

Abstract
Background and Purpose
Ischemia/hypoxia induces de novo expression of the sulfonylurea receptor 1-regulated NC(Ca-ATP) channel. In rodent models of ischemic stroke, early postevent administration of the sulfonylurea, glibenclamide, is highly effective in reducing edema, mortality, and lesion volume, and in patients with diabetes presenting with ischemic stroke, pre-event plus postevent use of sulfonylureas is associated with better neurological outcome. However, the therapeutic window for treatment with glibenclamide has not been studied.
Methods
We examined the effect of low-dose (nonhypoglycemogenic) glibenclamide in 3 rat models of ischemic stroke, all involving proximal middle cerebral artery occlusion (MCAo): a thromboembolic model, a permanent suture occlusion model, and a temporary suture occlusion model with reperfusion (105 minutes occlusion, 2-day reperfusion). Treatment was started at various times up to 6 hours post-MCAo. Lesion volumes were measured 48 hours post-MCAo using 2,3,5-triphenyltetrazolium chloride.
Results
Glibenclamide reduced total lesion volume by 53% in the thromboembolic MCAo model at 6 hours, reduced corrected cortical lesion volume by 51% in the permanent MCAo model at 4 hours, and reduced corrected cortical lesion volume by 41% in the temporary MCAo model at 5.75 hours (P<0.05 for all 3). Analysis of pooled data from the permanent MCAo and temporary MCAo series indicated a sigmoidal relationship between hemispheric swelling and corrected cortical lesion volume with the half-maximum cortical lesion volume being observed with 10% hemispheric swelling.
Conclusions
Low-dose glibenclamide has a strong beneficial effect on lesion volume and has a highly favorable therapeutic window in several models of ischemic stroke.
Keywords: cerebral ischemia, glibenclamide, stroke, SUR1, SUR1-regulated NCCa-ATP channel
The sulfonylurea receptor 1 (SUR1)-regulated NCCa-ATP channel is a 30 pS channel that conducts monovalent but not divalent cations.1 This channel is not constitutively expressed but is newly upregulated in neurons, astrocytes, and capillary endothelial cells after central nervous system ischemia or injury. Channel opening, which is triggered by ATP depletion, has been implicated in oncotic cell swelling and oncotic cell death.2–4 Block of the channel by glibenclamide and other SUR1 inhibitors soon after an insult results in significant improvements in rodent models of ischemic stroke and spinal cord injury.4,5
Unlike most putative neuroprotective drugs, available evidence suggests that glibenclamide may benefit humans with ischemic stroke. A retrospective study has shown that, in patients with diabetes mellitus who present with an ischemic stroke, those already taking glibenclamide or another sulfonylurea drug and who continue on it fare significantly better than matched patients with diabetes not on a sulfonylurea drug.6
To date, no study has examined the therapeutic window for glibenclamide. We examined the effect of glibenclamide administered with a delay of 2 to 6 hours after onset of focal cerebral ischemia. Because humans with ischemic stroke have various sorts of insults, including thromboembolic strokes, permanent occlusions as well as transient occlusions, we sought to evaluate the effect of glibenclamide in models relevant to each of these conditions. We examined the efficacy of delayed administration of low-dose glibenclamide in 3 models of stroke with assessment of infarct volumes 48 hours after onset of ischemia. Our data indicate that treatment with nonhypoglycemogenic doses of glibenclamide is beneficial in a variety of models of ischemic stroke and with a very favorable therapeutic window.
Methods
Rat Models of Ischemic Stroke
All surgical procedures were approved by the Institutional Animal Care and Use Committee of the University of Maryland. Two investigators who were blinded to the treatment and who did not evaluate outcome performed all of the surgical procedures. Fasted male Wistar rats (250 to 300 g; Harlan, Indianapolis, Ind) were anesthetized (60 mg/kg ketamine and 7.5 mg/kg xylazine intraperitoneally) and allowed to ventilate air spontaneously. Temperature was maintained at 37°C using a heating pad regulated by rectal temperature (Harvard Apparatus, Holliston, Mass). For all 3 models, the right carotid sheath was exposed through a ventral midline incision, the common, external, and internal carotid arteries (CCA, ECA, ICA) were dissected and the pterygopalatine artery was ligated. Blood gases (i-STAT; Heska Corp, Ft Collins, Col), generally sampled from either the ECA or CCA shortly before middle cerebral artery occlusion (MCAo), were (mean±SD): pO2, 90±14 mm Hg; pco2, 42±5 mm Hg; and pH, 7.38±0.04; glucose, 195±43 (n=61). Additional measurements of serum glucose were obtained at later times by bleeding the tip of the tail.
Thromboembolic Middle Cerebral Artery Occlusion Model
Thrombi used for embolization were prepared as described.7 PE50 tubing loaded with 6 thrombi approximately 1.0 mm in length that appeared white and that did not easily fragment was used to catheterize the ECA. Thrombi were flushed into the ICA using approximately 200 μL of normal saline (NS). The stump of the ECA was ligated and flow was restored in the CCA/ICA.
