

Abstract
Topoisomerase IIα (topo IIα) is exported from the nucleus of human myeloma cells by a CRM1-dependent mechanism at cellular densities similar to those found in patient bone marrow. When topo IIα is trafficked to the cytoplasm, it is not in contact with the DNA; thus topo IIα inhibitors are unable to induce DNA-cleavable complexes and cell death. Using a CRM1 inhibitor or a CRM1-specific small interfering RNA (siRNA), we were able to block nuclear export of topo IIα as shown by immunofluorescence microscopy. Human myeloma cell lines and patient myeloma cells isolated from bone marrow were treated with a CRM1 inhibitor or CRM1-specific siRNA and exposed to doxorubicin or etoposide (VP-16) at high cell densities. CRM1-treated cell lines or myeloma patient cells were fourfold more sensitive to topo II poisons, as determined by activated caspase assay. Normal cells were not significantly affected by CRM1-topo II combination treatment. Cell death was correlated with increased DNA double-strand breaks as shown by the comet assay. Band depletion assays of CRM1 inhibitor-exposed myeloma cells demonstrated increased topo IIα covalently bound to DNA. Topo IIα knockdown by a topo IIα-specific siRNA abrogated the CRM1-topo II therapy synergistic effect. These results suggest that blocking topo IIα nuclear export sensitizes myeloma cells to topo II inhibitors. This method of sensitizing myeloma cells suggests a new therapeutic approach to multiple myeloma.
Keywords: topoisomerase, multiple myeloma, CRM1, doxorubicin, etoposide, ratjadone
Introduction
Drug resistance is a major obstacle in the treatment of multiple myeloma, including resistance to topoisomerase (topo II) inhibitors. Topo II poisons that are used in the treatment of multiple myeloma include doxorubicin and etoposide. Several mechanisms of resistance to topo IIα inhibitors have been described (reviewed previously (1-5)). Cell adhesion-mediated drug resistance and stromal cell adherence are important parameters in the local bone marrow environment in patients with multiple myeloma and appear to be major determinants of drug resistance (6-9).
Our laboratory has previously shown that human multiple myeloma cell density is a determinant of sensitivity to topo IIα inhibitors (8-10). At increased cell densities, a considerable fraction of nuclear topo IIα is exported to the cytoplasm (>90%), resulting in reduced sensitivity to etoposide and doxorubicin. This appears to occur both in human myeloma cell lines and in CD-138-positive cells isolated from patients with multiple myeloma (9). We have reported that the nuclear export of topo IIα may contribute to drug resistance (9), and our data suggest that resistance is not due to differences in drug uptake, cell cycle, or cellular topo IIα protein levels (8-10). In a previous report, our group defined the nuclear export signals for topo IIα at amino acids 1017-1028 and 1054-1066 (10). Export by both signals was blocked by treatment of the cells with leptomycin B, indicating that a CRM1-dependent pathway mediates export (10). In the present study, we show that inhibition of CRM1-mediated export of topo IIα may render myeloma cells both in vitro and ex vivo more sensitive to topo IIα -targeted chemotherapy.
Use of CRM1 inhibition in cancer therapy has met with limited success. The first CRM1 inhibitor, leptomycin B, was found to efficiently inhibit nuclear export (11). However, leptomycin was found to have acute relative toxicities, both in a human phase I trial (12) and in vitro (13). Leptomycin B in vitro studies found acute toxicity at concentrations less than 5 nM for 1 hour (13). Therefore, in this study, we used the CRM1 inhibitor ratjadone C (RDC) (14-18). RDC has been found to inhibit nuclear export without producing apoptosis or necrosis at concentrations up to 300 nM for 48 hours in an in vitro assay (18). However, RDC prevents nuclear export of topo IIα; in this study, we demonstrate that RDC also acutely sensitizes myeloma cells to the topo II inhibitors doxorubicin and VP-16. Additional low-toxicity CRM1 inhibitors are also being investigated by other laboratories and may become available for pre-clinical studies (13).
Materials and Methods
Cell Lines
Human myeloma cell lines NCI-H929 (H929) and RPMI-8226 (8226) were newly obtained from the American Type Culture Collection (Manassas, VA). U266B1 (U266) human myeloma cell lines were provided by Dr. Lori Hazlehurst at the Moffitt Cancer Center (Tampa, FL). All cell lines were grown in RPMI 1640 media containing 100 U/mL penicillin, 100 μg/mL streptomycin (Invitrogen, Carlsbad, CA), and 10% fetal bovine serum (Hyclone, Logan, UT) at 37°C and 5% CO2. H929 cell media required the addition of 0.025% β-mercaptoethanol (Sigma, St. Louis, MO).
