

Abstract
Integrating quantitative proteomic and transcriptomic datasets promises valuable insights in unraveling the molecular mechanisms of the brain. We concentrate on recent studies using mass spectrometry and microarray data to investigate transcript and protein abundance in normal and diseased neural tissues. Highlighted are dual spatial maps of these molecules obtained using voxelation of the mouse brain. We demonstrate that the relationship between transcript and protein levels displays a specific anatomical distribution, with greatest fidelity in midline structures and the hypothalamus. Genes are also identified that have strong correlations between mRNA and protein abundance. In addition, transcriptomic and proteomic analysis of mouse models of Parkinson's disease are discussed.
Keywords: brain mapping, in situ hybridization, mass spectrometry, microarray, mouse model, neurodegeneration, Parkinson's disease, proteomics, transcriptomic, voxelation
The mammalian brain is a complex organ exhibiting a rich variety of gene expression patterns across a broad range of cell types. In-depth multimodal study is required to understand this complex network of cells and their associated transcripts and proteins. Recent advances in the quantitative detection of mRNA and proteins on a genomic scale permit localization of these gene products onto high-quality maps of the brain. Integrating this diverse quantitative information in a spatially resolved manner will be a potent tool in unraveling the molecular mechanisms of the brain, while shedding light on the etiology and pathology of disease. For reasons of accessibility and cost, the mouse (Mus musculus) is commonly used for these studies.
A number of methods are currently being employed to map and visualize gene products in the mouse brain. A recent large-scale effort by the Allen Institute for Brain Science utilized in situ hybridization (ISH) to localize transcripts on sections of the mouse brain [1]. This approach produced single-cell-resolution images of gene expression patterns in a quantitative fashion for the entire genome. Although advances in automation have increased the throughput of ISH, this commonly employed and informative method can still be laborious and costly. Sample variability also remains a concern. Quantitative comparison of expression between genes may be problematic, since mRNA levels for each gene are not measured in the same sample. Furthermore, anatomical analysis of normal and disease models would be impractical using ISH, as the interrogation is serial in nature and the cost and labor increases linearly with the number of transcripts evaluated.
The Gene Expression Nervous System Atlas (GENSAT) visualizes gene expression in the brain using large stretches (~200 kb) of the mouse genome cloned into bacterial artificial chromosomes. These clones are sufficiently large enough to cover the coding regions of genes along with many of their regulatory elements. A gene coding sequence in a bacterial artificial chromosome is replaced with enhanced green-fluorescent protein [2] and injected into mouse eggs, creating transgenic mice. Patterns of neural gene expression are then traced by localizing fluorescence in tissue sections. The process is also performed serially and is highly labor intensive. In addition, the approach does not work well with large genes (>250 kb).
High-throughput localization of protein abundance in the brain is more difficult than transcripts. This is largely owing to proteins displaying diverse chemical properties that cannot be manipulated using the generic approaches useful with nucleic acids. In principle, antibodies can be used to generate protein-abundance maps. However, this would mean producing antibodies to all known proteins - a daunting task.
Mass spectrometry (MS), an analytical technique used to identify chemical properties of unknown compounds using separation by ionization, shows much promise in elucidating regulatory mechanisms of protein abundance [3]. This method can be applied to dissected brain regions, providing crude spatial maps. An interesting approach giving higher-resolution spatial maps uses MALDI MS [4]. A fresh frozen brain section is mounted on a stainless-steel target plate, which is then coated with a matrix solution that aids in energy absorption from a laser beam while protecting the proteins. The laser beam rasters across a region of interest, vaporizing peptides and proteins that can be detected and quantified using a mass spectrometer. By collecting this information at each position across the section, 2D images of peptide localization can be reconstructed at a resolution that is only limited by the number of data points that are collected (typically 50 μm between spots). The identity of the detected peptides can be determined using automated protein database searching. As well as in the brain, the technique has been used in the relatively rapid identification of cancer-specific markers in dissected tumors [5].
Significant advantages of this MS imaging approach include its unbiased nature, high multiplexing capability and ability to detect high-molecular-weight polypeptides of up to 300 kDa in size. However, data acquisition using this approach can be time-consuming. Variability in sample preparation combining the matrix and the analyte and inconsistency in laser-impact angle can also lead to preferential ionization of soluble proteins and inconsistent results. As is the case for many imaging modalities, detection sensitivity comes at the expense of resolution.