Permanent Middle Cerebral Artery Occlusion Model
Thread occluders were manufactured using 4–0 monofilament nylon with occluder tips of 0.35×5 mm molded from silicone polymer (Sylgard 184; Dow Corning Corp, Midland, Mich) using PE20 tubing.8 The CCA was catheterized and flushed with 200 μL of warm NS. The occluder was introduced into the CCA, was advanced into the ICA approximately 18 mm from the CCA bifurcation until resistance was felt, and was secured in place.
Temporary Middle Cerebral Artery Occlusion Model
Relative cerebral blood flow (rCBF) was measured using a laser Doppler flowmeter (Moor Instruments, Axminster, UK). After thinning the skull 2 mm caudal and 4 mm lateral to bregma, the laser Doppler probe was affixed to the skull using α-cyanoacrylate adhesive. The ICA was flushed with 200 μL of warm NS and MCAo was performed by inserting the occluder, manufactured as previously, retrograde into the ECA stump, and advancing it under guidance of the flowmeter. Animals with a drop in rCBF <75% were not further studied. After 105 minutes, the occluder was removed, the ECA stump was ligated, and flow was restored in the CCA/ICA. The rat was maintained under anesthesia throughout the period of occlusion, and rCBF was monitored continuously, including for 10 minutes after reperfusion. rCBF values were calculated as a percentage of the immediate preocclusion baseline.
Treatment
Stock solutions of glibenclamide (Sigma, St Louis, Mo) were prepared in dimethylsulfoxide (5 mg/mL). Solutions for delivery were prepared by adding stock solution to NS and clarifying the solution as needed using a minimum amount of NaOH to a pH approximately 8 to 8.5. Solutions prepared in this way and stored at 37°C for 48 hours retained >80% efficacy as measured in patch clamp experiments on KATP channels in insulinoma cells. Infusion doses were delivered using a subcutaneously implanted miniosmotic pump (Model 2001; 1.0 μL/h; Durect Corp, Cupertino, Calif), which was always implanted immediately after MCAo. For delayed treatment, pumps were fitted with a catheter containing air (“spacer substance”) to produce the desired delay (see Supplemental Material, available online at http://stroke.ahajournals.org). Loading doses, which were used only with delayed treatment, were delivered intraperitoneally at the designated time. For the thromboembolic MCAo series, we used an infusion dose of 75 ng/h and, for delayed treatments, a loading dose of either 3.3 μg/kg or 33 μg/kg. For the permanent MCAo series, we used an infusion dose of 200 ng/h and a loading dose of 33 μg/kg. For the temporary MCAo series, we used an infusion dose of 200 ng/h and a loading dose of 10 μg/kg. Controls were administered vehicle (NS plus dimethylsulfoxide) in the same way.
Exclusion Criteria
Animals were excluded for: subarachnoid hemorrhage (9% to 10%/treatment group); total lesion volume <10% before correction; malpositioned occluder (permanent MCAo model); rCBF reduction <75% or inadequate reperfusion (temporary MCAo model). Deaths (<5%/group), typically occurring within 4 hours of MCAo, were attributable to anesthesia or surgery with no stroke-related deaths encountered.
Immunohistochemistry
Some rats from the temporary MCAo series were used for immunohistochemistry. After transcardiac perfusion/fixation with paraformaldehyde and cryoprotection with 30% sucrose, cryosections were immunolabeled using primary antibodies directed against SUR1 (C-16; Santa Cruz Biotechnology, Santa Cruz, Calif; diluted 1:200; 1 hour at room temperature, 48 hours at 4°C) or IgG (Santa Cruz). Alexa Fluor 550- or fluorescein isothiocyanate-conjugated secondary antibodies (Jackson Immunoresearch, West Grove, Pa) were used. Slides were coverslipped with ProLong Gold antifade reagent containing 4′,6-diamino-2-phenylindole for nuclear staining (P36935; Invitrogen, Carlsbad, Calif).
Stroke Outcome Measures and Data Analysis
All rats used for assessment of lesion size were euthanized 48 hours post-MCAo; 2-mm coronal sections were immersed in 2% 2,3,5-tri-phenyltetrazolium chloride (Sigma-Aldrich) in NS for 20 minutes at 37°C. Images of 2,3,5-triphenyltetrazolium chloride-stained sections, obtained using a flatbed scanner, were analyzed using Photoshop (Adobe), which allowed tracings to be stored for later review by a different investigator. Area measurements made in pixels on 5 consecutive slices were converted to mm3 or percent hemisphere. For the temporary MCAo series, the measurements collected consisted only of the size of the 2,3,5-triphenyltetrazolium chloride(-) lesion and of the ipsilateral hemisphere, which allowed calculation of the total uncorrected lesion volume. For the permanent MCAo and the temporary MCAo series, the data collected included the size of the cortical 2,3,5-triphenyltetrazolium chloride(-) lesion of the ipsilateral and contralateral cortex and of the ipsilateral and contralateral hemisphere, which allowed calculation of the cortical lesion volume corrected for swelling and of hemispheric swelling.