Cell Density and Drug Treatment
The model used to assay density-dependent protein trafficking involved incubating cells at high- and low-density culture conditions. Our group has previously shown that cells grown at different densities exhibit specific characteristics such as drug resistance and nuclear-cytoplasmic trafficking of topo IIα (8-10). Human myeloma cell lines (8226, H929, U266) grown at 2 ×105 cells/mL were defined as low density (log phase), and cells grown at 2 ×106 cells/mL were defined as high density (plateau phase). Cell lines were placed at log- and plateau-density conditions and incubated with and without the highly specific CRM1 inhibitor RDC (14-18) (Department of Chemical Biology, HZI/Helmholtz Centre for Infection Research, Braunschweig, Germany) or were transfected with CRM1 200 nM small interfering RNA (siRNA) (Dharmacon, catalog no. M00303-01). Rajadones are naturally occurring antibiotics isolated from myxobacteria (16). RDC is a potent inhibitor of CRM1 and prevents nuclear export by alkylating the active site Cys528 amino acid residue of CRM1 (17). RDC has been shown to have anti-cancer properties and has reduced toxicity in vitro as compared to leptomycin B (14, 16, 18). To determine the concentration of RDC to use, we titrated RDC to find the lowest concentration that would inhibit nuclear export of topo IIα in myeloma cell lines H929 and 8226. The concentration arrived at was approximately 5 nM, all RDC experiments used this concentration. Cells were treated with RDC for 16 hours followed by doxorubicin (2 μM; Sigma) for 4 hours or etoposide (VP-16; 10 μM; Sigma) for 7 hours and assayed for apoptosis by either anti-caspase 3 or TUNEL staining (BD Pharmingen).
CRM1 Inhibitor Sensitizes Patient Myeloma Cells to Topo IIα Drugs
Human bone marrow aspirates obtained from multiple myeloma patients were collected using a protocol approved by the University of South Florida Institutional Review Board. Patient samples were assayed for percentage of plasma cells by toluidine blue staining and microscopy. Cells used for assay consisted of between 75% and 90% plasma cells isolated by Ficoll gradient centrifugation. Cells (4 × 106 cells/mL) were treated with RDC (5 nM) for 16 hours followed by doxorubicin (2 μM) for 4 hours and assayed for apoptosis by anti-caspase 3 (BD Pharmingen).
siRNA Knockdown and Western Blot
All electroporation transfections were performed in a freshly made transfection buffer containing 120 mM potassium chloride (Sigma), 0.15 mM calcium chloride, 10 mM potassium phosphate (pH 7.6), 25 mM HEPES, 2 mM EGTA (pH 7.6), 5 mM magnesium chloride, 2 mM ATP (pH 7.6), 5 mM glutathione, 1.25% dimethyl sulfoxide (DMSO), and 50 mM trehalose (Sigma). Each transfection used 3 × 106 myeloma cells. Cells were washed two times in phosphate-buffered saline (PBS) and placed in a 200 μl volume of transfection buffer. CRM1-specific siRNA (Dharmacon, catalog no. M00303-01), scramble control siRNA (Dharmacon, catalog no. D001210-02), or topo IIα-specific siRNA (Ambion, catalog no. AM51333) was added (200 nM); the sample was then placed in a 2-mm electroporation cuvette and transfected at 140 V and 975 μF in a Bio-Rad GenePulser Xcell electroporation unit (Bio-Rad, Hercules, CA). Transfected cells were incubated in the cuvette for 15 minutes at 37°C in a 5% CO2 incubator and transferred to a sterile T25 tissue culture flask, and 10 mL of fresh media were added. After 48 hours, the transfected cells were harvested by centrifugation at 500 × g for 5 minutes, washed with cold PBS, and lysed by sonication (40% duty cycle, 7 bursts) in SDS buffer (2% SDS, 10% glycerol, 60 mM Tris; pH 6.8). Protein from 2 × 105 cells per lane was separated on 8% SDS-PAGE gels and transferred to PVDF membranes (Amersham, Piscataway, NJ) overnight (30 V at 4°C) with the use of a Bio-Rad Mini-Transblot apparatus. Membranes were blocked for 1 hour at ambient temperature in a blocking buffer containing 0.1 M Tris-HCl, 0.9% NaCl, and 0.5% Tween 20 (TBST) and 5% non-fat dry milk. CRM1 was identified by incubation in a 1:1000 dilution of H-300 antibody (Santa Cruz Biotechnology, Santa Cruz, CA) in blocking buffer overnight at 4°C. Membranes were washed three times for 10 minutes in TBST and incubated for 1 hour with goat anti-rabbit polyclonal IgG antibody linked to a horseradish peroxidase antibody (Sigma) in blocking buffer at a 1:2000 dilution. Antibody binding was visualized by enhanced chemiluminescence (Amersham) on autoradiography film (Kodak). Transfected cells were treated with doxorubicin (2 μM) for 4 hours and assayed for apoptosis by annexin V-FITC staining (BD Pharmingen).
Immunofluorescent Microscopy
Multiple myeloma cells (1 × 105) were plated on double cytoslides (Shandon, Waltham, MA) by cyto-centrifugation at 500 rpm for 3 minutes and fixed with 1% paraformaldehyde (Fisher Scientific, Suwanee, GA) on ice for 30 minutes. Permeabilization of cells was performed with 0.5% Triton X-100 (Sigma) in PBS at room temperature for 60 minutes. Cells were stained with a polyclonal antibody against topo IIα, which was produced in our laboratory (PAB454) (19). The topo IIα antibody was diluted 1:100 in a buffer containing 1% bovine serum albumin (Sigma) and 0.1% IGEPAL CA-630 (Sigma) in PBS and incubated for 1 hour at room temperature. After three washes with PBS, slides were incubated with a secondary anti-rabbit Alexa Fluor 594 (Invitrogen) in addition to a cytoskeletal protein stain, phalloidin-Alexa Fluor 488 conjugate (Invitrogen). Each was diluted 1:1000 in 1% bovine serum albumin and 0.1% IGEPAL CA-630 in PBS and incubated for 40 minutes at room temperature. Slides were washed four times in PBS and once in distilled water, and the nuclei were stained with diamindino-2-phenylindole dihydrochloride hydrate (DAPI; Vector Laboratories, Burlingame, CA). Immunofluorescence was observed with the Zeiss Axio Imager Z1 microscope (Carl Zeiss Microimaging, Thornwood, NY) with an Axiocam MRm camera (Carl Zeiss Microimaging). Two experiments were performed with 50 cells assayed per experiment. Cells were chosen randomly and were scored as nuclear or cytoplasmic when ≥90% of the fluorescence was in the respective cellular compartment.
