factors that were reflected in the risk for MD in relatives particularly manifest themselves in early-onset forms of MD. Of special interest, and examining this for the first time to our knowledge, we found a significant positive relationship between AAO in depressed twins and the risk for objectively determined VD in their cotwin. That is, the familial/genetic factors that were reflected in the risk for VD in relatives particularly manifest themselves in late-onset forms of MD.
Furthermore, when we determined the optimal piecewise regression model for each of these relationships, the relationship between AAO and risk for MD in the cotwin fell relatively steeply from ages 13 to 23 years and then declined at a much more gradual pace. In this sample, onsets of MD in adolescence and early adulthood carry particularly high familial loading for MD. We found an opposite pattern in the relationship between AAO for MD and risk for VD in the cotwin. An increase in AAO for MD from 13 to 46 years was accompanied by a moderate change in risk for VD in the cotwin. After age 46, the relationship between increasing AAO and elevated risk for VD in the cotwin approximately doubled. Consistent with the concept of vascular depression (14), onsets of MD in this sample from the late 40s onward was associated with particularly elevated familial risk for VD.
These findings have two implications for further investigations into the genetics of MD. First, they provide firm evidence in support of the commonly held view that MD is etiologically and genetically heterogeneous. Second, these results indicate that this familial/genetic heterogeneity can, with some power, be indexed by AAO. Our results would predict that interactions would be seen between AAO and various molecular variants in predicting risk for MD. This interaction would be negative if the variant reflected familial risk for MD and positive if it reflected familial risk for VD.
Limitations
These results should be interpreted in the context of three potential methodologic limitations. First, results are limited to Swedish twins and may not extrapolate to other ethnic groups. Second, although we were confident about the completeness and objectivity of our VD data, all information about MD was based on personal interviews, which, in community samples, have only moderate reliability (33,34). Third, despite our large total sample size, we did not have sufficient power to discriminate the relationship between AAO of MD and risk in cotwin for MD and VD in MZ versus DZ twins to determine whether it resulted from genetic or familial-environmental factors. Given prior evidence from this sample that twin resemblance for both MD (25) and cardiovascular illness (19; K.S. Kendler, et al., unpublished data, 2008) results entirely from genetic factors, we suspect that genetic mechanisms are at work. Fourth, most editions of the ICD codes do not permit the separation of thrombotic from embolic stroke. Whereas the former arises from VD, the latter has heterogeneous etiologies some of which are unrelated to VD.
Acknowledgments
This work was supported in part by National Institute of Health Grant Nos. MH-49492 and AG-08724, the Swedish Scientific Council, and the Swedish Department of Higher Education. We thank Ulf de Faire, M.D., and Helena Chui, M.D., for consultation on the system of coding coronary artery disease and nonhemorrhagic stroke, respectively, used in this study.
Footnotes
Drs. Kendler, Fiske, Gardner and Gatz report no biomedical financial interests or potential conflicts of interest.
