

Abstract
PURPOSE
Elevated metabolic activity of ovarian cancer cells causes increased ubiquitin-proteasome-system (UPS) stress, resulting in their greater sensitivity to the toxic effects of proteasomal inhibition. The proteasomes and a potentially compensatory histone deacetylase 6 (HDAC6)-dependent lysosomal pathway mediate eukaryotic protein turnover. We hypothesized that up-regulation of the HDAC6-dependent lysosomal pathway occurs in response to UPS stress and proteasomal inhibition, and thus ovarian cancer cell death can be triggered most effectively by co-inhibition of both the proteasome- and HDAC6-dependent protein degradation pathways.
EXPERIMENTAL DESIGN
To address this hypothesis we examined HDAC6 expression patterns in normal and cancerous ovarian tissues, and utilized a novel HDAC6-specific inhibitor, NK84, to address HDAC6 function in ovarian cancer.
RESULTS
Abnormally high levels of HDAC6 are expressed by ovarian cancer cells in situ and in culture relative to benign epithelium and immortalized ovarian surface epithelium respectively. Specific HDAC6 inhibition acts in synergy with the proteasome inhibitor Bortezomib (PS-341) to cause selective apoptotic cell death of ovarian cancer cells at doses that do not cause significant toxicity when used individually. Levels of UPS stress regulate the sensitivity of ovarian cancer cells to proteasome/HDAC6 inhibition. Pharmacologic inhibition of HDAC6 also reduces ovarian cancer cell spreading and migration consistent with its known function in regulating microtubule polymerization via deacetylation of α-tubulin.
CONCLUSION
Our results suggest the elevation of both the proteasomal and alternate HDAC6-dependent proteolytic pathways in ovarian cancer and the potential of combined inhibition of proteasome and HDAC6 as a therapy for ovarian cancer.
Introduction
The Ubiquitin-Proteasome-System (UPS) and the HDAC6-dependent lysosomal pathway are two major pathways for protein turn over within eukaryotic cells (1). The proteasome inhibitor Bortezomib (PS-341) has recently been licensed for the treatment of refractory multiple myeloma and mantle cell lymphoma and it is currently being examined as a treatment for several cancer types including ovarian carcinoma (2–4). PS-341 exhibits selective anti-tumor activity against ovarian cancer cells in vitro, but in a xenograft model only slowed ovarian tumor growth (5). Accumulating evidence suggests that the lysosomal pathway can compensate for intracellular poly-ubiquitinated protein degradation when UPS activity is insufficient (6–9). A critical component of the lysosomal protein degradation pathway is a microtubule-associated deacetylase, histone deacetylase 6 (HDAC6), that directly interacts with misfolded and/or poly-ubiquitinated proteins to target them for lysosome-mediated protein degradation via aggresome formation/autophagy (10–12). Because misfolded and ubiquitinated proteins are degraded via both proteasomes and HDAC6-dependent autophagy, simultaneous inhibition of proteasome and HDAC6 has been proposed as a new strategy to synergistically induce cell death in multiple myeloma and pancreatic cancer settings (6, 13). Since we previously found that ovarian cancer cells exhibit significant UPS stress (5), here we examine the potential of inhibiting both the proteasomal and HDAC6-dependent protein degradation pathways as new approach for ovarian cancer treatment. Herein we show that ovarian cancer cells are selectively sensitive to combined inhibition of proteasome and HDAC6-dependent protein degradation pathways, and the potential of this approach for treatment of ovarian cancer.
Materials and Methods
Human Specimens and Cell Lines
Studies using human tissue were performed with the approval of the Johns Hopkins Institutional Review Board. Fresh and archival tissues were obtained from the Department of Pathology of the Johns Hopkins Hospital and the latter assembled in tissue microarrays by a core facility. IOSE-29 and IOSE-397, were kindly provided by Nelly Auesperg (University of British Columbia, Vancouver, British Columbia, Canada) and cultured in Medium 199 and MCDB105 (1:1) with 10% fetal bovine serum and 50μg/mL gentamycin (Invitrogen). SKOV-3 and ES-2 and TOV-21G were obtained from American Type Culture Collection (Manassas, VA) and cultured in DMEM medium containing 10% fetal bovine serum and 50μg/mL gentamycin (Invitrogen).
Preparation of Bone Marrow Samples and Isolation of CD43+ Cells
Bone marrow aspirate was obtained from individuals who gave written informed consent in accordance with the Johns Hopkins Institutional Review Board. Under sterile conditions, samples were processed through Ficoll-density gradient for isolation of mononuclear cells (MNCs) as described previously (14). To purify CD34+ cells, MNCs were resuspended in 500 μl of binding buffer containing PBS+0.5% BSA. The cell suspension was incubated with 100 μl of human CD34 MicroBeads (Miltenyi Biotech, Auburn, CA), for 30 min at 4°C. After incubation, the cells were washed and processed through a MACS magnetic separator column (Miltenyi Biotech, Auburn, CA). Cells labeled with microbeads were passed through a column placed in a magnetic field and the target cells retained. After washing the column the target cells were recovered by removing the magnetic field and flushing the column with PBS+0.5% BSA. Recovered cells were resuspended in DMEM medium containing 10% fetal bovine serum and their viability evaluated by trypan blue dye exclusion.
Immunohistochemistry of Tissue Microarrays
Immunohistochemical analysis of paraffin-embedded tissues was done as previously described (5). Staining was diffuse across the cancer tissues and therefore was scored only on the basis of intensity. The staining was scored by three observers blind to specimen identity to obtain a consensus. Staining intensity was scored as negative (0), weak (1), intermediate (2) or strong (3).