Band Depletion Assay
Band depletion assays were performed as described by Xiao et al. (20). Briefly, 5 × 105 cells were lysed in 50 μL of alkaline lysis solution for 30 minutes on ice (200 mM NaOH, 2 mM EDTA), and the lysate was neutralized by the addition of 4 μL of both 1 M HCl and 1.2 M Tris (pH 8.0). The lysate was then mixed with 30 μL of 3× SDS sample buffer (150 mM Tris-HCl, pH 6.8, 6 mM EDTA, 45% sucrose, 9% SDS, and 10% β-mercaptoethanol) and separated on 8% SDS-PAGE gels.
Comet Assay
Log-density H929 myeloma cells were plated at a concentration of 2 × 105 cells/mL, and plateau-density cells were plated at 2 × 106 cells/mL. All cells were grown in 24-well plates (Falcon) with 1 mL of sample per well. Drug treatment groups were vehicle only (1 μL/mL DMSO), 10 μM etoposide, 5 nM RDC, or a combination of 10 μM etoposide and 5 nM RDC. Cells that were treated with RDC were first plated at log or plateau density and incubated for 16 hours with RDC or vehicle, after which etoposide was added for 1 hour. After the 1 hour of etoposide exposure, the comet assay was performed as described by Kent et al. (21) and modified by Chen et al. (22). To ensure random sampling, 50 images were captured per slide on a Leica fluorescent microscope and quantified with ImageQuant software (Molecular Dynamics, Sunnyvale, CA). The average comet moment value obtained from vehicle control samples was subtracted from the average comet moment of each drug treatment sample. The data shown are means and standard deviations of two separate experiments.
Additional Drug Combinations in H929 Human Myeloma Cell Lines
H929 human myeloma cell lines were placed at high-density (4 × 106) with and without 5 nM RDC and incubated for 16 hours. Cells were then incubated for 4 hours with one of the following: 2 μM doxorubicin, 10 μM VP-16, 10 μM melphalan, or 10 μM velcade. Cells were assayed for apoptosis by TUNEL staining.
Results
Log- and Plateau-Density Myeloma Cells
The myeloma cell lines 8226, H929, and U266 were grown at log-density (2 × 105 cells/mL media) and at plateau-density (2 × 106 cells/mL) growth conditions for 16 hours. Cells were then treated with the topo II inhibitor doxorubicin (2 μM) for 4 hours and assayed for apoptosis by activated caspase 3 expression. We found that cells grown at plateau densities and treated with 2 μM doxorubicin had extremely low levels of apoptosis compared with results shown for log-phase cells (Fig. 1A). These data confirmed previous results that showed that cells grown at different densities exhibit specific characteristics, such as drug resistance and nuclear-cytoplasmic trafficking of topo IIα (8-10). The topo IIα isozyme, topoisomerase IIβ, is not exported from the nucleus in human myeloma cells (8).
Figure 1.

Intracellular trafficking of topoisomerase (topo) IIα in log- and plateau-density myeloma cells. A, H929, U266, and 8226 human myeloma cells grown at plateau phase (high density) export topo IIα, whereas cells grown at log phase (low density) maintain topo IIα in the nucleus. Cells were grown for 16 hours at log or plateau densities and treated with 2 μM doxorubicin for 4 hours (n = 2). Apoptosis was determined by caspase 3 staining with the use of flow cytometry (10,000 cells). Cells that maintained nuclear topo IIα were more sensitive to topo IIα-targeted chemotherapy. B, Cells grown at log- or plateau-phase conditions or cells treated with RDC (100 cells/experiment) were stained for topo IIα by fluorescence microscopy (n = 2). Myeloma cells grown at log-phase conditions had the majority (≥90%) of the topo IIα in the nucleus, whereas plateau-phase cells exported topo IIα into the cytoplasm. The CRM1 inhibitor RDC (5 nM) was found to block export of topo IIα in cells grown in plateau-phase conditions.
Intracellular Trafficking of Topo IIα
Cells grown at log and plateau densities and RDC-treated cells (100 cells/experiment) were scored as “nuclear” or “cytoplasmic” if ≥90% of topo IIα was in that compartment as determined by fluorescence microscopy (Fig. 1B). For myeloma cells grown at log-phase concentrations, 73% to 87% of cells had ≥90% of the topo IIα in the nucleus; for cells grown at plateau-phase concentrations, 85% to 93% of cells had ≥90% of the topo IIα in the cytoplasm. CRM1 inhibition by RDC was found to block export of topo IIα in cells grown in plateau-phase conditions (Fig. 1B).
Topo IIα Trafficking and CRM1 Inhibition
H929 human myeloma cells were grown at log and plateau densities and stained for cytoskeletal protein (phalloidin, green), topo IIα (red), and DNA (DAPI, blue). Results indicate that topo IIα was present in the nucleus of log-density cells and was exported from the nucleus in plateau-density cells (Fig. 2). Nuclear export was blocked in plateau cells by the CRM1 inhibitor RDC and by transfection with a CRM1-specific siRNA. Under the conditions of this experiment, CRM1 siRNA knockdown was 69%. For RDC-treated plateau-density cells (Fig. 1B), approximately 70% of the cells had ≥90% topo IIα in the nucleus for each myeloma cell line.