References
- 1.Sullivan PF, Neale MC, Kendler KS. Genetic epidemiology of major depression: review and meta-analysis. Am J Psychiatry. 2000;157:1552–1562. doi: 10.1176/appi.ajp.157.10.1552. [DOI] [PubMed] [Google Scholar]
- 2.Akiskal HS, McKinney WT., Jr Depressive disorders: Toward a unified hypothesis. Science. 1973;182:20–29. doi: 10.1126/science.182.4107.20. [DOI] [PubMed] [Google Scholar]
- 3.Whybrow P, Akiskal H, McKinney WT., Jr . Mood Disorders: Toward a Psychobiology. New York: Plenum Press; 1984. Toward a psychobiological integration; pp. 173–203. [Google Scholar]
- 4.Kendler KS, Gardner CO, Prescott CA. Toward a comprehensive developmental model for major depression in women. Am J Psychiatry. 2002;159:1133–1145. doi: 10.1176/appi.ajp.159.7.1133. [DOI] [PubMed] [Google Scholar]
- 5.Krishnan V, Nestler EJ. The molecular neurobiology of depression. Nature. 2008;455:894–902. doi: 10.1038/nature07455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Levinson DF, Evgrafov OV, Knowles JA, Potash JB, Weissman MM, Scheftner WA, et al. Genetics of recurrent early-onset major depression (GenRED): Significant linkage on chromosome 15q25-q26 after fine mapping with single nucleotide polymorphism markers. Am J Psychiatry. 2007;164:259–264. doi: 10.1176/ajp.2007.164.2.259. [DOI] [PubMed] [Google Scholar]
- 7.Boomsma DI, Willemsen G, Sullivan PF, Heutink P, Meijer P, Sondervan D, et al. Genome-wide association of major depression: description of samples for the GAIN Major Depressive Disorder Study: NTR and NESDA biobank projects. Eur J Hum Genet. 2008;16:335–342. doi: 10.1038/sj.ejhg.5201979. [DOI] [PubMed] [Google Scholar]
- 8.Levinson DF. The genetics of depression: A review. Biol Psychiatry. 2006;60:84–92. doi: 10.1016/j.biopsych.2005.08.024. [DOI] [PubMed] [Google Scholar]
- 9.Weissman MM, Wickramaratne P, Merikangas KR, Leckman JF, Prusoff BA, Caruso KA, et al. Onset of major depression in early adulthood. Increased familial loading and specificity. Arch Gen Psychiatry. 1984;41:1136–1143. doi: 10.1001/archpsyc.1984.01790230022003. [DOI] [PubMed] [Google Scholar]
- 10.Kendler KS, Gatz M, Gardner CO, Pedersen NL. Age at onset and familial risk for major depression in a Swedish national twin sample. Psychol Med. 2005;35:1573–1579. doi: 10.1017/S0033291705005714. [DOI] [PubMed] [Google Scholar]
- 11.Van der KK, van Hout H, Marwijk H, Marten H, Stehouwer C, Beekman A. Depression and the risk for cardiovascular diseases: Systematic review and meta analysis. Int J Geriatr Psychiatry. 2007;22:613–626. doi: 10.1002/gps.1723. [DOI] [PubMed] [Google Scholar]
- 12.Jiang W, Glassman A, Krishnan R, O'Connor CM, Califf RM. Depression and ischemic heart disease: What have we learned so far and what must we do in the future? Am Heart J. 2005;150:54–78. doi: 10.1016/j.ahj.2005.04.012. [DOI] [PubMed] [Google Scholar]
- 13.Newberg AR, Davydow DS, Lee HB. Cerebrovascular disease basis of depression: Post-stroke depression and vascular depression. Int Rev Psychiatry. 2006;18:433–441. doi: 10.1080/09540260600935447. [DOI] [PubMed] [Google Scholar]
- 14.Alexopoulos GS, Meyers BS, Young RC, Campbell S, Silbersweig D, Charlson M. “Vascular depression” hypothesis. Arch Gen Psychiatry. 1997;54:915–922. doi: 10.1001/archpsyc.1997.01830220033006. [DOI] [PubMed] [Google Scholar]
- 15.de Groot JC, de Leeuw FE, Oudkerk M, Hofman A, Jolles J, Breteler MMB. Cerebral white matter lesions and depressive symptoms in elderly adults. Arch Gen Psychiatry. 2000;57:1071–1076. doi: 10.1001/archpsyc.57.11.1071. [DOI] [PubMed] [Google Scholar]