Another drawback of MALDI MS tissue imaging is that it is less sensitive in detecting noncovalent interactions [6] between proteins than electrospray ionization, a flow-based method often paired with MS for protein detection. Electrospray ionization is not used for imaging purposes but may serve as a complementary measure of protein composition in dissected brain regions. Standard methods, including immunohistochemistry and immunofluorescence, can be used to validate MS tissue imaging findings at single-cell resolution.
Automated, multidimensional fluorescence microscopy using multiepitope-ligand cartography (MELC) is a recently heralded technique to generate spatial maps of protein localization [7]. The system incorporates cycles of affinity agent-based fluorescence tagging, imaging and bleaching in situ, and is capable of localizing hundreds of proteins in tissue sections with cellular resolution. Interestingly, hierarchical clusters of protein colocalization and organization (toponome) can be established using this method. However, this method requires libraries of affinity-based recognition agents, such as monoclonal antibodies, limiting the discovery of new proteins. In addition, the collected data are robust but binary rather than quantitative in nature.
High-resolution voxelation
Another approach giving dual transcript and protein maps in the brain is voxelation. This discovery-driven approach involves dividing the mouse brain into spatially registered voxels or cubes. Analyses of the voxels using microarrays or MS allows the reconstruction of spatial images with quantitative information on transcripts or proteins, respectively, in parallel.
A recent study voxelated a coronal section (bregma = 0.02 mm; interaural = 3.82 mm) from adult C57BL/6J mouse brains at a resolution of 1 mm [8]. For each replicate, voxels from approximately 20 mice were pooled to obtain sufficient mRNA for a linear-labeling reaction that was subsequently applied to microarrays. Three biological replicates were obtained and gene expression levels measured using custom cDNA microarrays incorporating a dye swap. This study allowed reconstruction of 20,000 2D images of gene expression.
A number of analyses confirmed that the voxelation data was of good quality. These included good concordance between replicates, a high degree of congruence between the left and right hemispheres, good agreement with quantitative reverse transcription PCR voxelation for selected genes, and strong spatial and quantitative concordance of the voxelation dataset with ISH-derived images from the Allen Brain Atlas.
Patterns of mRNA expression revealed by voxelation
A clustering analysis of gene expression patterns from the mouse voxelation data revealed four distinct groups of genes corresponding to different mapping patterns. The first cluster consisted of genes expressed in the cortex, the second cluster contained genes showing a shallow dorsal/ventral gradient, the third cluster showed genes expressed in the hypothalamus, while the fourth cluster revealed genes with expression in the striatum and corpus callosum. Although the resolution of this method is not sufficient to localize gene products at a cellular level, clustering algorithms can provide the means to detect unanticipated patterns of regionally specific gene expression in the voxelation data. Interestingly, the cluster of genes expressed in a dorsal/ventral gradient was not restricted to a known anatomical distribution. One of the genes in the cluster, CBS, was implicated previously in dorsal neural tube defects in humans. These observations suggest that the cluster may be involved in specifying the dorsal/ventral axis of the mammalian brain.
Localization of protein abundance using voxelation
A parallel study with the same brain coordinates used voxelation at a resolution of 1 mm in combination with capillary liquid chromatography (LC) Fourier transform ion cyclotron resonance (FTICR) MS to create spatial maps of relative protein abundance [9,10]. LC-FTICR MS is a powerful method for determining the proteomic differences between normal brains and disease models and makes integration of information on transcript and protein abundance practical and feasible. In this investigation, trypsin catalyzed C-terminal isotopic labeling of peptides with 16O and 18O isotopes was used to quantify relative protein levels. FITCR MS offers superior sensitivity, mass-measurement accuracy and dynamic range compared with other MS approaches, albeit at higher cost and operational complexity.
The study successfully quantitated more than 1000 relative peptide abundances. To validate the observed data, protein abundance images were compared with existing mRNA expression data from the Allen Brain Atlas and GENSAT. Although there was not a simple direct correspondence between mRNA and protein levels, good agreement was found between the datasets, lending credence to this high-throughput proteomics methodology.