Reagents
The proteasome inhibitor PS-341was provided by Millenium Pharmaceuticals Inc., (Cambridge, MA). The HDAC6-specific inhibitors Tubacin and NK84 were obtained from the Broad Institute and the Massachusetts Institute of Technology. Cycloheximide was purchased from SigmaAlderich (St. Louis, MO).
Antibodies and Western Blot Analysis
Total cellular protein (10–20 μg) from each sample was subjected to standard Western blot analysis. Antibodies were obtained from the following vendors: anti-HDAC6 (Cell Signaling, Beverly, MA), anti-ubiquitin and anti-vimentin (Santa Cruz Biotechnology, Santa Cruz, CA), anti-β-actin (Sigma, St. Louis, MO), anti-PARP (BD biosciences, Franklin Lakes, NJ),, anti-Hsp90 (StressGene, Victoria, British Columbia, Canada), anti- acetyl-lysine, (Cell Signalling, Beverly, MA), anti-cortactin and anti-acetyl-α-tubulin (Abcam, Cambridge, MA), peroxidase-linked anti-rabbit or anti-mouse Immunoglobulin G (Amersham, Piscataway, NJ), Texas red-labeled goat anti-mouse Immunoglobulin G (Molecular Probes, Eugene, OR), Fluorescein-labeled horse anti-rabbit Immunoglobulin G (Vector Laboratories Burlingame, CA). Antibodies were utilized at the concentration recommended by the manufacturer.
Immunoprecipitation of Hsp90 or Cortactin and Immunoblot analyses
ES-2 ovarian cancer cells treated or not with Tubacin or NK84 were lysed in RIPA buffer (Thermo Scientific, Rockford, IL) containing complete protease inhibitor cocktail (Roche, Basel, Switzerland) for 15 min on ice and subsequently centrifuged to remove nuclear and cellular debris. 200 μg of cell lysates were incubated with Hsp90 or Cortactin specific monoclonal antibodies for 1 hour at 4°C. The following day protein G-agarose beads were added and the mixture incubated at 4° C overnight. The immunoprecipitates were washed and the proteins eluted with SDS sample loading buffer prior immunoblot analysis with specific antibodies against Hsp90, Cortactin and Acetyl-Lysine. (15, 16).
XTT Assay
Cell viability/proliferation was determined by 2,3-bis-[2-methoxy-4-nitro-5-sulfophenil]-2H-tetrazolium-5-carboxanilide inner salt (XTT) assay (Roche Diagnostics GmbH, Mannheim, Germany) as previously described (5). All the experiments were performed in triplicates.
Histone Deacetylase Biochemical Assay
To measure the inhibitory effect on HDAC6 function in vitro, an optimized, continuous kinetic biochemical assay was employed. Purified, full-length HDAC protein (HDAC1 3.33 ng/μL, HDAC2 1 ng/μL, HDAC3/NCor2 0.17 ng/μL, HDAC6 1.3 ng/μL; BPS Biosciences) was incubated with a commercially-available fluorophore conjugated substrate at a concentration equivalent to the substrate Km (Upstate 17–372; 6 μM for HDAC1, 3 μM for HDAC2, 6 μM for HDAC3 and 16 μM for HDAC6). Reactions were performed in assay buffer (50 mM HEPES, 100 mM KCl, 0.001% Tween-20, 0.05% BSA, pH 7.4) and followed for fluorigenic release of 7-amino-4-methylcoumarin from substrate upon deacetylase and trypsin enzymatic activity. Fluorescence measurements are obtained as replicates in real-time on a Varioskan microtiter plate reader (Thermo). Calculation of Ki is determined using a derivation of the standard formula Ki = [Inhibitor]/((V0/Vi)*(1+S/Km))-[Substrate]/Km)-1.
In Vitro Cell Spreading and Migration Assays
SKOV-3 confluent cultures were scratched by using a sterile pipette tip and examined by phase-contrast microscopy with Spot 3.5.8 acquisition software. For migration chamber assay (2.5 × 104) SKOV-3 or ES-2 mock-treated or NK84-treated cells were seeded in the upper well of a 24-well transwell migration chamber (Costar, Lowell, MA) previously coated with Matrigel. Higher serum concentration was added to the lower well as chemo-attractant. Eight hours after seeding, the filter separating the chambers was removed and its upper surface swabbed to remove Matrigel and non-migrating cells. The migrating cells on the lower surface of the membrane migration chambers were fixed and stained with Hematoxylin and Eosin and counted. Each assay was performed in triplicates.
Immuno-fluorescence analysis
For analysis of HDAC6, ubiquitin and vimentin sub-cellular localization, cultures of SKOV-3, IOSE-29 and ES-2 were grown as described in Lab-Tek II chambered coverglass (Nalge Nunc International, Rochester, NY). At the indicated times, cells were fixed and permeabilized with methanol and incubated with the indicated primary antibodies. Fluorescent secondary antibodies were used to visualize protein localization and nuclei were visualized by 4,6-diamidino-2-phenylindole (DAPI) staining. Mounted samples were viewed under a Nikon Eclipse TE 2000E inverted microscope and images captured with Spot 3.5.8 acquisition software (Diagnostic Instruments, Sterling Heights, MI).