- 16.Kim JM, Stewart R, Kim SW, Yang SJ, Shin IS, Yoon JS. Vascular risk factors and incident late-life depression in a Korean population. Br J Psychiatry. 2006;189:26–30. doi: 10.1192/bjp.bp.105.015032. [DOI] [PubMed] [Google Scholar]
- 17.Thomas AJ, O'Brien JT, Davis S, Ballard C, Barber R, Kalaria RN, Perry RH. Ischemic basis for deep white matter hyperintensities in major depression: A neuropathological study. Arch Gen Psychiatry. 2002;59:785–792. doi: 10.1001/archpsyc.59.9.785. [DOI] [PubMed] [Google Scholar]
- 18.Salloway S, Malloy P, Kohn R, Gillard E, Duffy J, Rogg J, et al. MRI and neuropsychological differences in early- and late-life-onset geriatric depression. Neurology. 1996;46:1567–1574. doi: 10.1212/wnl.46.6.1567. [DOI] [PubMed] [Google Scholar]
- 19.Zdravkovic S, Wienke A, Pedersen NL, de Faire U. Genetic susceptibility of myocardial infarction. Twin Res Hum Genet. 2007;10:848–852. doi: 10.1375/twin.10.6.848. [DOI] [PubMed] [Google Scholar]
- 20.Mayer B, Erdmann J, Schunkert H. Genetics and heritability of coronary artery disease and myocardial infarction. Clin Res Cardiol. 2007;96:1–7. doi: 10.1007/s00392-006-0447-y. [DOI] [PubMed] [Google Scholar]
- 21.Humphries SE, Morgan L. Genetic risk factors for stroke and carotid atherosclerosis: Insights into pathophysiology from candidate gene approaches. Lancet Neurol. 2004;3:227–236. doi: 10.1016/S1474-4422(04)00708-2. [DOI] [PubMed] [Google Scholar]
- 22.Pedersen NL, Lichtenstein P, Svedberg P. The Swedish Twin Registry in the third millennium. Twin Res. 2002;5:427–432. doi: 10.1375/136905202320906219. [DOI] [PubMed] [Google Scholar]
- 23.World Health Organization. International Classification of Diseases. 10th. Geneva: World Health Organization; 1992. [Google Scholar]
- 24.National Board of Health and Welfare. Klassiftkation av kirurgiska atgarder 1997: Reviderad november 2004. [Classification of surgical procedures: Revised November 2004]. Lindesberg, Sweden: National Board of Health and Welfare; 2004. [Google Scholar]
- 25.Kendler KS, Gatz M, Gardner C, Pedersen N. A Swedish national twin study of lifetime major depression. Am J Psychiatry. 2006;163:109–114. doi: 10.1176/appi.ajp.163.1.109. [DOI] [PubMed] [Google Scholar]
- 26.American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 4th. Washington, DC: American Psychiatric Association; 1994. [Google Scholar]
- 27.Kessler RC, Andrews G, Mroczek DK, Ustun B, Wittchen HU. The World Health Organization Composite International Diagnostic Interview Short Form (CIDI-SF) Int J Meth Psychiatr Res. 1998;7:171–185. [Google Scholar]
- 28.World Health Organization. International Classification of Diseases. 9th rev. Geneva: World Health Organization; 1977. [Google Scholar]
- 29.World Health Organization. International Classification of Diseases. 8th rev. World Health Organization; 1968. [Google Scholar]
- 30.World Health Organization. International Classification of Diseases. 7th rev. Geneva: World Health Organization; 1955. [Google Scholar]
- 31.Cox DR. Regression models and life tables (with discussion) J Roy Stat Soc Ser B. 1972;34:187–220. [Google Scholar]
- 32.Binder DA. Fitting Cox proportional hazards models from survey data. Biometrika. 1992;79:139–147. [Google Scholar]
- 33.Bromet EJ, Dunn LO, Connell MM, Dew MA, Schulberg HC. Long-term reliability of diagnosing lifetime major depression in a community sample. Arch Gen Psychiatry. 1986;43:435–440. doi: 10.1001/archpsyc.1986.01800050033004. [DOI] [PubMed] [Google Scholar]
- 34.Kendler KS, Neale MC, Kessler RC, Heath AC, Eaves LJ. The lifetime history of major depression in women. Reliability of diagnosis and heritability. Arch Gen Psychiatry. 1993;50:863–870. doi: 10.1001/archpsyc.1993.01820230054003. [DOI] [PubMed] [Google Scholar]