Integrating transcriptomic & proteomic data with spatial information
Integrating data from the aforementioned voxelation-based mRNA and protein-abundance maps provided the opportunity to examine the relationship between these two molecular domains across a coronal section of the brain. Permutation testing revealed significant correlation between relative transcript and protein levels [8]. Examples of agreement between ISH images from the Allen Atlas and voxelation maps for both microarrays and MS are shown in Figure 1.
Figure 1. Voxelation maps of protein and mRNA levels for selected genes and corresponding ISH images from the Allen Brain Atlas.

ISH: In situ hybridization; MS: Mass spectroscopy.
To create a spatial map of the fidelity between transcript and protein levels across the coronal section, we used the Pearson correlation coefficient to evaluate concordance between relative transcript levels and protein abundances (Figure 2A). The p-value was determined empirically for each voxel by resampling the protein level data and the mRNA-expression level data 100,000 times to create a null distribution of r-values. The proportion of the null distribution that was more extreme than the actual observed r-value was taken as the p-value. Significance is shown as -log10 p-value corrected for false-discovery rates [11].
Figure 2. Relationship between mRNA and protein levels.

(A) Spatial map of the correlation between detected mRNA and protein levels in a coronal section of the brain. Significance denoted by -log10 p-value of the Pearson correlation coefficient corrected for FDR. (B) Distribution of -log10 p-values for each gene across all voxels corrected for FDR. FDR: False-discovery rate.
The central axis of the section showed the highest correlation between mRNA and protein levels, with pronounced bilateral similarity in the hypothalamus. This pattern of transcript/protein correlation may reflect the greater nuclear heterogeneity of the hypothalamus. These findings are consistent with a recent study in 3-month postnatal mice showing high levels of correlation between transcript and protein levels across different individuals using Affymetrix microarrays and 2D gel electrophoresis, respectively [12]. This work also used MELC to show high stability of protein levels in the stratum pyrimidale and stratum radiale of the hippocampus within samples and between individuals.
Conversely, we looked at the correlation of transcript and protein levels across all 71 voxels of the coronal slice for each gene. Figure 2B represents a histogram of the -log10 (p-values) of the Pearson correlation for each gene across all voxels across the 2D slice. Again, p-values were determined empirically for each gene by resampling the protein levels and mRNA gene-expression levels 10,000 times to obtain a null distribution of the r-test statistic. The p-values represent the proportion of null r-values more extreme than the observed.
Table 1 shows genes with high levels of correlation between mRNA and protein level across the coronal slice (corrected p ≤ 0.0068). These may represent genes with little translational control. By contrast, for less well-correlated genes, transcriptional and post-translational regulatory mechanisms, including variations in molecular stability, may contribute to discrepancies between detected mRNA and protein levels. A recent study integrating transcriptome and proteome data from mouse brain tissue at two different embryonic stages showed good concordance between the two datasets when compared at the level of expression ratios (E13.5 vs E9.5) of significant differentially expressed proteins and genes [13]. However, absolute expression values showed a reduced correlation that could be attributed to ambiguities in the classification of genes that give rise to multiple transcripts and protein variants. Thus, alternative splicing in mammalian cells can add substance to transcriptome and proteome comparisons, and examining gene expression at the exon instead of the gene level could improve the quality of overlap analysis. Additionally, proteins that are more prominent in the toponomic hierarchy may correlate more highly with transcript levels than those that are lower.
Table 1.
Genes with high correlation between mRNA and protein levels across the coronal slice.