Statistical significance was assessed using either Student t test (permanent MCAo series) or one-way analysis of variance (thromboembolic MCAo and temporary MCAo series) with Bonferroni group comparisons. To analyze the relationship between hemispheric swelling and cortical lesion volume, data from the permanent MCAo and temporary MCAo series were pooled and were fit to a sigmoid function, LV=LVmax/(1 +e−B), where LV=lesion volume, LVmax=maximum lesion volume, and B=(HS-HShalf)/s with HS=hemisphere swelling, HShalf=the value of HS at which LV is half maximum, and s is a measure of the steepness of the curve.
Maps of the spatial density of the probability of infarction (POI) were prepared using sections from each of the 3 groups of rats in the temporary MCAo series (CTR and 2 treatment groups). The 2,3,5-triphenyltetrazolium chloride image from each rat at −2 mm from bregma was processed (Abode Photoshop). The “green channel” of each RGB image had its “opacity” set by an amount equal to the reciprocal of the number of sections in the group. Processed sections for each group were overlaid to generate a “pseudoaverage” image. The resulting 3 images, one for each group, were then processed identically to display the spatial density of POI with white pseudocolor denoting a probability of 1 and black pseudocolor denoting a probability of 0.
Results
Lesion Volume
For the thromboembolic MCAo series, total lesion volumes with no delay in glibenclamide treatment (no loading dose) and with 2-hour delay (loading dose, 3.3 μg/kg) were significantly smaller than in controls (Figure 1A). The benefit of glibenclamide was lost when treatment was started at 4 or 6 hours post-MCAo with a low loading dose (3.3 μg/kg) but was regained with a higher loading dose (33 μg/kg). The data with 4-hour delay showed a trend that was not statistically significant (P=0.07), whereas the data with 6-hour delay were statistically different from controls.
Figure 1.
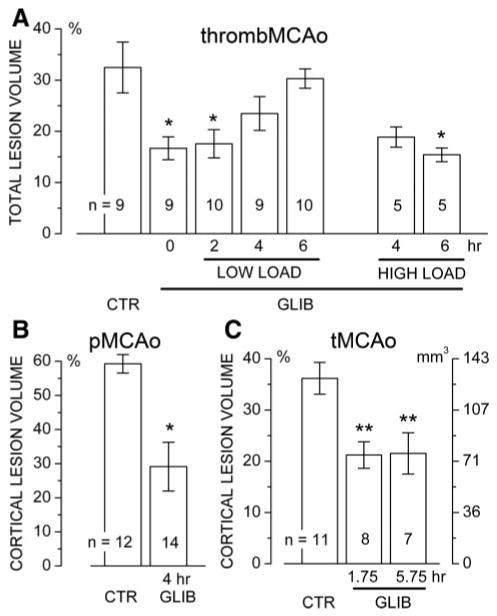
Glibenclamide reduces lesion volume. A–C, Lesion volumes are given for the thromboembolic MCAo model (A), the permanent MCAo model (B), and the temporary MCAo model (C) with vehicle versus glibenclamide administered at the indicated times; the number of rats in each group is indicated; values in B and C are for the “corrected” cortical lesion volume; the relationship between “percent hemisphere” and actual volume (mm3) is shown in C. *P<0.05; **P<0.01.
For the permanent MCAo series, corrected cortical lesion volumes with 4-hour delay were significantly smaller than in controls (Figure 1B).
For the temporary MCAo series, values for the reduction in rCBF were not different between groups (82±2, 81 ±2, and 84±2 for controls and the groups with 1¾-hour and 5¾-hour delays, respectively). Corrected cortical lesion volumes with 1¾-hour and 5¾-hour delays were both significantly smaller than in controls (Figure 1C).
Swelling
A scatterplot of the pooled data from both the permanent MCAo and temporary MCAo series, both with and without treatment, demonstrated a homogeneous sigmoidal relationship between hemispheric swelling and corrected cortical infarct volume (Figure 2). Fit of the data with a sigmoid function indicated that half-maximum cortical lesion volumes occurred with approximately 10% hemispheric swelling. This analysis also showed that the data with glibenclamide treatment (red symbols) occupied predominantly the lower end of the curve, consistent with better outcomes.
Figure 2.
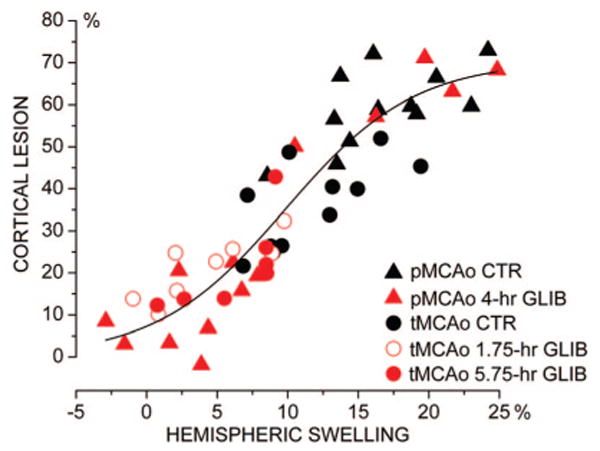
Hemispheric swelling predicts cortical lesion volume. Scatterplot of the corrected cortical lesion volume versus hemispheric swelling for treated and untreated rats in the permanent MCAo and temporary MCAo series (n=52). Fit to a sigmoid function indicated half-maximum cortical lesion (35%) occurring with 9.8% hemispheric swelling (maximum cortical lesion volume, 70%).