Statistical analysis
Results are reported as mean ± SD. Unless otherwise indicated, statistical significance of difference was assessed by two-tailed Student’s t using Prism (V.4 Graphpad, San Diego, CA) and Excel. The level of significance was set at p<0.05. The combination index of PS-341 and Tubacin was calculated by the method of Chou and Talalay (17).
Results
HDAC6 over-expression in ovarian cancer cells and tissues
To determine whether HDAC6 expression is altered in ovarian carcinogenesis we assessed HDAC6 expression patterns in benign ovarian lesions and ovarian serous carcinoma by semi-quantitative immunohistochemical staining of tissue microarrays. HDAC6 expression levels were higher in low-grade and high-grade ovarian carcinomas as compared to benign lesions (serous cystadenoma and serous adenofibroma) (Fig. 1a). Consistent with the immunohistochemical study, immunoblot analysis shows higher levels of HDAC6 in ovarian carcinomas than the benign cystadenoma (Fig. 1b). HDAC6 levels were assessed in a panel of ovarian cancer cell lines (SKOV-3, TOV-21G, ES-2) and in immortalized ovarian surface epithelial cell lines (IOSE-29 and IOSE-397). Consistent with the in vivo profile of HDAC6 over-expression, the ovarian cancer cell lines tested showed consistently higher levels of HDAC6 as compared to IOSE cell lines (Fig. 1c).
Figure 1. HDAC6 over-expression in ovarian carcinoma and cell lines.

A, left panel,immunohistochemical staining of HDAC6 in ovarian tumors. Representative examples of intense HDAC6 staining of high-grade and low-grade ovarian serous carcinomas and weaker staining in serous adenofibroma and serous cystadenoma (imaging with X40 objective), right panel, staining intensity for each case was graded as 0 (no staining), 1 (weak staining), 2 (moderate staining) and 3 (intense staining), and the statistical significance of differences in staining intensities among indicated groups was calculated using the Mann-Whitney U test. Error bars indicate ± SD. B, Western blot analysis of HDAC6 in clinical specimens of serous cystadenoma (lanes 1–4) and serous carcinoma (lanes 5–10). Equal loading was verified by using an antibody directed against β-actin. C, Western blot analysis of HDAC6 immortalized ovarian surface epithelial cells (IOSE-29 and IOSE-397) and ovarian cancer cell lines (SKOV-3, ES-2, TOV-21G). Equal protein loading in each lane was verified by using an antibody directed against β-actin.
HDAC6 inhibition specifically inhibits growth of ovarian cancer cells in vitro
We recently reported (5) that over-expression of proteasomes in ovarian cancer correlates with increased sensitivity of ovarian cancer cells to the proteasome inhibitor PS-341. Given our observation of increased HDAC6 expression in ovarian cancer cells, we examined if HDAC6 activity is important for normal growth/survival of ovarian cancer cells by comparing the relative sensitivity of a panel of ovarian cancer cell lines (SKOV-3, TOV-21G, ES-2) and IOSE cell lines to selective 1,3-dioxane-based HDAC6 inhibitors, Tubacin (6) and NK84 (18). Tubacin and NK84 are potent inhibitors of HDAC6 (HDAC6 Ki 20μM and 2.5μM, respectively), which demonstrate a 10-fold to more than 100-fold window of selectivity over other Class I and Class II deacetylases (J.E.B. and R.M., unpublished data). While minimal cell death is observable after 24 hours of NK84 treatment in all cell lines (Fig. 2a), 48 hours of NK84 treatment severely compromised the viability of ovarian cancer cell lines in a dose-dependent fashion sparing the immortalized counterpart. (Fig. 2b). Similar results were obtained when using the previously characterized HDAC6 specific inhibitor Tubacin (Supplemental Fig. 1). The toxicity profile of NK84 and Tubacin for ovarian cancer cells is consistent with their greater dependence upon HDAC6 activity.
Figure 2. NK84 treatment reduces the viability ovarian cancer cells while sparing IOSEs.

A, Ovarian cancer cell lines (SKOV-3, ES-2, TOV-21G) and immortalized ovarian surface epithelial cells (IOSE-29 and IOSE-397) were treated with the HDAC6 inhibitor NK84 (A,B) at the concentrations indicated. Cell viability was measured by XTT assay after culturing the cells for 24 hours (A) or 48 hours (B) in presence of NK84 inhibitor. Error bars indicate ± SD.
Synergistic killing of ovarian cancer cells by NK84 and PS-341
The up-regulation of both proteasome and HDAC6 in ovarian cancer, together with the selective cytotoxicity of individual treatment with either proteasome or HDAC6 inhibitors, suggested that combined inhibition of both proteasome and HDAC6-assisted proteolytic pathways might represent an effective treatment for ovarian carcinoma. To test this hypothesis, we compared the effects of combined treatment with PS-341 and NK84 on a panel of ovarian cancer cell lines and IOSEs. Fig. 3a and b show that sub-maximal doses of inhibitors act synergistically to cause dramatic cytotoxicity in the ovarian cancer cells (ES-2 and TOV-21G). Combination indices (CI) of 0.3 and 0.5 were observed when combining 10μM NK84 with either 5nM or 10nM of PS-341 respectively (17). Similar data were obtained with the several ovarian cancer cell lines we tested (data not shown) and also using the HDAC6-specific inhibitor Tubacin. Significantly, the cytotoxicity achieved using the combination of individually non-toxic doses of PS-341 (5nM) and NK84 (5μM) was comparable to that achieved with the highest dose of PS-341 or PS-341/NK84 in combination. This apparent saturation of cytotoxicity suggests that both compounds are acting on the same pathway to cause cell death. In contrast to the results with cancer cells, the combination of PS-341 and NK84 does not affect cell viability of either non-tumorigenic IOSE cell lines or CD34+ bone marrow derived progenitor cells (Supplemental Fig. 2), indicating a potential host sparing effect of HDAC6/proteasome combination.