| Gene name | Unigene ID | GenBank ID | FDR |
|---|---|---|---|
| Mus musculus α-globin gene and flanking regions (DNA) | Mm.196110 | H3125H07 | 0.0056 |
| M. musculus Gap43 (mRNA) | Mm.1222 | H3157F06 | 0.0058 |
| M.musculus α-globin gene and flanking regions (DNA) | Mm.196110 | H3140G04 | 0.0058 |
| M.musculus Gi2α (CBA/J, cochlea, mRNA partial, 1548 nt) | Mm.196464 | H3120G02 | 0.0060 |
| M. musculus Sdfr1 (mRNA) | Mm.15125 | H3138G03 | 0.0061 |
| M. musculus α-globin gene and flanking regions (DNA) | Mm.196110 | H3112E02 | 0.0065 |
| M. musculus Wdr1 (mRNA) | Mm.2654 | H3046H04 | 0.0065 |
| M. musculus creatine kinase B gene, complete cds | Mm.16831 | H3007E10 | 0.0065 |
| M. musculus Ldh2 (mRNA) | Mm.9745 | H3128G04 | 0.0065 |
| M. musculus Prdx3 (mRNA) | Mm.29821 | H3140D07 | 0.0068 |
FDR: False-discovery rate.
Transcriptomic & proteomic analysis of targeted brain regions
Another strategy for spatially resolving neural gene and protein expression involves the transcriptomic and proteomic analysis of dissected brain regions. One recent study combined microarray analysis of the striatum with LC-FTICR MS [14].
Two different toxicological mouse models of Parkinson's disease (PD) were used, both of which recapitulate the hallmark loss of dopaminergic neurons in the substantia nigra pars compacta. These neurons project to the striatum, and their loss is thought to be responsible for the characteristic akinesia, rigidity and tremor of PD. The first model used 1-methyl-4-phenyl-1,2,3,6-tetra-hydro pyridine (MPTP) [15], while the second model employed toxic doses of methamphetamine (METH) [16].
Using Affymetrix 430A 2.0 microarrays to evaluate relative transcript changes between experimental and control samples, the MPTP-treated striata showed 34 significantly upregulated and 29 downregulated genes, while the METH-treated striata showed 51 significantly upregulated and 40 downregulated (all with false-discovery rate < 0.05). There was significant overlap between the two disease models, with 17 upregulated and ten downregulated genes in common (χ2 = 104; df = 1; p < 0.001). This reproducibility of relative expression levels extended to all genes (r = 0.718; p < 10-16) in addition to the significantly regulated genes.
The shared genes may represent a common response of the striatum to the loss of dopaminergic afferents induced by the two different neurotoxins, while the unique genes represent idiosyncratic responses to each of the drugs. The shared genes in the two PD models implicated pathways involved in oxidative stress, mitochondrial dysfunction and proapoptotic activity. By contrast, Cyc1 and Gapdh were found with increased abundance in only the MPTP-treated mice, suggesting higher levels of cell death and oxidative damage in this model.
The MS investigation used 16O/18O labeling in conjunction with LC-FTICR MS. The analysis identified approximately 4600 unique peptides corresponding to 1614 proteins. There was good reproducibility between relative protein abundances in the biological replicates (r = 0.94 ± 0.02 within METH and MPTP models). There was also a high degree of similarity between the METH and MPTP models, with a Pearson correlation coefficient of r = 0.89 ± 0.03, suggesting that common pathways may be utilized by the two neurotoxins. The METH and MPTP models identified 149 and 199 proteins (Student's t-test) respectively, with log2 ratios significantly different from zero (p < 0.05). After further requiring a greater than 25% relative change in order to reduce false-positive results, 86 of these proteins were in common between both lists.
Only two proteins, GFAP and GPX4, showed significant relative changes at both the transcriptomic and proteomic levels in each of the neurotoxin models. Similarly, there was no significant concordance between relative protein and transcript changes for either drug (MPTP: r = 0.034, p = 0.333; METH: r = 0.042, p = 0.216). It is possible that neither arrays nor MS offers sufficient reproducibility to reliably quantitate comparatively small relative changes (<30%). Translational and post-translational regulation may also complicate the relationship of relative values between transcripts and proteins.
By contrast, comparing absolute intensity for transcript abundances across all experiments with absolute protein levels, (estimated using spectral counts) revealed a significant positive relationship (r = 0.2889; p < 10-21) [17]. These results indicate that absolute transcript abundance can predict absolute protein abundance, even in the presence of post-translational regulation.