Glucose
The glycemic response to anesthesia plus surgery was similar in vehicle-treated controls and in rats treated with glibenclamide (Figure 3A). Higher doses of glibenclamide than those used for treatment did not reduce serum glucose below 100 mg/dL (Figure 3B).
Figure 3.
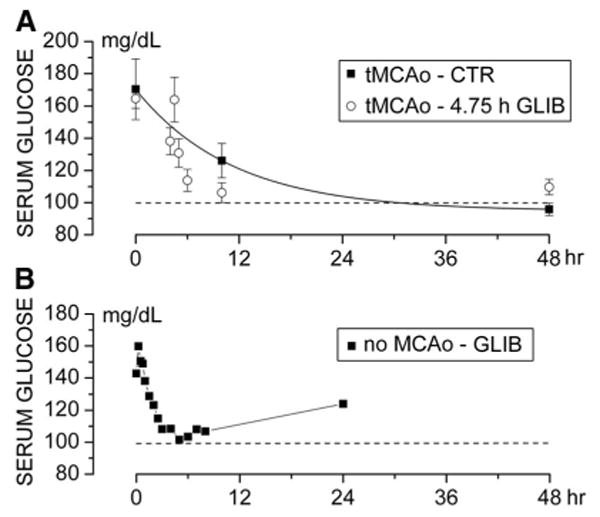
The doses of glibenclamide used are not hypoglycemogenic. A–B, Serum glucose levels at various times for vehicle-treated rats and rats treated with glibenclamide 5¾ hours post-MCAo (temporary MCAo series; loading dose, 10 μg/mL; infusion dose, 200 ng/h; A) and for sham rats (no MCAo) administered a higher dose (loading dose, 80 μg/mL; infusion dose, 600 ng/h; B).
Probability of Infarction
Maps of the spatial density of POI, constructed using data from the temporary MCAo series, showed that the cortex could be divided into 4 relatively distinct regions where POI correlated with proximity to the middle cerebral artery origin (Figure 4): Region 1, dorsal/medial cortex, supplied by the anterior cerebral artery with POI approximately 0; Region 2, dorsal/lateral cortex, the most-distal middle cerebral artery territory with POI approximately 1; Region 3, lateral cortex, the intermediate middle cerebral artery territory with POI intermediate; and Region 4, inferior/lateral cortex, the most-proximal portion of the middle cerebral artery territory with POI lowest. Glibenclamide had no effect on Regions 1 or 2, whereas it reduced the POI by approximately half in Regions 3 and 4 (Figure 4D).
Figure 4.
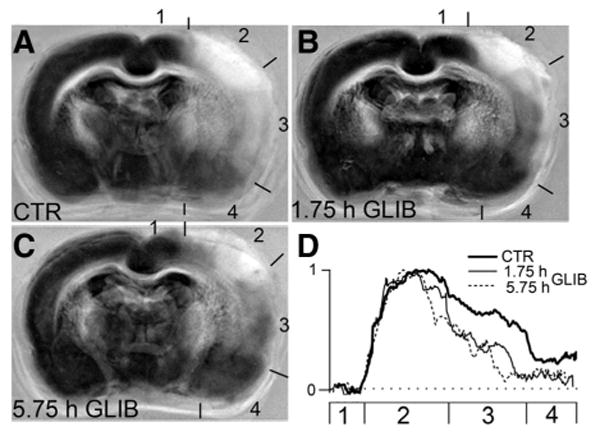
Glibenclamide reduces the POI in the lateral and inferior/lateral cortex. A–C, Maps of the spatial density of POI were constructed using images from the control group (A), the group treated 1¾ hours post-MCAo (B), and the group treated 5¾ hours post-MCAo (C); temporary MCAo series; POI=1 is shown in white pseudocolor and POI=0 is shown in black pseudocolor; numbers indicate cortical Regions 1 to 4 (see text). D, The POI in cortical Regions 1 to 4 for the 3 groups, as indicated.
Delayed Sulfonylurea Receptor 1 Expression Post–Middle Cerebral Artery Occlusion
We hypothesized that the beneficial effect of glibenclamide observed in Regions 3 and 4, despite a delay in treatment, might be related to delayed upregulation of its target, SUR1, because in a model of malignant cerebral edema, maximal expression of SUR1 occurs only at 8 hours.4 We assessed SUR1 expression and IgG labeling, with the latter serving as a marker of vasogenic edema and cellular demise.9 In Regions 3 and 4 at 5¾ hours, relatively little SUR1 expression was evident and IgG labeling was largely absent (Figure 5). In the same regions at 24 hours, however, both SUR1 expression and IgG labeling were prominent throughout the cortex, including in neurons and microvessels, consistent with delayed SUR1 expression and worsening tissue demise after 5¾ hours (Figure 5).
Figure 5.