Figure 3. Simultaneous inhibition of HDAC6 and proteasome activity induces synergistic killing of ovarian cancer cells.
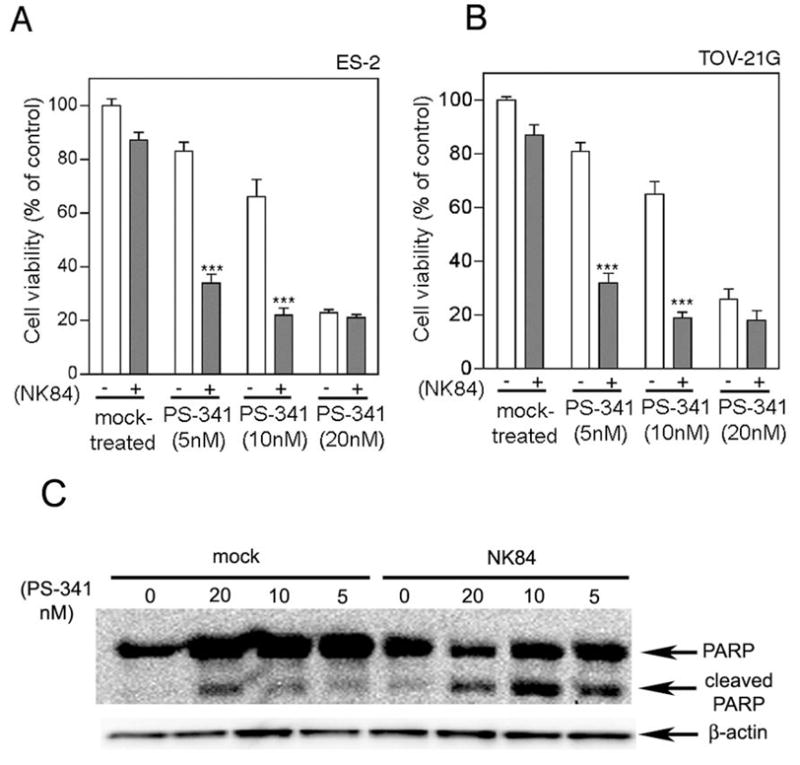
Dose-dependent inhibition of the cell viability of ES-2 (A) and TOV-21G (B) in the absence (−) or in the presence (+) of 5μM NK84 HDAC6 inhibitor and PS-341 at the indicated concentration. Cell viability was measured after a 24 hours incubation by XTT assay and the percentage of viable cells is presented relative to mock-treated controls. ***P < 0.001. Error bars indicate ± SD. Lysate of ES-2 cell line treated with NK84 (5 μm) and/or PS-341 at the indicated concentrations was immunoblotted with an antibody recognizing the full-length and cleaved forms of PARP. Equal protein loading was verified by using an antibody directed against β-actin.
NK84 is a derivative of the previously identified HDAC6-specific inhibitor Tubacin. To provide direct evidence that NK84 specifically inhibits HDAC6 we show that NK84 treatment induces α-tubulin hyper-acetylation in cultured ovarian cancer cells (Supplemental Fig. 2a). Because cortactin and Hsp90 are additional known substrates for HDAC6 activity, we tested whether HDAC6 inhibition via Tubacin or NK84 treatment induces heat shock protein 90 and/or cortactin hyper-acetylation in ovarian cancer cells. Our data show no change in the levels of acetylated cortactin or Hsp90 following NK84 treatment (Supplemental Fig. 2b,c). These results are in line with previous reports (19, 20) indicating that while de-acetylation of Hsp90 and cortactin is HDAC6-mediated, both Tubacin and NK85 only affect the α-tubulin deacetylase (TDAC) domain of HDAC6. As a further evidence that the synergistic effect upon PS-341/NK84 inhibition does not occur via Hsp90 hyper-acetylation, we failed to observe synergy for the combination of PS-341 and the Hsp90 inhibitor Geldanomycin (21) for killing of ovarian cancer cell lines (data not shown).
To assess whether the loss of viability occurs via apoptosis, we measured the levels of PARP cleavage in ovarian cancer cells receiving NK84 and/or PS-341 treatment. As shown in Fig. 3c, high levels of cleaved PARP are apparent when using the combination of individually non-toxic doses of PS-341 (5nM) and NK84 (5μM), indicating caspase-3 activation and the onset of apoptosis.
Synergy of NK84 and PS-341 is dependent upon proliferation rate and UPS stress
We have shown that the sensitivity of ovarian cancer cells to PS-341 is dependent upon their metabolic rate and degree of UPS stress (22). Thus we reasoned that the requirement of ovarian cancer cell lines for proteasome and HDAC6 activity may also depend upon their levels of metabolic rate.