Codon usage & translational efficiency
Based on the availability of both absolute protein and mRNA levels, it was possible to examine whether codon usage affected translational efficiency. Proteins were divided into two groups, efficiently and inefficiently translated, depending on their position in the regression scatterplot relating absolute transcript and protein levels in the striatum. Figure 3 plots the percent usage difference between the efficiently and inefficiently translated proteins in the striatum for each codon, with codons ranked from most to least common. As expected, the data showed that proteins with frequent codons tended to be efficiently translated and vice versa. As transcriptome and proteome analyses improve in sensitivity and reliability, analyses such as these can give insights into the regulation of molecular and translational processes in the cell.
Figure 3. Relationship between codon-use frequency and translation efficiency.
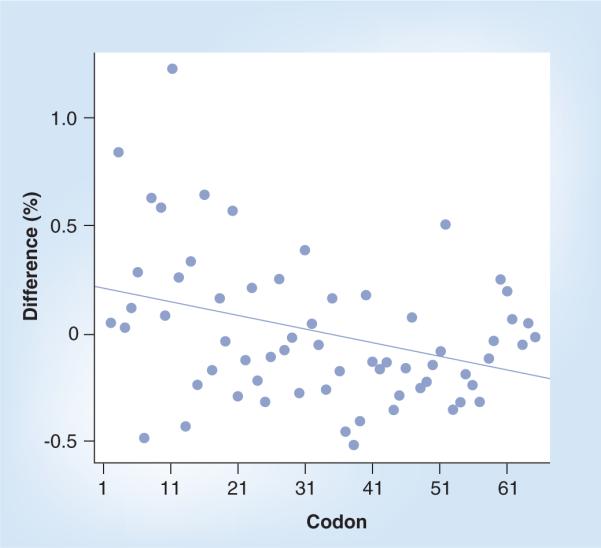
Percent usage difference shown between the efficiently and inefficiently translated proteins for each codon. Codons ranked from most to least commonly used in the mouse genome.
Expert commentary
Developing technology for high-throughput detection and quantification of proteins presents greater challenges than for transcripts. Each method for spatial mapping of proteins and transcripts in the brain offers compelling information but also suffers from significant drawbacks. Integrating data from complementary imaging modalities can offer a more comprehensive view of the brain. ISH is expensive and time-consuming but offers a single-cell-resolution view of transcript expression. Similar strengths and weaknesses apply to immunohistochemistry for visualization of proteins with the additional expense of creating antibodies. Tissue imaging using MALDI provides good resolution (500 nm) but is slow in data acquisition. Technical problems with matrix and sample preparation, along with laser angle, can lead to less-reliable results. MELC is an innovative new imaging approach with cellular resolution and the power to determine toponomic hierarchies but protein localization data is binary. Voxelation allows high-throughput acquisition of both transcript and protein mapping data and provides intermediate resolution. For all methods, cost is the major limitation, although at different steps of the analysis. For example, the expense of capital equipment can be daunting in MS, while personnel and consumables offer the greater burden in ISH. Perhaps some judicious combination of the available methods will allow optimal in-depth study of the normal and diseased brain in the future.
Information from disease models using all mapping approaches is lacking. Databases integrating many modalities of imaging across diverse disease states at differing time points will produce a clearer picture and understanding of neural disease. Currently, these resources are independently maintained but combining their strengths can prove a potent tool for the modern biologist to develop novel hypotheses. Further efforts to integrate different sources of information must be initiated and implemented.
Five-year view
Using voxelation to combine gene expression data collected using microarrays with protein abundance data, (obtained using high-throughput LC-FITCR MS) will serve as a potent method to help unravel post-transcriptional and translational control in the brain. Integrating these complementary biological datasets will help identify brain regions where translational regulation varies, leading to novel biological hypotheses. These datasets will also offer the possibility of interrogating gene expression at various stages along the continuum of disease progression. Additional advances in transcript and protein detection will be required to decrease the size of voxels and improve resolution. Given the current rapid progress in transcriptomic profiling and MS, it seems probable that these improvements will be imminent.
Voxelation can be extended to chart gene expression in three dimensions. Reconstruction of these data combined with other 3D imaging modalities, such as MRI, will allow for a much clearer understanding of neural signaling at the transcript and protein level. Voxelation and precision laser microscopic dissection of neural cellular structures can also be paired with deep-sequencing technologies, which promise greatly improved sensitivity and robustness compared with current microarray platforms [1]. Absolute rather than relative transcript abundance can be assessed while remaining nearly completely unbiased, allowing for detection of novel transcripts and spliceoforms without a priori knowledge of sequence information.
