

Abstract
Surprising findings about human germline mutation come from applying new technologies to detect rare mutations in germline DNA and from analyzing DNA sequence divergence between human and closely related species as well as human polymorphic variation. In this Review we discuss how these approaches affect our current understanding of the roles that sex, age, mutation hot spots, germline selection and genomic factors play in determining human base substitution mutation patterns and frequencies. To enhance progress, more extensive molecular data on the human germline with regard to mutation origin, DNA repair, epigenetic status and the consequences to gamete development of newly arisen mutations are needed.
INTRODUCTION
In humans, a sizable burden of inherited mutation contributes to ill health throughout the world1. The bulk of this morbidity and mortality derives from transmission of mutations from affected or carrier parents to their children. Some affected individuals also arise each generation as a result of inheriting a de novo autosomal dominant or X-linked germline mutation. Although new mutations are relatively rare, they are the source of the heritable genetic variation that contributes both to adaptive evolution and our species' genetic burden.
Human germline mutations are incredibly varied: they include alterations in the number of chromosomes (aneuploidy2, 3 and triploidy4) and changes in gene dosage due to duplications or deletions, ranging in size from a few base pairs to megabases. Duplications and deletions can involve microsatellites5, 6, minisatellites and expanded simple tandem repeats7, and so called “genomic disorders”8-13 including copy number variation. Remodeling of chromosomes by retrotransposition14-16, translocation17, 18 or inversions8, 17 is also observed. In this review we concentrate, for the most part, on nucleotide substitution mutations and the factors that influence their frequency and other characteristics (nucleotide changes that arise by gene conversion-like events is reviewed elsewhere19). Space restrictions virtually preclude any discussion of the important contributions made by studies in model organisms.
In the past, mutation frequency estimates (see BOX 1) were based on directly measuring the fraction of individuals born with a new disease mutation or using data on disease prevalence in the population in conjunction with estimates of the reproductive fitness of affected individuals (see20, 21). Recently, it has become possible to make experimental estimates of human disease mutation frequencies at specific nucleotides based on direct DNA analysis of male germline cells from normal individuals and some of these studies have come to the surprising conclusion that certain new human mutations provide the premeiotic germ cell with a selective advantage over non-mutant cells. Features of human germline mutation can also be inferred using computational methods to analyze large DNA sequence data sets. In this review we integrate new results from both experimental and computational methods into the broader context of what we know about human germline mutation from work in human medical genetics, molecular biology and population genetics.
BOX 1. Mutation frequency.
The term “mutation frequency” refers to the proportion of all gametes containing a new (spontaneous or de novo) mutation. This proportion should be approximately equivalent to, for example, the proportion of children born to unaffected parents (sporadic cases) for a trait inherited in an autosomal dominant fashion. At one extreme, the term could refer to the sum of all mutations of any type in a particular gene leading to the same recognizable phenotype (the gene mutation frequency); at the other extreme it can refer just to the frequency of a particular type of mutation at a specific nucleotide site. In either case “mutation frequency” is commonly used interchangeably with “mutation rate per generation”. The latter is often shortened to “mutation rate” (as we sometimes do in this paper) but should be distinguished from the “mutation rate per cell generation” (also often shortened to “mutation rate”, but not by us in this review) that can be measured in experiments on single cell organisms. In multicellular organisms both the number of germline cell generations and the time in development when the mutation arose (early mutations can inflate estimates of the number of independent mutation events) cannot be precisely established so estimated mutation rates per cell division are likely to be imprecise.
We initially describe direct and indirect methods to assess the mutation frequency. Then we consider the example of several disease mutations which arise much more frequently than would be expected and examine whether these elevated frequencies result from an increased chance of substitution at these sites. In addition to elevated frequencies at individual nucleotides, other forms of mutation rate variation have been observed; next we explore whether any genomic factors can explain this variation. Finally we consider the impact of parental origin and age. All together these new findings provide a broader understanding of germline mutation dynamics and suggest how the direction of future data collection could help us better define the factors that influence mutation frequency.
METHODS TO MEASURE SUBSTITUTION FREQUENCIES
In order to understand the implications of nucleotide substitution mutations for human disease and genetic variation it is essential to measure the frequency of substitutions at different sites in the genome. One current approach is to make direct measurements using germline DNA. Another is to make indirect measurements by comparing human DNA sequences to those of other species. The principles of these methods are described below.
Direct measurements using human germline DNA
To obtain quantitative estimates of new mutation frequencies using germline DNA requires availability of germline cells in numbers that exceed the reciprocal of the mutation frequency. Human semen is an ideal source of genetic material as a single sample can contain over 108 sperm. Obtaining comparable numbers of mature female gametes from any mammal is not feasible.
One way to make direct estimates of nucleotide substitution frequencies using germline cells from normal individuals is to employ PCR-based methods designed to only amplify molecules containing the specific substitution mutation under study22, 23. Alternatively, restriction enzymes may be found that destroy a sufficient proportion of the wild type (but not mutant) genomic template. PCR across the chosen nucleotide site follows the restriction digest and, subsequently, the product is subjected to DNA purification and sequencing24 or ligase chain reaction25. In both cases the mutation frequency at the site can be directly quantified. In a third approach26, 27 a dilution is found where no more than a single mutant molecule (on average) is expected in any DNA sample. The number of samples containing a mutation is counted using a highly sensitive modification of PCR28 and divided by the total number of genome equivalents in all the samples to estimate the frequency. The first of these three approaches has the highest false positive rate and the last the lowest. The second method requires the mutation of interest to eliminate an already existing restriction enzyme site and has an intermediate false positive rate.