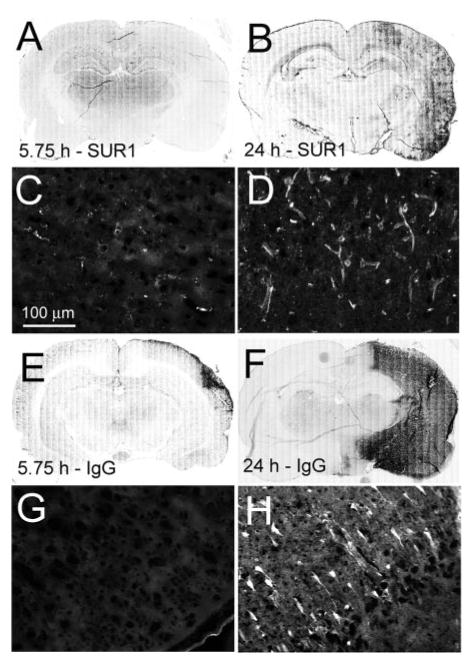
SUR1 expression, protein extravasation, and cellular damage continue beyond 5¾ hours after temporary MCAo. A–D, Immunolabeling for SUR1 at 5¾ hours (A, C) and at 24 hours (B, D) post-MCAo (temporary MCAo model with no treatment); C and D are from Region 4, as defined in Figure 4. E–H, Immunolabeling for IgG at 5¾ hours (E, G) and at 24 hours (F, H) post-MCAo; G and H are from Region 4. The data shown are representative of findings in 3 rats at each time.
Discussion
The assessment of purported neuroprotective agents should include studies with comorbidities and testing in a wide scope of models including in vitro and in vivo models, with the latter including transient and permanent occlusion and testing of efficacy with delayed treatment.10–12 The studies presented here and previously have begun to address these objectives for glibenclamide.
Comorbidity: Hyperglycemia
Admission hyperglycemia complicates approximately one third of acute ischemic strokes and is associated with a worse clinical outcome.13 Our use of ketamine/xylazine anesthesia produced hyperglycemia with mean serum glucose levels approximately 200 mg/dL at the time of MCAo,14 which could adversely affect outcome.15 Our data indicated that glibenclamide was effective in reducing mortality4 as well as lesion volume (this study) despite the presence of hyperglycemia at the time of onset of ischemia. Notably, the loading doses of glibenclamide used in both of our studies were 40 to 400 times less than typically used to induce hypoglycemia in rats.16 However, the data of Figure 3 suggest that glucose levels fell somewhat more rapidly in rats treated with glibenclamide compared with controls. Although no literature suggests that such small differences would account for the magnitude of protection observed, nevertheless, additional study of this question may be warranted.
Scope of Testing: Cell Systems, Animal Models, and Humans
Glibenclamide reduces depolarization, oncotic cell swelling, and oncotic cell death induced by ATP depletion in isolated neurons, astrocytes, and central nervous system endothelial cells that express the SUR1-regulated NCCa-ATP channel,3–5 providing important information about cellular and molecular mechanisms of drug action.
We recently reported that contemporaneous use of sulfonylureas in human diabetics presenting with ischemic stroke is associated with significantly better outcomes measured as an improvement in National Institutes of Health Stroke Scale and in modified Rankin scale at approximately 2 weeks.6 We have now studied the effect of the sulfonylurea, glibenclamide, in 4 different models of ischemic stroke: a model of malignant cerebral edema,4 a thromboembolic model, a permanent occlusion model, and a temporary occlusion model with reperfusion. The study in humans suggested that sulfonylurea are beneficial mainly in the context of large vessel disease. Our work with animal models, all of which have involved proximal large vessel occlusions, reaffirms this. Another important variable that affects stroke outcome is the presence or absence of reperfusion after an ischemic event.17 Two of our 4 models involved permanent MCAo without reperfusion, and 2 involved temporary MCAo with reperfusion. The 2 reperfusion models included a thromboembolic model associated with spontaneous reperfusion occurring at undetermined times18,19 and a standard temporary suture occlusion model with predetermined 105-minute occlusion and reperfusion that were strictly documented with LDF. Glibenclamide showed a strong beneficial effect in all 4 models; it reduces edema and mortality in the malignant cerebral edema model,4 it improves rCBF in the thromboembolic MCAo model,4 and we show that it significantly reduced lesion volume in the 3 nonlethal models. Overall, our data indicate that glibenclamide confers significant protection from ischemic injury in a variety of models relevant to the human condition.
One limitation of the present study is that end points were measured 48 hours after onset of ischemia with treatment continued throughout. Additional studies will be required to determine the optimal duration of treatment required and the durability of treatment.