Proteomic approaches face a multitude of future challenges, but the field continues to evolve. Enhancements in sensitivity and dynamic range owing to better LC separations and MS instrumentation will offer increased coverage of complex proteomes. Improved bioinformatic algorithms will facilitate comparison of transcript abundance and protein levels across all imaging modalities, leading to more reliable datasets and further biological insights on a systems level. For instance, combining ISH transcript data from the Allen Brain Atlas with protein-localization data using MELC would be an intriguing prospect, as both methods provide cellular resolution. In addition, future work will incorporate functional information of genes and their co-localization in brain structures. Integrating these large datasets will lead to an increasingly systems-level view of neural biology. We can, therefore, expect forthcoming work in neurogenomics, and neuroproteomics will yield increasingly detailed signatures of disease, leading to improved characterization, diagnosis and treatment.
Key issues.
Comprehensive multimodal mapping of transcript and protein abundance will become more important in understanding of the brain.
Voxelation combined with mass spectrometry and microarray analysis is a highly parallel and scalable method for mapping proteins and transcripts in normal and diseased brains.
Technical challenges in sample preparation, throughput and cost must be overcome to improve resolution.
Significant advances in deep-sequencing technology paired with higher-resolution voxelation and precision laser microscopic dissection of neural cellular structures promise improvements in the quality of transcript localization data in the mouse brain.
Integration of datasets from protein and transcript maps incorporating refinements in bionfomatics methology will lead to a systems-level view of neural biology.
Acknowledgments
Financial & competing interests disclosure Portions of the research were supported by the NIH National Center for Research Resources (RR18522 to R.D.S.) and NIH grant R01 NS050148 to Desmond Smith. ISH images downloaded from the Allen Brain Atlas [Internet]. Seattle (WA, USA): Allen Institute for Brain Science. © 2008. Available from: www.brain-map.org. Proteomic analyses were performed in the Environmental Molecular Sciences Laboratory, a US Department of Energy (DOE) national scientific user facility located at the Pacific Northwest National Laboratory (PNNL) in Richland, DC, USA. PNNL is a multiprogram national laboratory operated by Battelle Memorial Institute for the DOE under Contract DE-AC05-76RL01830. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Footnotes
No writing assistance was utilized in the production of this manuscript.
References
Papers of special note have been highlighted as:
• of interest
•• of considerable interest
- 1.Lein ES, Hawrylycz MJ, Ao N, et al. Genome-wide atlas of gene expression in the adult mouse brain. Nature. 2007;445(7124):168–176. doi: 10.1038/nature05453. [DOI] [PubMed] [Google Scholar]
- 2•.Gong S, Zheng C, Doughty ML, et al. A gene expression atlas of the central nervous system based on bacterial artificial chromosomes. Nature. 2003;425(6961):917–925. doi: 10.1038/nature02033. [DOI] [PubMed] [Google Scholar]; Complete in situ hybridization atlas of all genes in the mouse brain.
- 3.Meng F, Forbes AJ, Miller LM, Kelleher NL. Detection and localization of protein modifications by high resolution tandem mass spectrometry. Mass Spectrom. Rev. 2005;24(2):126–134. doi: 10.1002/mas.20009. [DOI] [PubMed] [Google Scholar]
- 4•.Stoeckli M, Chaurand P, Hallahan DE, Caprioli RM. Imaging mass spectrometry: a new technology for the analysis of protein expression in mammalian tissues. Nat. Med. 2001;7(4):493–496. doi: 10.1038/86573. [DOI] [PubMed] [Google Scholar]; Use of MALDI to create 2D protein‑abundance maps from brainsections.