To test this hypothesis, ES-2 ovarian cancer cells were induced into quiescence by treatment with the translation inhibitor cyclohexamide (CHX). Cellular quiescence was accompanied with decrease of the UPS stress levels in CHX treated cells versus control (Fig. 4a). Quiescent ES-2 ovarian cancer cell lines were tested for their sensitivity to the combination of PS-341 and NK84. The viability in CHX treated cells was significantly higher as compared to mock-treated cells, consistent with the hypothesis of a greater requirement of metabolically active cells for proteasome and HDCA6 activity (Fig. 4b). Because a decrease in proliferation rate of ovarian cancer cells is accompanied by a reduction in UPS stress (5), the proliferation of ES-2 and TOV-21G cells was slowed by contact-mediated growth inhibition (Fig. 4c) and their sensitivity to combination of NK84 and PS-341 treatment was tested. Consistent with the hypothesis, NK84/PS-341 combination treatment severely compromised the viability of exponentially growing cells, while sparing contact-inhibited cell cultures (Fig. 4d).
Figure 4. Synergistic activity of PS-341 and NK84 on ovarian cancer cells is dependent upon level of metabolic activity.

A, Immunoblot analysis of the levels of poly-ubiquitinated proteins in cyclohexamide-exposed (24h) ES-2 ovarian cancer cells. Equal loading was verified by using an antibody directed against β-actin. B, ES-2 ovarian cancer cells pre-exposed to 0, 1 or 5 mg/ml CHX (24 hours) subsequently received treatment with of PS-341 (1nM) and NK84 (5 μM) or mock treatment. Cell viability was as evaluated by XTT assay after 24 hours of treatment. Percentage of viable cells is relative to mock-treated control cells is presented. ** P < 0.02. Error bars indicate ± SD. C, proliferation rate of confluent (+) or sub-confluent (−) ES-2 and TOV-21G ovarian cancer cell lines was measured by XTT assay. Each assay was performed in triplicate. Shown are bars ± SD of proliferative activity measured in terms of optical density at 450 nm on each given day. D, cell viability of ES-2 and TOV-21G ovarian cancer cell lines was evaluated by XTT assay in sub-confluent (−) versus confluent (+) cultures in the presence of PS-341 1nM and NK84 5 μM. Percentage of viable cells is relative to mock-treated control cells. ***P < 0.001. Error bars indicate ± SD.
Formation of aggresomes in response to UPS stress in prevented by NK84
Previously reported data suggest that the formation of aggresomes following proteasome inhibition is a cytoprotective event to limit proteasome inhibitor-induced cell death (13), while the cytotoxic effects of HDAC6 inhibition seems to be linked to inhibition of aggresome formation. Consistent with this view, immunofluorescence analysis of the poly-ubiquitinated proteins in ovarian cancer cells treated with a low, sub-toxic dose of PS-341 (5nM) revealed the presence of vimentin-caged, ubiquitin-positive aggresomes in a half of the PS-341 treated cells. Conversely, simultaneous inhibition of both proteasome and HDAC6 function caused the appearance of poly-ubiquitinated proteins at multiple punctate sites throughout the cytoplasm (Fig. 5a). Interestingly, IOSEs failed to form aggresome-like structures even at the highest dose (20nM) of PS-341 (Fig. 5b).
Figure 5. HDAC6 inhibition prevents aggresome formation in UPS stressed ovarian cancer cells.

A, ES-2 ovarian cancer cells were incubated with 5nM PS-341, 10 μM NK84 or the combination of both for 24 hours before fixation and immuno-fluorescent staining of DNA (blue), ubiquitin (green) and vimentin (red) and imaging (X60 objective). B, IOSE-29 cells were incubated with 20nM PS-341 for 24 hours before fixation and immuno-fluorescent staining of DNA (blue), ubiquitin (green) and vimentin (red) and imaging (X60 objective). C, Immunoblot analysis of ubiquitinated protein in ES-2 cells (left panel) or IOSE-29 cells (right panel) 24h after treatment with or without 10 μM NK84 and the indicated concentrations of PS-341. Equal protein loading in each lane was verified by using an antibody directed against β-actin.
We then examined the impact of proteasome and HDAC6 inhibition on accumulation of poly-ubiquitinated proteins by immunoblot analysis in ES-2 ovarian cancer cell and IOSE-29. Poly-ubiquitinated proteins accumulate in ES-2 cells upon treatment with PS-341, whereas treatment with 10μM NK84 does not lead to accumulation of poly-ubiquitinated proteins. However, simultaneous inhibition of proteasome and HDAC6 activity results in massive accumulation of poly-ubiquitinated proteins and cell toxicity (Fig. 5c left panel). This suggests that while the HDAC6-dependent pathway is a minor protein degradation pathway under normal conditions, it becomes crucial in compensating for high levels of UPS stress generated by proteasomal inhibition in ovarian cancer cells. Surprisingly, while PS-341 treatment of IOSE-29 cells does trigger the accumulation of poly-ubiqutinated protein, simultaneous inhibition of proteasome and HDAC6 activity does not further increase in levels of poly-ubiquitinated protein as compared to PS-341 treatment alone (Fig. 5c right panel). This finding and the failure of IOSE-29 cell to form aggresome-like structures even at higher level proteasome inhibition (20 nM PS-341) both suggest that ovarian cancer cells and IOSEs differ in their capacity to form aggresomes following proteasome blockade and are consistent with the low level of HDAC6 expression in IOSE.