Figure 2.
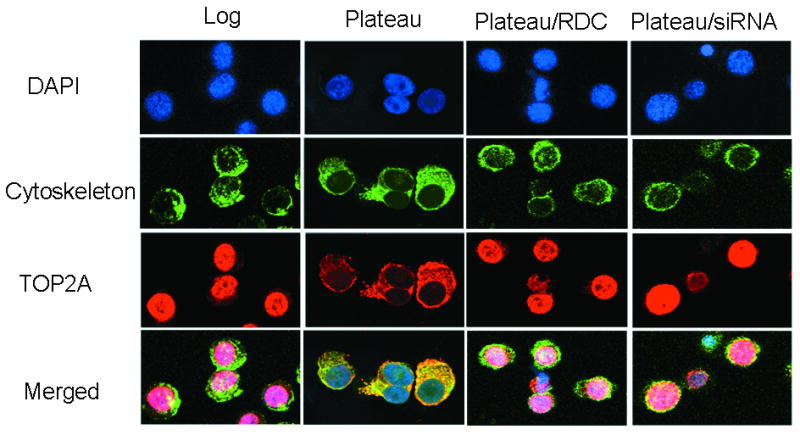
NCI-H929 topo IIα immunofluorescence. H929 human myeloma cells were grown at log and plateau densities, fixed with 4% paraformaldehyde, permeabilized with 0.25% Triton X-100, and stained for cytoskeletal protein (phalloidin, green), topo IIα (red), and DNA [blue with diamindino-2-phenylindole dihydrochoride hydrate (DAPI)]. Results indicate that topo IIα is present in the nucleus of log-density cells and is exported from the nucleus in plateau-density cells. However, nuclear export is blocked in plateau cells by the CRM1 inhibitor RDC and by transfection with CRM1-specific siRNA. Under the conditions of this experiment, CRM1 siRNA knockdown was 69%.
CRM1 Inhibitor and Topo IIα Inhibitor Synergy
The myeloma cell lines 8226, H929, and U266 were grown for 16 hours at plateau densities in the presence of RDC (5 nM). The cells were then treated with doxorubicin (2 μM) for 4 hours and assayed for apoptosis by caspase 3 staining with the use of flow cytometry (n = 3 for each cell line). Fig. 3A demonstrates that myeloma cells were rendered more sensitive to topo IIα inhibitor by inhibition of CRM1 export with RDC. Figs. 3A, 3B, and 3C illustrate that high-cell density-induced drug resistance is reversed by the CRM1 inhibitor RDC. RDC is potent inhibitor of CRM1 and prevents nuclear export by alkylating the active site Cys528 amino acid residue of CRM1 (17). RDC has been shown to have anti-cancer properties and has reduced toxicity in vitro as compared to leptomycin B when used at low dosages (14, 16, 18).
Figure 3.

CRM1 inhibitor sensitizes myeloma cells to doxorubicin. A, Human myeloma cell lines 8226, H929, and U266 (2 × 106 cells/mL) were incubated with the CRM1 inhibitor RDC (5 nM) for 16 hours. Cells were then treated with doxorubicin (2 μM) for 4 hours and assayed for caspase 3 staining by flow cytometry. Cell lines were assayed in triplicate, and data from each drug combination were pooled. RDC versus control samples are statistically different (P = 0.006) in A (control = 1.50% and RDC = 3.84%). RDC was found to significantly (P = 0.00005) sensitize cells to doxorubicin. B, CRM1 inhibitor sensitizes ex vivo patient myeloma cells to doxorubicin. Bone marrow aspirates (n = 7) obtained from multiple myeloma patients were treated with RDC (5 nM) for 16 hours followed by doxorubicin (2 μM) for 4 hours and assayed for cleaved caspase 3 to determine apoptosis. Cells (4 × 106 cells/mL) treated with RDC only were significantly different (P = 0.03) from untreated cells (control = 4.97% and RDC = 14.31%). Cells (4 × 106 cells/mL) treated with both RDC and doxorubicin were significantly (P = 0.0003) more sensitive to doxorubicin (threefold) than doxorubicin alone. C, Mechanism of CRM1 and topo IIα inhibitor synergy. Human myeloma cell lines (n = 9) were incubated at high density (2 × 106/mL) for 16 hours with the CRM1 inhibitor RDC (5 nM). Cell cultures were then exposed to the topo IIα-targeted agents etoposide (VP-16; 10 μM) for 7 hours or doxorubicin (2 μM) for 4 hours and assayed for apoptosis with the TUNEL assay (BD Pharmingen). CRM1 inhibition by RDC increased the effectiveness of DNA-damaging agents to induce apoptosis. This effect was significant for doxorubicin/RDC (P = 0.00019) and VP-16/RDC (P = 0.0185). When topo IIα protein was reduced by topo IIα siRNA knockdown, apoptosis was substantially decreased, suggesting that apoptosis is topo IIα dependent.