- 5.Schwartz SA, Weil RJ, Thompson RC, et al. Proteomic-based prognosis of brain tumor patients using direct-tissue matrix-assisted laser desorption ionization mass spectrometry. Cancer Res. 2005;65(17):7674–7681. doi: 10.1158/0008-5472.CAN-04-3016. [DOI] [PubMed] [Google Scholar]
- 6.Peschke M, Verkerk UH, Kebarle P. Features of the ESI mechanism that affect the observation of multiply charged noncovalent protein complexes and the determination of the association constant by the titration method. J. Am. Soc. Mass Spectrom. 2004;15(10):1424–1434. doi: 10.1016/j.jasms.2004.05.005. [DOI] [PubMed] [Google Scholar]
- 7.Schubert W, Bonnekoh B, Pommer AJ, et al. Analyzing proteome topology and function by automated multidimensional fluorescence microscopy. Nat. Biotechnol. 2006;24(10):1270–1278. doi: 10.1038/nbt1250. [DOI] [PubMed] [Google Scholar]
- 8.Chin MH, Geng AB, Khan AH, et al. A genome-scale map of expression for a mouse brain section obtained using voxelation. Physiol. Genomics. 2007;30(3):313–321. doi: 10.1152/physiolgenomics.00287.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9••.Petyuk VA, Qian WJ, Chin MH, et al. Spatial mapping of protein abundances in the mouse brain by voxelation integrated with high-throughput liquid chromatography-mass spectrometry. Genome Res. 2007;17(3):328–336. doi: 10.1101/gr.5799207. [DOI] [PMC free article] [PubMed] [Google Scholar]; Voxelation maps of proteins for a coronal section of the mouse brain at 1-mm resolution.
- 10.Qian WJ, Camp DG, 2nd, Smith RD. High-throughput proteomics using Fourier transform ion cyclotron resonance mass spectrometry. Expert Rev. Proteomics. 2004;1(1):87–95. doi: 10.1586/14789450.1.1.87. [DOI] [PubMed] [Google Scholar]
- 11.Benjamini Y, Hochberg Y. Controlling the false-discovery rate - a practical and powerful approach to multiple testing. J. R Stat. Soc. Series B. 1995;57(1):289–300. [Google Scholar]
- 12.Bode M, Irmler M, Friedenberger M, et al. Interlocking transcriptomics, proteomics and toponomics technologies for brain tissue analysis in murine hippocampus. Proteomics. 2008;8(6):1170–1178. doi: 10.1002/pmic.200700742. [DOI] [PubMed] [Google Scholar]
- 13.Irmler M, Hartl D, Schmidt T, et al. An approach to handling and interpretation of ambiguous data in transcriptome and proteome comparisons. Proteomics. 2008;8(6):1165–1169. doi: 10.1002/pmic.200700741. [DOI] [PubMed] [Google Scholar]
- 14••.Chin MH, Qian WJ, Wang H, et al. Mitochondrial dysfunction, oxidative stress, and apoptosis revealed by proteomic and transcriptomic analyses of the striata in two mouse models of Parkinson's disease. J. Proteome Res. 2008;7(2):666–677. doi: 10.1021/pr070546l. [DOI] [PMC free article] [PubMed] [Google Scholar]; Combined transcriptome and proteome analysis of the striatum in a Parkinson's disease model.
- 15.Jackson-Lewis V, Przedborski S. Protocol for the MPTP mouse model of Parkinson's disease. Nat. Protoc. 2007;2(1):141–151. doi: 10.1038/nprot.2006.342. [DOI] [PubMed] [Google Scholar]
- 16.Sonsalla PK, Jochnowitz ND, Zeevalk GD, Oostveen JA, Hall ED. Treatment of mice with methamphetamine produces cell loss in the substantia nigra. Brain Res. 1996;738(1):172–175. doi: 10.1016/0006-8993(96)00995-x. [DOI] [PubMed] [Google Scholar]
- 17.Qian WJ, Jacobs JM, Camp DG, 2nd, et al. Comparative proteome analyses of human plasma following in vivo lipopolysaccharide administration using multidimensional separations coupled with tandem mass spectrometry. Proteomics. 2005;5(2):572–584. doi: 10.1002/pmic.200400942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.'t Hoen PA, Ariyurek Y, Thygesen HH, et al. Deep sequencing-based expression analysis shows major advances in robustness, resolution and inter-lab portability over five microarray platforms. Nucleic Acids Res. 2008;36(21):e141. doi: 10.1093/nar/gkn705. [DOI] [PMC free article] [PubMed] [Google Scholar]