HDAC6 inhibition impedes cell spreading and migration of ovarian cancer cells
Pharmacological inhibition of HDAC6 activity and knockdown of HDAC6 protein levels have been shown to hinder the motility of fibroblasts (23, 24), T-cells (25) and breast cancer cell lines (26). Because cell motility has clear relevance to cancer cell invasion and metastasis, we assessed whether HDAC6 function is important for motility/migration of ovarian cancer cells. Immuno-staining analysis reveals that HDAC6 is mainly localized in punctate structures around the nuclei in non-migrating cells, whereas it localizes in the leading edges of migrating ovarian cancer cells (Supplemental Fig. 3), suggesting a role for HDAC6 during ovarian cancer cell motility. To investigate the potential role for HDAC6 during cell movement, the effect of HDAC6 inhibition on ovarian cancer cell spreading was tested using scratch assays. As shown in Fig. 6a, spreading of SKOV-3 ovarian cancer cells across a gash in a monolayer occurs at a retarded rate upon NK84 treatment as compared to mock treated cells (Fig. 6a, b).
Figure 6. Pharmacologic inhibition of HDAC6 impedes cell motility and migration of ovarian cancer cells.

A Plates of confluent SKOV-3 cells either mock or NK84 treated (10 μM) were examined by phase-contrast microscopy at the time of removal by scratching (0 hours) and 5 hours later. B, Analysis of the number of SKOV-3 cells spreading across the wound; results are means ± SD of three independent experiments. C, an equivalent number (2.5 × 104) of SKOV-3 or ES-3 cells mock-treated or treated with 10 μM of NK84 HDAC6 inhibitor were seeded in migration chambers and migrating cells were counted per each condition 8 hours thereafter. Three different fields of cells for each condition were counted at 40X. Each assay was performed in triplicate. Shown are means ± SD of migrating cells per microscopic field. D, representative example of migration assay conducted on mock or NK84-treated SKOV-3 cells.
We further investigated the effect of HDAC6 inhibition upon motility in a trans-matrigel migration assay based upon chemo-attraction to higher serum levels. As shown in Fig. 6c, the ability of ES-2 and SKOV-3 ovarian cancer cell lines to invade the matrigel substratum is inhibited in NK84-treated as compared to control cells. A representative example of these trans-matrigel migration assays with or without NK84 treatment is given in Fig. 6d. The rapid re-distribution of HDAC6 during ovarian cancer cell motility and the retardation of movement by pharmacologic inhibition of HDAC6 deacetylase activity are consistent with its pivotal role in controlling the levels of cortactin (27) and acetylated α-tubulin in dynamic cellular structures (23, 24).
Discussion
In this study we show that up-regulation of HDAC6 occurs in ovarian cancer cell lines and tissues likely in response to UPS stress. Consistent with the hypothesis of higher demand for HDAC6 activity by ovarian cancer cells, the HDAC6-specific inhibitor NK84 selectively induces cytotoxicity in ovarian cancer cells while sparing IOSE cell lines. Importantly, we find that inhibition of HDAC6 synergistically enhances the cytotoxic effect caused by proteasomal inhibition in a UPS stress-dependent manner. Finally we show that HDAC6 plays a critical role in ovarian cancer cell motility and migration.
As a result of a tightly regulated process necessary to maintain homeostasis, proteins within cells are constantly synthesized and then degraded via two pathways; the proteasomal and the lysosomal pathway. Importantly, the HDAC6-dependent lysosomal pathway seems to become a critical pathway for proteasomal degradation under circumstances of aberrant accumulation of aggregation-prone protein or upon inhibition of proteasome function, situations leading to increased and toxic levels of UPS stress.
We have previously shown that aberrant high expression of proteasomes naturally occurs in ovarian cancer in vivo and that their protein levels follow the demand/offers rule as they can be manipulated in vitro by changing the level of metabolic activity (5). In this report we show that ovarian cancer cells also aberrantly express HDAC6 in vivo and in vitro, suggesting that the concomitant up-regulation of conventional and alternative protein degradation pathway may be required in ovarian cancer cells to ensure homeostasis. Other investigators have reported up-regulation of HDAC6 in oral squamous cell carcinoma (28), suggesting that this may be a common phenomenon. Further evidence for up-regulation of the alternate pathway in ovarian cancer cells is provided by enhanced basal autophagic activity, their selective loss of viability upon HDAC6 inhibition (with and without concomitant proteasomal inhibition) and their propensity to form aggresomes upon proteasomal treatment as compared with IOSE.
Specific small molecule inhibitors represent a powerful tool for investigating the effect of inhibition of HDAC6 in cancer setting (29–31). Indeed Tubacin, the first HDAC6-specific inhibitor discovered, has been successfully evaluated for its anti-tumor activity in several cancer types, including multiple myeloma, pancreatic cancer, lung cancer and breast cancer-derived cell lines (6, 13, 32). Our study extends these previous findings by showing that the cytotoxic effect of HDAC6 inhibition occurs in ovarian cancer cells while sparing their immortalized normal counterpart. Significantly, increased levels of acetylation of α-tubulin are already observable after only 8 hours of NK84 exposure (data not shown). This result confirms efficient and rapid inhibition of HDAC6 activity by NK84, while the cytotoxicity requires prolonged exposure to HDAC6 inhibition.
Leading edges of cells are highly dynamic structures. Within eukaryotic cells, motility requires tight regulation of the levels of acetylated α-tubulin and cortactin. Various lines of evidence indicate that HDAC6 also plays an important role in cell motility. Indeed, its effect on the levels of α-tubulin acetylation at the leading edges of fibroblasts ensures tight regulation of microtubule polymerization. Notably, over-expression of HDAC6 has been shown to promote chemotactic cell movement, and here we show that its activity is required for chemotaxis. These findings suggest that the combination therapy may have benefits beyond simply killing ovarian cancer cells, possibly by retarding metastatic spread. Efforts to determine the pharmacodynamics of HDAC6 inhibitors are underway with a view to preclinical testing of these hypotheses.