Treatment Window
Our data suggest that glibenclamide has a favorable window of treatment. A significant beneficial effect on stroke outcome was observed in humans already on (“pretreated with”) sulfonylureas at the time of their ischemic event.6 In our previous report on the malignant cerebral edema model,4 as well as here with the thromboembolic MCAo model, we studied the effect of glibenclamide when drug was administered with nominally no delay in treatment. In these cases, treatment was effected by implanting a miniosmotic pump shortly after MCAo with no loading dose administered. Pharmacokinetic analysis (clearance, 5.8 mL/min/kg)16 indicated that the infusion rates used (75 or 200 ng/h) would require approximately 3 hours to reach 90% of steady-state levels, rendering these experiments, in actuality, studies of delayed treatment. We also examined the effect of a deliberate delay in treatment out to 6 hours after onset of ischemia. Strong protection was found when delayed treatment included use of a sufficient loading dose. Because of uncertainties with thromboembolic models, specifically with delayed reperfusion at unpredictable times,18,19 we place less emphasis on our data with the thromboembolic MCAo model. However, our data with the temporary MCAo model are robust with clear documentation of the degree (>75% reduction in rCBF) and duration (105 minutes) of ischemia. With the temporary MCAo model, treatment beginning 5¾ hours after onset of ischemia showed a strong protective effect. The therapeutic window for glibenclamide compares favorably with other treatments; human albumin is beneficial when administered out to 4 hours, but not 5 hours after onset of ischemia (120-minute temporary MCAo/3-day reperfusion)20; minocycline is beneficial at 4 hours (90-minute temporary MCAo/3-day reperfusion)21 and at 5 hours but not at 6 hours (90-minute temporary MCAo/1-day reperfusion).22
The favorable treatment window observed with glibenclamide probably relates to the fact that SUR1-regulated NCCa-ATP channels are not constitutively expressed, but are expressed de novo over the course of several hours postinsult. In the malignant cerebral edema model, SUR1 is upregulated early (<3 hours) in the core and then disappears as necrosis sets in, whereas in penumbra, SUR1 is upregulated later with maximum expression at 8 hours.4 In the temporary MCAo model, an important part of the salvageable penumbra was the inferior/lateral cortex (Regions 3 and 4), where at 5¾ hours, IgG labeling for leakage and cellular damage was largely absent and SUR1 upregulation was relatively modest compared with sharp increases in both at 24 hours. SUR1 expression, vascular leakage, and cellular uptake of IgG were dynamic processes that progressed gradually over many hours postevent, consistent with the observation of a prolonged window of time during which a beneficial effect of glibenclamide could be observed.
Pathophysiology
Several mechanisms by which glibenclamide could potentially act to effect protection can be excluded. Ischemic tolerance is unlikely to have been important, because ischemic tolerance induced by various treatments including adenosine23 and hypothermia24 is abolished by glibenclamide. Induction of hypoglycemia is unlikely to have been important, because the doses of glibenclamide used were not hypoglycemogenic (Figure 3), suggesting minimal action on either pancreatic β cells or hypothalamic KATP channels that are important for glycemic control. An effect on temperature is unlikely, because glibenclamide does not affect hypothalamic temperature regulation and does not protect neuronal activity from the effects of an increase in temperature.25 Finally, augmentation of rCBF is unlikely, because glibenclamide does not affect CBF normally,26 and it attenuates compensatory increases in CBF associated with hypoxia27 and hypercapnia.28
Our analysis indicated that cortical lesion size was closely related to hemispheric swelling (Figure 2).29 Hemispheric swelling of 10% yielded half-maximum cortical infarction, and hemispheric swelling of 20% or more yielded maximum cortical infarction. For a rat hemisphere with a volume of 600 μL, 10% to 20% swelling would add 60 to 120 μL, which, if abruptly infused, would be enough to increase intracranial pressure above mean venous pressure or even above mean arterial pressure30 and thereby impair tissue perfusion. Our analysis also indicated that the apparent probability of infarcting a given region of cortex correlated with how distal that territory was from the middle cerebral artery origin (Figure 4). Overall, these findings are not unexpected; relative flow in vessels decreases as they branch ever more distantly from their source, and flow decreases with both increasing tissue pressure and with increasing vascular resistance (Darcy's law,31 F=ΔP/R, where F is blood flow, ΔP is mean arterial pressure minus venous pressure, with venous pressure determined by tissue pressure, and R is vascular resistance). Swelling thus preferentially affects more distal middle cerebral artery territories as tissue pressure from edema increases and as resistance of pial vessels increases due to their being forced against the inner table of the skull.
The data presented in Figure 2 demonstrate that glibenclamide treatment was associated with a reduction in hemispheric swelling, reaffirming previous findings that glibenclamide is highly effective at blocking cellular edema and ionic edema,2–4 which together are the main determinants of brain swelling.32 Brain swelling is probably a more important contributor to tissue loss in ischemic stroke than is generally recognized. In addition to its effect on swelling, glibenclamide is also highly effective at blocking oncotic death of cells that express the SUR1-regulated NCCa-ATP channel.4 Effective prevention of swelling, coupled with direct prevention of cell death, may account for the strong protective effect of glibenclamide.
Supplementary Material
Figure I. A–B, Photographs of “Lynch coils” attached to miniosmotic pumps obtained at approximately 1.5 hours (A) and approximately 4 hours (B) after start of the experiment; asterisks are next to small air bubbles being extruded from the tip of the “Lynch coils”; arrows point to the level of the phenol red solution in the “Lynch coils” at the indicated times. C, The volume of solution released from the pump as a function of time; n=3.
Acknowledgments
Sources of Funding: This work was supported by grants (to J.M.S.) from the National Heart, Lung and Blood Institute (HL051932, HL082517), the National Institute of Neurological Disorders, and Stroke (NS048260).
Footnotes
Disclosures: J.M.S. holds a US patent (7285574), “A novel nonselective cation channel in neural cells and methods for treating brain swelling.”