CRM1 Inhibitor Sensitizes Patient Myeloma Cells to Topo IIα Drugs
Human bone marrow aspirates were obtained from multiple myeloma patients and purified by Ficoll-paque gradient. Bone marrow samples with greater than 75% plasma cells, as determined by toluidine blue staining and microscopy, were used for each assay (n = 7). Cells were placed at plateau concentration (4 × 106 cells/mL), treated with RDC (5 nM) for 16 hours followed by doxorubicin (2 μM) for 4 hours, and assayed for cleaved caspase 3 to determine apoptosis. Cells treated with RDC were significantly (P = 0.0003) more sensitive to doxorubicin (threefold) than doxorubicin alone (Fig. 3B). Normal cells, including flow 2000 cells and peripheral blood mononuclear cells (PBMCs) (n = 5), were not sensitized by the CRM1 inhibitor (P = 0.22). Figure 3B shows PBMCs only.
CRM1 Inhibitor and Topo IIα Poisons
The human myeloma cell lines 8226, H929, and U266 were incubated at high density (2 × 106 cells/mL) for 16 hours in the presence of the CRM1 inhibitor RDC (5 nM). Cell cultures were then exposed to the topo IIα-targeted agents etoposide (VP-16; 10 μM) for 8 hours or doxorubicin (2 μM) for 4 hours and assayed for apoptosis with a caspase 3 assay (BD Pharmingen). In addition, to show whether the RDC/doxorubicin and the RDC/etoposide synergistic activities were because of topo IIα nuclear localization, cells were transfected with a siRNA to knockdown topo IIα expression (Fig. 3C). Topo IIα knockdown was >90% when assayed by Western blot (Fig. 3C, inset). In myeloma cell lines, with both topo II inhibitors (doxorubicin and etoposide), we found that knockdown of topo IIα protein expression reversed the synergistic effect and reduced apoptosis to untreated levels (4% to 12% apoptosis) (Fig. 3C).
CRM1 siRNA Sensitizes Myeloma Cells to Topo IIα Poisons
In addition to CRM1 pharmacologic modification by a chemical agent, we used a CRM1-specific siRNA to determine whether we could reproduce the observed synergistic activity in another model system. H929 cells were transfected by electroporation with a CRM1-specific siRNA (Fig. 4). After transfection, the cells were incubated at log-phase densities for 24 hours to allow CRM1 siRNA-mediated knockdown and then concentrated at plateau-phase conditions for an additional 16 hours. CRM1 knockdown cells grown at high density for 16 hours were then treated with the topo II inhibitor doxorubicin (2 μM) for 4 hours and assayed for apoptosis by annexin V staining using flow cytometry (Fig. 4). CRM1 knockdown was found to increase the effectiveness of doxorubicin. Myeloma cell line, 8226, demonstrated similar results (data not shown). To demonstrate efficient siRNA knockdown, SDS lysates of equal cell numbers were assayed for CRM1 by Western blot (Fig. 4, inset).
Figure 4.

CRM1 knockdown using siRNA makes myeloma cells more sensitive to the topo II poison doxorubicin. H929 cells were transfected by electroporation with a CRM1-specific siRNA or scrambled siRNA (scram) control. After transfection, the cells were incubated at log-phase density for 24 hours to allow CRM1 siRNA-mediated knockdown and concentrated at the plateau-phase condition for an additional 16 hours. CRM1 knockdown cells grown at high density for 16 hours were then treated with the topo II inhibitor doxorubicin (2 μM) for 4 hours and assayed for apoptosis by annexin V/PI (propidium iodide) staining with the use of flow cytometry. CRM1 knockdown rendered plateau-density cells more sensitive to topo IIα inhibitors. Inset: Western blot data for siRNA transfection. Percent knockdown, compared to control siRNA, ranged between 60 and 65%.
Increase in Cleavable Complex Formation by CRM1 Inhibition
Etoposide stabilizes DNA-topo IIα adducts, resulting in double-stranded DNA breaks. We speculated that, when topo IIα is kept in the nucleus by RDC, a greater number of DNA-topo IIα complexes would be observed. To assay this potential effect, several band depletion assays were performed (n = 3). Band depletion assay can assess the amount of topo IIα-DNA covalent complexes formed in intact cells. Large covalent complexes will have decreased migration into a SDS-PAGE gel; therefore, the amount of topo IIα, as measured by Western blot, will be depleted. Etoposide (VP-16) alone did not produce significant band depletion; however, when used together with RDC, we saw a large depletion at both 25 and 50 μM VP-16 concentrations (Fig. 5A). These data indicate that blocking nuclear export of topo IIα will increase the effectiveness of etoposide and induce apoptosis by increased cleavable complexes.
Figure 5.

Band depletion and comet assay. A, Band depletion assay of the combination of RDC and etoposide produced more DNA-topo IIα complexes, depleting the topo IIα band in the Western blot analysis. These data indicate that blocking nuclear export of topo IIα will increase the effectiveness of etoposide and induce apoptosis. B, Comet assay of plateau-density H929 cells treated with RDC (5 nM) for 16 hours and then with etoposide (10 μM) for 60 minutes. The comet moment of cells (50/experiment) were assayed in 2 separate experiments for each drug combination and control samples. Comet assay is a measure of DNA cleavage (22). The CRM1 inhibitor RDC increased DNA cleavage as compared to the topo IIα inhibitor etoposide alone (P = 0.0006).
Comet Assay
Plateau-density H929 cells were treated with 5 nM RDC for 16 hours and then with 10 μM etoposide for 60 minutes. DNA fragmentation was measured by the neutral comet assay. The CRM1 inhibitor RDC increased the DNA cleavage induced by the topo IIα inhibitor etoposide (Fig. 5B). Increased DNA fragmentation led to increased apoptosis in cells treated with both etoposide and RDC.