Supplementary Material
Acknowledgments
Grant support was provided by the US PHS Grants RO1CA122581 (RBSR), P50 CA098252 (RBSR, REB and M-CW), and the HERA foundation (MB, ZL, AS). We thank the Tissue bank of the Johns Hopkins SPORE in Cervical Cancer for tissue specimens, and Drs. Ivan Borrello and Kimberly Noonan for providing us with bone marrow aspirates. We thank Sean Patrick for her encouragement and fund raising activities.
Abbreviations
- UPS
ubiquitin-proteasome-system
- HDAC6
histone-deacetylase 6
- MM
multiple myeloma
- CHX
cyclohexamide
- XTT
2,3-bis-[2-methoxy-4-nitro-5- sulfophenil]-2H-tetrazolium-5-carboxanilide inner salt
- IOSE
immortalized ovarian surface epithelial
Footnotes
Conflict of Interest: The authors declare none.
Statement of Cancer Relevance: We show that levels of Ubiquitin Proteasome System (UPS) stress regulate the sensitivity of ovarian cancer cells to combined inhibition HDAC6 with a novel compound and proteasome function with a licensed agent, Bortezomib/PS-341. We also show that inhibition of HDAC6, which is known to regulate tubulin acetylation and microtubule polymerization, reduces ovarian cancer cell spreading and migration. Our results support the development of combined inhibition of proteasome and HDAC6 as a therapy for ovarian cancer. Our findings are significant because of the novel mechanistic insights into the aberrant regulation of HDAC6 and the proteolytic machinery in ovarian cancer, and the potential value of inhibitors in these pathways as a new approach that is urgently needed to treat ovarian cancer patients.
References
- 1.Ciechanover A. Intracellular protein degradation: from a vague idea thru the lysosome and the ubiquitin-proteasome system and onto human diseases and drug targeting. Cell Death Differ. 2005;12:1178–90. doi: 10.1038/sj.cdd.4401692. [DOI] [PubMed] [Google Scholar]
- 2.Aghajanian C. Clinical update: novel targets in gynecologic malignancies. Semin Oncol. 2004;31:22–6. doi: 10.1053/j.seminoncol.2004.10.015. discussion 33. [DOI] [PubMed] [Google Scholar]
- 3.Richardson PG, Mitsiades C, Hideshima T, Anderson KC. Proteasome inhibition in the treatment of cancer. Cell Cycle. 2005;4:290–6. [PubMed] [Google Scholar]
- 4.Rajkumar SV, Richardson PG, Hideshima T, Anderson KC. Proteasome inhibition as a novel therapeutic target in human cancer. J Clin Oncol. 2005;23:630–9. doi: 10.1200/JCO.2005.11.030. [DOI] [PubMed] [Google Scholar]
- 5.Bazzaro M, Lee MK, Zoso A, et al. Ubiquitin-proteasome system stress sensitizes ovarian cancer to proteasome inhibitor-induced apoptosis. Cancer Res. 2006;66:3754–63. doi: 10.1158/0008-5472.CAN-05-2321. [DOI] [PubMed] [Google Scholar]
- 6.Hideshima T, Bradner JE, Wong J, et al. Small-molecule inhibition of proteasome and aggresome function induces synergistic antitumor activity in multiple myeloma. Proc Natl Acad Sci U S A. 2005;102:8567–72. doi: 10.1073/pnas.0503221102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pandey UB, Batlevi Y, Baehrecke EH, Taylor JP. HDAC6 at the Intersection of Autophagy, the Ubiquitin-Proteasome System and Neurodegeneration. Autophagy. 2007:3. doi: 10.4161/auto.5050. [DOI] [PubMed] [Google Scholar]
- 8.Pandey UB, Nie Z, Batlevi Y, et al. HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature. 2007;447:859–63. doi: 10.1038/nature05853. [DOI] [PubMed] [Google Scholar]
- 9.Iwata A, Riley BE, Johnston JA, Kopito RR. HDAC6 and microtubules are required for autophagic degradation of aggregated huntingtin. J Biol Chem. 2005;280:40282–92. doi: 10.1074/jbc.M508786200. [DOI] [PubMed] [Google Scholar]
- 10.Hook SS, Orian A, Cowley SM, Eisenman RN. Histone deacetylase 6 binds polyubiquitin through its zinc finger (PAZ domain) and copurifies with deubiquitinating enzymes. Proc Natl Acad Sci U S A. 2002;99:13425–30. doi: 10.1073/pnas.172511699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kawaguchi Y, Kovacs JJ, McLaurin A, et al. The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell. 2003;115:727–38. doi: 10.1016/s0092-8674(03)00939-5. [DOI] [PubMed] [Google Scholar]
- 12.Boyault C, Zhang Y, Fritah S, et al. HDAC6 controls major cell response pathways to cytotoxic accumulation of protein aggregates. Genes Dev. 2007;21:2172–81. doi: 10.1101/gad.436407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nawrocki ST, Carew JS, Pino MS, et al. Aggresome disruption: a novel strategy to enhance bortezomib-induced apoptosis in pancreatic cancer cells. Cancer Res. 2006;66:3773–81. doi: 10.1158/0008-5472.CAN-05-2961. [DOI] [PubMed] [Google Scholar]
- 14.Narasipura SD, Wojciechowski JC, Charles N, Liesveld JL, King MR. P-Selectin coated microtube for enrichment of CD34+ hematopoietic stem and progenitor cells from human bone marrow. Clin Chem. 2008;54:77–85. doi: 10.1373/clinchem.2007.089896. [DOI] [PubMed] [Google Scholar]