References
- 1.Simard JM, Woo SK, Bhatta S, Gerzanich V. Drugs acting on SUR1 to treat CNS ischemia and trauma. Curr Opin Pharmacol. 2008;8:42–49. doi: 10.1016/j.coph.2007.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen M, Simard JM. Cell swelling and a nonselective cation channel regulated by internal Ca2+ and ATP in native reactive astrocytes from adult rat brain. J Neurosci. 2001;21:6512–6521. doi: 10.1523/JNEUROSCI.21-17-06512.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen M, Dong Y, Simard JM. Functional coupling between sulfonylurea receptor type 1 and a nonselective cation channel in reactive astrocytes from adult rat brain. J Neurosci. 2003;23:8568–8577. doi: 10.1523/JNEUROSCI.23-24-08568.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Simard JM, Chen M, Tarasov KV, Bhatta S, Ivanova S, Melnitchenko L, Tsymbalyuk N, West GA, Gerzanich V. Newly expressed SUR1-regulated NC(Ca-ATP) channel mediates cerebral edema after ischemic stroke. Nat Med. 2006;12:433–440. doi: 10.1038/nm1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Simard JM, Tsymbalyuk O, Ivanov A, Ivanova S, Bhatta S, Geng Z, Woo SK, Gerzanich V. Endothelial sulfonylurea receptor 1-regulated NC Ca-ATP channels mediate progressive hemorrhagic necrosis following spinal cord injury. J Clin Invest. 2007;117:2105–2113. doi: 10.1172/JCI32041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kunte H, Schmidt S, Eliasziw M, del Zoppo GJ, Simard JM, Masuhr F, Weih M, Dirnagl U. Sulfonylureas improve outcome in patients with type 2 diabetes and acute ischemic stroke. Stroke. 2007;38:2526–2530. doi: 10.1161/STROKEAHA.107.482216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Toomey JR, Valocik RE, Koster PF, Gabriel MA, McVey M, Hart TK, Ohlstein EH, Parsons AA, Barone FC. Inhibition of factor IX(a) is protective in a rat model of thromboembolic stroke. Stroke. 2002;33:578–585. doi: 10.1161/hs0202.102950. [DOI] [PubMed] [Google Scholar]
- 8.Spratt NJ, Fernandez J, Chen M, Rewell S, Cox S, van RL, Hogan L, Howells DW. Modification of the method of thread manufacture improves stroke induction rate and reduces mortality after thread-occlusion of the middle cerebral artery in young or aged rats. J Neurosci Methods. 2006;155:285–290. doi: 10.1016/j.jneumeth.2006.01.020. [DOI] [PubMed] [Google Scholar]
- 9.Remmers M, Schmidt-Kastner R, Belayev L, Lin B, Busto R, Ginsberg MD. Protein extravasation and cellular uptake after high-dose human-al-bumin treatment of transient focal cerebral ischemia in rats. Brain Res. 1999;827:237–242. doi: 10.1016/s0006-8993(99)01304-9. [DOI] [PubMed] [Google Scholar]
- 10.Stroke Therapy Academic Industry Roundtable. Recommendations for standards regarding preclinical neuroprotective and restorative drug development. Stroke. 1999;30:2752–2758. doi: 10.1161/01.str.30.12.2752. [DOI] [PubMed] [Google Scholar]
- 11.Hoyte L, Kaur J, Buchan AM. Lost in translation: taking neuroprotection from animal models to clinical trials. Exp Neurol. 2004;188:200–204. doi: 10.1016/j.expneurol.2004.05.008. [DOI] [PubMed] [Google Scholar]
- 12.O'Collins VE, Macleod MR, Donnan GA, Horky LL, van der Worp BH, Howells DW. 1026 experimental treatments in acute stroke. Ann Neurol. 2006;59:467–477. doi: 10.1002/ana.20741. [DOI] [PubMed] [Google Scholar]
- 13.Martini SR, Kent TA. Hyperglycemia in acute ischemic stroke: a vascular perspective. J Cereb Blood Flow Metab. 2007;27:435–451. doi: 10.1038/sj.jcbfm.9600355. [DOI] [PubMed] [Google Scholar]
- 14.Saha JK, Xia J, Grondin JM, Engle SK, Jakubowski JA. Acute hyperglycemia induced by ketamine/xylazine anesthesia in rats: mechanisms and implications for preclinical models. Exp Biol Med (Maywood) 2005;230:777–784. doi: 10.1177/153537020523001012. [DOI] [PubMed] [Google Scholar]
- 15.Kawai N, Keep RF, Betz AL. Hyperglycemia and the vascular effects of cerebral ischemia. Stroke. 1997;28:149–154. doi: 10.1161/01.str.28.1.149. [DOI] [PubMed] [Google Scholar]
- 16.bd Elaziz MA, Al-Dhawailie AA, Tekle A. The effect of stress on the pharmacokinetics and pharmacodynamics of glibenclamide in diabetic rats. Eur J Drug Metab Pharmacokinet. 1998;23:371–376. doi: 10.1007/BF03192296. [DOI] [PubMed] [Google Scholar]
- 17.Aronowski J, Strong R, Grotta JC. Reperfusion injury: demonstration of brain damage produced by reperfusion after transient focal ischemia in rats. J Cereb Blood Flow Metab. 1997;17:1048–1056. doi: 10.1097/00004647-199710000-00006. [DOI] [PubMed] [Google Scholar]