RDC Sensitized H929 Human Myeloma Cell Lines to Other Myeloma Drugs
H929 myeloma cells were exposed to RDC and various additional drug combinations (Fig. 6), including the alkylating agent melphalan and proteosome inhibitor velcade. We found that CRM1 inhibition will sensitize myeloma cells to the topo IIα inhibitors doxorubicin (P = 0.00005) and VP-16 (P = 0.02), but it did not significantly sensitizes myeloma cells treated with melphalan (P = 0.35) or velcade (P = 0.30).
Figure 6.
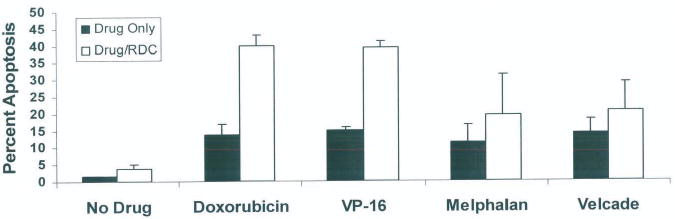
Additional drug combinations in H929 and 8226 human myeloma cell lines. H929 and 8226 human myeloma cell lines were placed at high density (4 × 106) with and without 5 nM RDC and incubated for 16 hours (n = 4 for each cell line). Cells were then incubated for 4 hours with one of the following: 2 μM doxorubicin, 10 μM VP-16, 10 μM melphalan, or 10 μM velcade. CRM1 inhibition by RDC sensitized myeloma cells to the topo IIα inhibitors doxorubicin (P = 0.00005) and VP-16 (P = 0.02), but not myeloma cells treated with melphalan (P = 0.35) or velcade (P = 0.30).
Discussion
The intracellular location of a protein may be at least as important as its expression. Diseases as dissimilar as cystic fibrosis (23), schizophrenia (24), nephrogenic diabetes insipidus (25), and many types of cancers, as reviewed previously (26, 27), may be caused by intracellular mislocalization of individual proteins. Specific examples of proteins that must be in the nucleus to prevent cancer initiation, progression, or chemotherapeutic response include p53 (reviewed previously (28)), galectin-3 (29), FOXO (30), INI1/hSNPF5 (31), p27Kip1 (32), p21Cip1 (33), and topo IIα (8-10). Mislocalization of a protein can render it ineffective as a tumor suppressor or as a target for chemotherapy. However, it is possible that blocking nuclear export of any or all of these proteins may induce tumor suppression or apoptosis or, in the case of topo IIα, may reverse drug resistance to topo IIα inhibitors. This may be true in multiple myeloma where the cells possess a CRM1-mediated mechanism in which topo IIα is exported from the nucleus and away from the DNA, rendering topo IIα inhibitors ineffective to produce cleavable complexes and DNA strand breaks.
In previous reports, our group has shown that myeloma cells, under high-density conditions, will export topo IIα into the cytoplasm both in vivo and in vitro (8-10). We found that nuclear export of topo IIα contributes to drug resistance (9) and that the resistance was not due to differences in drug uptake, cell cycle, or total cellular topo IIα protein levels. In addition, topo IIα nuclear export has been shown to be CRM1 mediated, and topo IIα protein has been found to contain two functional nuclear export signals at amino acids 1017-1028 and 1054-1066 (10). Export by both signals was blocked by treatment of the cells with leptomycin B, indicating that a CRM1-dependent pathway mediates export (10). Leptomycin B has been shown to be too toxic for clinical use (12); however, less toxic CRM1 inhibitors such as RDC and others compounds, as identified in a recent publication, may soon become available (13).
In this study, we demonstrated that myeloma cells grown at high density are highly resistant to topo II-directed chemotherapeutic drugs (Fig. 1A) and that drug resistance correlated with nuclear export of topo IIα (Figs. 1B and 2). Based on these data, we proposed that blocking CRM1-mediated export of topo IIα may make myeloma cells more sensitive to topo II-active agents. To evaluate whether blocking topo IIα export would sensitize cells, we knocked down CRM1 mRNA and protein expression in cells by transfection with CRM1-specific siRNAs and by using the CRM1-inhibiting drug RDC. RDC was used in this study because it is a potent inhibitor of CRM1, has been shown to have anti-cancer properties, and has reduced toxicity in vitro as compared to leptomycin B when used at low dosages (14, 16-18). CRM1 inhibition by siRNA and RDC in human myeloma cells was found to prevent nuclear export of topo IIα in plateau-density cell cultures (Figs. 1B and 2). Depletion or inhibition of CRM1 by siRNA or RDC caused high-density myeloma cells to become fourfold more sensitive to the topo II inhibitors doxorubicin and etoposide, as measured by apoptosis (Fig. 3A-C). Depletion of topo IIα protein by specific topo IIα siRNA knockdown reversed this synergistic effect, indicating that topo IIα was the targeted molecule for CRM1 synergistic activity (Fig. 3B). In addition, we found that blocking CRM1-mediated export sensitized patient myeloma cells obtained from bone marrow aspirates to the topo IIα poison doxorubicin. Normal PBMCs were not sensitized by CRM1 inhibition. It is likely that these cells were not sensitized because they are not replicating at a high rate, unlike the myeloma cells, which double approximately every 24 hours. In addition, normal cells do not export topo IIα, and therefore RDC treatment would not affect intracellular localization.