- 15.Nimmanapalli R, Fuino L, Bali P, et al. Histone deacetylase inhibitor LAQ824 both lowers expression and promotes proteasomal degradation of Bcr-Abl and induces apoptosis of imatinib mesylate-sensitive or -refractory chronic myelogenous leukemia-blast crisis cells. Cancer Res. 2003;63:5126–35. [PubMed] [Google Scholar]
- 16.Guo F, Sigua C, Tao J, et al. Cotreatment with histone deacetylase inhibitor LAQ824 enhances Apo-2L/tumor necrosis factor-related apoptosis inducing ligand-induced death inducing signaling complex activity and apoptosis of human acute leukemia cells. Cancer Res. 2004;64:2580–9. doi: 10.1158/0008-5472.can-03-2629. [DOI] [PubMed] [Google Scholar]
- 17.Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984;22:27–55. doi: 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
- 18.Sternson SM, Wong JC, Grozinger CM, Schreiber SL. Synthesis of 7200 small molecules based on a substructural analysis of the histone deacetylase inhibitors trichostatin and trapoxin. Org Lett. 2001;3:4239–42. doi: 10.1021/ol016915f. [DOI] [PubMed] [Google Scholar]
- 19.Bali P, Pranpat M, Bradner J, et al. Inhibition of histone deacetylase 6 acetylates and disrupts the chaperone function of heat shock protein 90: a novel basis for antileukemia activity of histone deacetylase inhibitors. J Biol Chem. 2005;280:26729–34. doi: 10.1074/jbc.C500186200. [DOI] [PubMed] [Google Scholar]
- 20.Rodriguez-Gonzalez A, Lin T, Ikeda AK, et al. Role of the aggresome pathway in cancer: targeting histone deacetylase 6-dependent protein degradation. Cancer Res. 2008;68:2557–60. doi: 10.1158/0008-5472.CAN-07-5989. [DOI] [PubMed] [Google Scholar]
- 21.Whitesell L, Mimnaugh EG, De Costa B, Myers CE, Neckers LM. Inhibition of heat shock protein HSP90-pp60v-src heteroprotein complex formation by benzoquinone ansamycins: essential role for stress proteins in oncogenic transformation. Proc Natl Acad Sci U S A. 1994;91:8324–8. doi: 10.1073/pnas.91.18.8324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rosenwald IB. The role of translation in neoplastic transformation from a pathologist’s point of view. Oncogene. 2004;23:3230–47. doi: 10.1038/sj.onc.1207552. [DOI] [PubMed] [Google Scholar]
- 23.Tran AD, Marmo TP, Salam AA, et al. HDAC6 deacetylation of tubulin modulates dynamics of cellular adhesions. J Cell Sci. 2007;120:1469–79. doi: 10.1242/jcs.03431. [DOI] [PubMed] [Google Scholar]
- 24.Hubbert C, Guardiola A, Shao R, et al. HDAC6 is a microtubule-associated deacetylase. Nature. 2002;417:455–8. doi: 10.1038/417455a. [DOI] [PubMed] [Google Scholar]
- 25.Cabrero JR, Serrador JM, Barreiro O, et al. Lymphocyte chemotaxis is regulated by histone deacetylase 6, independently of its deacetylase activity. Mol Biol Cell. 2006;17:3435–45. doi: 10.1091/mbc.E06-01-0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Saji S, Kawakami M, Hayashi S, et al. Significance of HDAC6 regulation via estrogen signaling for cell motility and prognosis in estrogen receptor-positive breast cancer. Oncogene. 2005;24:4531–9. doi: 10.1038/sj.onc.1208646. [DOI] [PubMed] [Google Scholar]
- 27.Gao YS, Hubbert CC, Lu J, et al. Histone deacetylase 6 regulates growth factor-nduced actin remodeling and endocytosis. Mol Cell Biol. 2007;27:8637–47. doi: 10.1128/MCB.00393-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sakuma T, Uzawa K, Onda T, et al. Aberrant expression of histone deacetylase 6 in oral squamous cell carcinoma. Int J Oncol. 2006;29:117–24. [PubMed] [Google Scholar]
- 29.Haggarty SJ, Koeller KM, Wong JC, Grozinger CM, Schreiber SL. Domain-selective small-molecule inhibitor of histone deacetylase 6 (HDAC6)-mediated tubulin deacetylation. Proc Natl Acad Sci U S A. 2003;100:4389–94. doi: 10.1073/pnas.0430973100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wong JC, Hong R, Schreiber SL. Structural biasing elements for in-cell histone deacetylase paralog selectivity. J Am Chem Soc. 2003;125:5586–7. doi: 10.1021/ja0341440. [DOI] [PubMed] [Google Scholar]
- 31.Itoh Y, Suzuki T, Kouketsu A, et al. Design, Synthesis, Structure-Selectivity Relationship, and Effect on Human Cancer Cells of a Novel Series of Histone Deacetylase 6-Selective Inhibitors. J Med Chem. 2007 doi: 10.1021/jm7009217. [DOI] [PubMed] [Google Scholar]
- 32.Marcus AI, Zhou J, O’Brate A, et al. The synergistic combination of the farnesyl transferase inhibitor lonafarnib and paclitaxel enhances tubulin acetylation and requires a functional tubulin deacetylase. Cancer Res. 2005;65:3883–93. doi: 10.1158/0008-5472.CAN-04-3757. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.