- 18.Niessen F, Hilger T, Hoehn M, Hossmann KA. Differences in clot preparation determine outcome of recombinant tissue plasminogen activator treatment in experimental thromboembolic stroke. Stroke. 2003;34:2019–2024. doi: 10.1161/01.STR.0000080941.73934.30. [DOI] [PubMed] [Google Scholar]
- 19.Wang CX, Yang T, Shuaib A. An improved version of embolic model of brain ischemic injury in the rat. J Neurosci Methods. 2001;109:147–151. doi: 10.1016/s0165-0270(01)00408-3. [DOI] [PubMed] [Google Scholar]
- 20.Belayev L, Liu Y, Zhao W, Busto R, Ginsberg MD. Human albumin therapy of acute ischemic stroke: marked neuroprotective efficacy at moderate doses and with a broad therapeutic window. Stroke. 2001;32:553–560. doi: 10.1161/01.str.32.2.553. [DOI] [PubMed] [Google Scholar]
- 21.Yrjanheikki J, Tikka T, Keinanen R, Goldsteins G, Chan PH, Koistinaho J. A tetracycline derivative, minocycline, reduces inflammation and protects against focal cerebral ischemia with a wide therapeutic window. Proc Natl Acad Sci U S A. 1999;96:13496–13500. doi: 10.1073/pnas.96.23.13496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xu L, Fagan SC, Waller JL, Edwards D, Borlongan CV, Zheng J, Hill WD, Feuerstein G, Hess DC. Low dose intravenous minocycline is neuroprotective after middle cerebral artery occlusion–reperfusion in rats. BMC Neurol. 2004;4:7. doi: 10.1186/1471-2377-4-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reshef A, Sperling O, Zoref-Shani E. Opening of K(ATP) channels is mandatory for acquisition of ischemic tolerance by adenosine. Neuroreport. 2000;11:463–465. doi: 10.1097/00001756-200002280-00007. [DOI] [PubMed] [Google Scholar]
- 24.Yuan HB, Huang Y, Zheng S, Zuo Z. Hypothermic preconditioning increases survival of Purkinje neurons in rat cerebellar slices after an in vitro simulated ischemia. Anesthesiology. 2004;100:331–337. doi: 10.1097/00000542-200402000-00023. [DOI] [PubMed] [Google Scholar]
- 25.Takeya M. Intrinsic factors involved in the depression of neuronal activity induced by temperature increase in rat hippocampal neurons. Kurume Med J. 2001;48:295–306. doi: 10.2739/kurumemedj.48.295. [DOI] [PubMed] [Google Scholar]
- 26.Esaki T, Itoh Y, Shimoji K, Cook M, Jehle J, Sokoloff L. Blockade of K(ATP) channels with glibenclamide does not alter functional activation of cerebral blood flow in the unanesthetized rat. Brain Res. 2002;948:56–63. doi: 10.1016/s0006-8993(02)02948-7. [DOI] [PubMed] [Google Scholar]
- 27.Tomiyama Y, Brian JE, Jr, Todd MM. Cerebral blood flow during hemodilution and hypoxia in rats: role of ATP-sensitive potassium channels. Stroke. 1999;30:1942–1947. doi: 10.1161/01.str.30.9.1942. [DOI] [PubMed] [Google Scholar]
- 28.Faraci FM, Breese KR, Heistad DD. Cerebral vasodilation during hypercapnia. Role of glibenclamide-sensitive potassium channels and nitric oxide. Stroke. 1994;25:1679–1683. doi: 10.1161/01.str.25.8.1679. [DOI] [PubMed] [Google Scholar]
- 29.Belayev L, Busto R, Zhao W, Clemens JA, Ginsberg MD. Effect of delayed albumin hemodilution on infarction volume and brain edema after transient middle cerebral artery occlusion in rats. J Neurosurg. 1997;87:595–601. doi: 10.3171/jns.1997.87.4.0595. [DOI] [PubMed] [Google Scholar]
- 30.Melton JE, Nattie EE. Intracranial volume adjustments and cerebrospinal fluid pressure in the osmotically swollen rat brain. Am J Physiol. 1984;246:R533–R541. doi: 10.1152/ajpregu.1984.246.4.R533. [DOI] [PubMed] [Google Scholar]
- 31.Darcy HPG. Les Fontaines Publiquesde la Ville de Dijon. Paris: Dalmont; 1856. [Google Scholar]
- 32.Simard JM, Kent TA, Chen M, Tarasov KV, Gerzanich V. Brain oedema in focal ischaemia: molecular pathophysiology and theoretical implications. Lancet Neurol. 2007;6:258–268. doi: 10.1016/S1474-4422(07)70055-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure I. A–B, Photographs of “Lynch coils” attached to miniosmotic pumps obtained at approximately 1.5 hours (A) and approximately 4 hours (B) after start of the experiment; asterisks are next to small air bubbles being extruded from the tip of the “Lynch coils”; arrows point to the level of the phenol red solution in the “Lynch coils” at the indicated times. C, The volume of solution released from the pump as a function of time; n=3.