When additional drugs were used in combination with RDC, we found that myeloma cells were sensitized to topo IIα inhibitors doxorubicin and VP-16, but not to the alkylating agent melphalan or to the proteosome inhibitor velcade (Fig. 6).
In conclusion, maintaining topo IIα in the nucleus by inhibition of CRM1 greatly enhanced the cytotoxic effect of the topo II inhibitors doxorubicin and etoposide in high-density myeloma cells. Band depletion assays indicated that more DNA-topo IIα complexes were stabilized in cells when CRM1 was inhibited (Fig. 5A), and these increased cleavable complexes resulted in increased strand breaks, as measured by the comet assay (Fig. 5B) and subsequent apoptosis. These findings may have potential therapeutic value in the treatment of multiple myeloma.
Acknowledgments
This work was supported by the National Cancer Institute (CA-82533). The work was also supported in part by the Flow Cytometry and Microscopy Core Facilities at the H. Lee Moffitt Cancer Center and Research Institute.
We thank Rasa Hamilton for her assistance in preparing this manuscript.
Footnotes
Conflict-of-interest disclosure: The authors declare no competing financial interest.
References
- 1.Valkov NI, Sullivan DM. Drug resistance to DNA topoisomerase I and II inhibitors in human leukemia, lymphoma, and multiple myeloma. Semin Hematol. 1997;34(4 Suppl 5):48–62. [PubMed] [Google Scholar]
- 2.Rasheed ZA, Rubin EH. Mechanisms of resistance to topoisomerase I-targeting drugs. Oncogene. 2003;22:7296–304. doi: 10.1038/sj.onc.1206935. [DOI] [PubMed] [Google Scholar]
- 3.Oloumi A, MacPhail SH, Johnston PJ, Banath JP, Olive PL. Changes in subcellular distribution of topoisomerase IIalpha correlate with etoposide resistance in multicell spheroids and xenograft tumors. Cancer Res. 2000;60:5747–53. [PubMed] [Google Scholar]
- 4.Sullivan DM, Chow KC, Ross WE. Topoisomerase II mediated mechanisms of drug resistance. Boca Raton, FL: CRC Press; 1989. [Google Scholar]
- 5.Sullivan DM, Ross WE. Resistance to inhibitors of DNA topisomerases. Boston, MA: Kluwer Academic Publishers; 1991. [DOI] [PubMed] [Google Scholar]
- 6.Hazlehurst LA, Valkov N, Wisner L, et al. Reduction in drug-induced DNA double-strand breaks associated with beta1 integrin-mediated adhesion correlates with drug resistance in U937 cells. Blood. 2001;98:1897–903. doi: 10.1182/blood.v98.6.1897. [DOI] [PubMed] [Google Scholar]
- 7.Hazlehurst LA, Argilagos RF, Emmons M, et al. Cell adhesion to fibronectin (CAM-DR) influences acquired mitoxantrone resistance in U937 cells. Cancer Res. 2006;66:2338–45. doi: 10.1158/0008-5472.CAN-05-3256. [DOI] [PubMed] [Google Scholar]
- 8.Valkov NI, Gump JL, Engel R, Sullivan DM. Cell density-dependent VP-16 sensitivity of leukaemic cells is accompanied by the translocation of topoisomerase IIalpha from the nucleus to the cytoplasm. Br J Haematol. 2000;108:331–45. doi: 10.1046/j.1365-2141.2000.01832.x. [DOI] [PubMed] [Google Scholar]
- 9.Engel R, Valkov NI, Gump JL, Hazlehurst L, Dalton WS, Sullivan DM. The cytoplasmic trafficking of DNA topoisomerase IIalpha correlates with etoposide resistance in human myeloma cells. Exp Cell Res. 2004;295:421–31. doi: 10.1016/j.yexcr.2004.01.012. [DOI] [PubMed] [Google Scholar]
- 10.Turner JG, Engel R, Derderian JA, Jove R, Sullivan DM. Human topoisomerase IIalpha nuclear export is mediated by two CRM-1-dependent nuclear export signals. J Cell Sci. 2004;117:3061–71. doi: 10.1242/jcs.01147. [DOI] [PubMed] [Google Scholar]
- 11.Hamamoto T, Seto H, Beppu T. Leptomycins A and B, new antifungal antibiotics. II. Structure elucidation. J Antibiot (Tokyo) 1983;36:646–50. doi: 10.7164/antibiotics.36.646. [DOI] [PubMed] [Google Scholar]
- 12.Newlands ES, Rustin GJ, Brampton MH. Phase I trial of elactocin. British J Cancer. 1996;74:648–9. doi: 10.1038/bjc.1996.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mutka SC, Yang WQ, Dong SD, et al. Identification of nuclear export inhibitors with potent anticancer activity in vivo. Cancer Res. 2009;69:510–7. doi: 10.1158/0008-5472.CAN-08-0858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Falini B, Bolli N, Shan J, et al. Both carboxy-terminus NES motif and mutated tryptophan(s) are crucial for aberrant nuclear export of nucleophosmin leukemic mutants in NPMc+ AML. Blood. 2006;107:4514–23. doi: 10.1182/blood-2005-11-4745. [DOI] [PubMed] [Google Scholar]
- 15.Kalesse M, Christmann M, Bhatt U, et al. The chemistry and biology of ratjadone. Chembiochem. 2001;2:709–14. doi: 10.1002/1439-7633(20010903)2:9<709::AID-CBIC709>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 16.Koster M, Lykke-Andersen S, Elnakady YA, et al. Ratjadones inhibit nuclear export by blocking CRM1/exportin 1. Exp Cell Res. 2003;286:321–31. doi: 10.1016/s0014-4827(03)00100-9. [DOI] [PubMed] [Google Scholar]
- 17.Meissner T, Krause E, Vinkemeier U. Ratjadone and leptomycin B block CRM1-dependent nuclear export by identical mechanisms. FEBS Lett. 2004;576:27–30. doi: 10.1016/j.febslet.2004.08.056. [DOI] [PubMed] [Google Scholar]
- 18.Burzlaff A, Kalesse M, Kasper C, Scheper T. Multi parameter in vitro testing of ratjadone using flow cytometry. Appl Microbiol Biotechnol. 2003;62:174–9. doi: 10.1007/s00253-003-1300-0. [DOI] [PubMed] [Google Scholar]
- 19.Sullivan DM, Latham MD, Rowe TC, Ross WE. Purification and characterization of an altered topoisomerase II from a drug-resistant Chinese hamster ovary cell line. Biochemistry. 1989;28:5680–7. doi: 10.1021/bi00439a051. [DOI] [PubMed] [Google Scholar]
- 20.Xiao H, Mao Y, Desai SD, et al. The topoisomerase IIbeta circular clamp arrests transcription and signals a 26S proteasome pathway. Proc Natl Acad Sci U S A. 2003;100:3239–44. doi: 10.1073/pnas.0736401100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kent CR, Eady JJ, Ross GM, Steel GG. The comet moment as a measure of DNA damage in the comet assay. Int J Radiat Biol. 1995;67:655–60. doi: 10.1080/09553009514550771. [DOI] [PubMed] [Google Scholar]
- 22.Chen Q, Van der Sluis PC, Boulware D, Hazlehurst LA, Dalton WS. The FA/BRCA pathway is involved in melphalan-induced DNA interstrand cross-link repair and accounts for melphalan resistance in multiple myeloma cells. Blood. 2005;106:698–705. doi: 10.1182/blood-2004-11-4286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Welsh MJ, Smith AE. Molecular mechanisms of CFTR chloride channel dysfunction in cystic fibrosis. Cell. 1993;73:1251–4. doi: 10.1016/0092-8674(93)90353-r. [DOI] [PubMed] [Google Scholar]
- 24.Karpa KD, Lin R, Kabbani N, Levenson R. The dopamine D3 receptor interacts with itself and the truncated D3 splice variant d3nf: D3-D3nf interaction causes mislocalization of D3 receptors. Mol Pharmacol. 2000;58:677–83. doi: 10.1124/mol.58.4.677. [DOI] [PubMed] [Google Scholar]
- 25.Edwards SW, Tan CM, Limbird LE. Localization of G-protein-coupled receptors in health and disease. Trends Pharmacol Sci. 2000;21:304–8. doi: 10.1016/s0165-6147(00)01513-3. [DOI] [PubMed] [Google Scholar]
- 26.Davis JR, Kakar M, Lim CS. Controlling protein compartmentalization to overcome disease. Pharm Res. 2007;24:17–27. doi: 10.1007/s11095-006-9133-z. [DOI] [PubMed] [Google Scholar]
- 27.Turner JG, Sullivan DM. CRM1-mediated nuclear export of proteins and drug resistance in cancer. Curr Med Chem. 2008;15:2648–55. doi: 10.2174/092986708786242859. [DOI] [PubMed] [Google Scholar]
- 28.Fabbro M, Henderson BR. Regulation of tumor suppressors by nuclear-cytoplasmic shuttling. Exp Cell Res. 2003;282:59–69. doi: 10.1016/s0014-4827(02)00019-8. [DOI] [PubMed] [Google Scholar]
- 29.Takenaka Y, Fukumori T, Yoshii T, et al. Nuclear export of phosphorylated galectin-3 regulates its antiapoptotic activity in response to chemotherapeutic drugs. Mol Cell Biol. 2004;24:4395–406. doi: 10.1128/MCB.24.10.4395-4406.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nakamura N, Ramaswamy S, Vazquez F, Signoretti S, Loda M, Sellers WR. Forkhead transcription factors are critical effectors of cell death and cell cycle arrest downstream of PTEN. Mol Cell Biol. 2000;20:8969–82. doi: 10.1128/mcb.20.23.8969-8982.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Craig E, Zhang ZK, Davies KP, Kalpana GV. A masked NES in INI1/hSNF5 mediates hCRM1-dependent nuclear export: implications for tumorigenesis. Embo J. 2002;21:31–42. doi: 10.1093/emboj/21.1.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Min YH, Cheong JW, Kim JY, et al. Cytoplasmic mislocalization of p27Kip1 protein is associated with constitutive phosphorylation of Akt or protein kinase B and poor prognosis in acute myelogenous leukemia. Cancer Res. 2004;64:5225–31. doi: 10.1158/0008-5472.CAN-04-0174. [DOI] [PubMed] [Google Scholar]
- 33.Keeshan K, Cotter TG, McKenna SL. Bcr-Abl upregulates cytosolic p21WAF-1/CIP-1 by a phosphoinositide-3-kinase (PI3K)-independent pathway. Br J Haematol. 2003;123:34–44. doi: 10.1046/j.1365-2141.2003.04538.x. [DOI] [PubMed] [Google Scholar]