Indirect measurements of mutation frequencies
The mutation frequency can be inferred indirectly by comparing aligned DNA sequences in different species. In addition to sequence data, this method requires estimates for the species divergence time, the generation times, and the effective population size of the common ancestral species (see BOX 2). Using human and chimpanzee data, Nachman and Crowell29 estimated the genome average neutral mutation rate to be 2.3×10-8 mutations per nucleotide site per generation in humans. Later datasets have given similar sequence divergence30-32, and therefore suggest similar mutation frequency estimates.
BOX 2. Divergence and diversity.
The divergence between two species is computed by counting the sequence differences between a representative of each species. The divergence can be computed for chromosomal blocks of various sequence lengths and at different positions. For neutrally evolving sequences, the average divergence equals 2ut+4Nau, where u is the mutation rate per generation, t is the divergence time measured in generations, and Na is the effective population size of the ancestral population common to the two species, (for example, see29). The mutation rate can be adjusted for the block sequence length to calculate an average per nucleotide site value. The 2ut term derives from the two branches in the separate species, while the 4Nau term comes from the coalescence time (time to find a common ancestor) in the ancestral species. Obviously there is considerable uncertainty in this latter term, and the two species are selected such that the former term is much larger. Moreover, since the coalescence time varies for different regions of the genome (due to recombination) some variation in divergence with chromosomal position is expected, however, the observed variation in divergence is greater than that expected from the variation in coalescence time30. Diversity is a measure of genetic variability within a population. The average diversity equals 4Neu, where Ne is the effective population size.
In addition to the divergence between species, the diversity within a single species is also related to the mutation rate per generation. Since the diversity formula is more strongly dependent on population genetics assumptions (which are most likely not true in natural populations) than the divergence formula, diversity is not often used to directly estimate mutation frequencies. However, both diversity and divergence are used to measure changes in mutation frequency with chromosomal position (Box 2).
MUTATION HOTSPOTS?
Substitution mutations that cause human disease are of course of great interest, as understanding the dynamics of these mutations helps us to understand the origin of many genetic disorders. In this section we discuss how direct experimental evidence has recently provided further insights into why some disease-causing mutations are seen more often than expected.
Apert syndrome and achondroplasia
Analyses of DNA sequence information29, 33 estimate the average substitution frequency per nucleotide site as 10-7-10-9. However, epidemiological analysis using hospital birth data on sporadic cases for two genetic conditions (achondroplasia and Apert syndrome) revealed high gene mutation frequencies: 10-4-10-5 for achondroplasia34, 35 and 10-5-10-6 for Apert syndrome36, 37. Surprisingly, analysis of blood DNA showed that virtually all individuals with achondroplasia have a substitution at the same nucleotide site of the fibroblast growth factor receptor 3 (FGFR3) gene38-40; and with Apert syndrome virtually all mutations arise at either one of two nucleotides in the fibroblast growth factor receptor 2 (FGFR2) gene41, 42. Therefore the Apert syndrome and achondroplasia birth data suggest a nucleotide substitution frequency higher by a factor of 100-1,000 than the average estimates29, 33. The standard explanation for this discrepancy is that these particular nucleotides have a much higher than average chance of undergoing a base substitution: they are mutation hot spots.
Testing the mutation hot spot model
Recently, new experimental approaches have been devised to test whether any particular nucleotide site is a mutation hot spot in the germline. One method26, 27 argues that the majority of base substitutions take place in the pre-meiotic self-renewing spermatogonial cell population (SrAp cells) of the testis that divides continuously throughout a man's life. SrAp cells lie uniformly scattered along the basal membrane of the seminiferous tubules that are themselves uniformly distributed throughout the testis FIGURE 1. It follows that if a nucleotide site is highly prone to mutation (a hot spot) and the mutation frequency is high enough, mutant SrAp (and their meiotic and post-meiotic descendents) will be found uniformly distributed throughout the testis FIGURE 2A.
Figure 1. Human testis and epididymis.
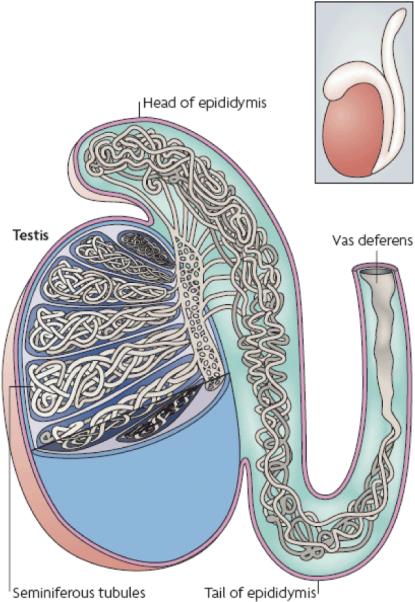
A whole testis with epididymis attached is shown in the inset box. A cross-section through the epididymis and partial exposure of the interior of the testis shows the seminiferous tubules. The testis is oriented with respect to the head and tail of the epididymis. The lumen of each tubule provides a pathway for sperm to reach the epididymis and the vas deferens.
Figure 2. Distribution of mutations at a single nucleotide site in the testis.

A. The distribution of mutations (orange dots) at a single nucleotide site in the testis expected according to the hot spot model; a uniform distribution in the tubules is predicted. B. The distribution of the Apert syndrome 755C>G or 758C>G mutants in one dissected human testis from a 62 year old donor. The testis was divided into six slices and each slice divided into the 32 pieces from which DNA was isolated. A separate aliquot was used to study each of the mutations. The orientation of the testis is relative to the head (on the left, slice 1) and tail (on the right, slice 6) of the epididymis whose long axis runs along the upper surface (slices 1-6). The mutation frequency is color-coded. (The distribution of high frequency pieces is not overly represented on the perimeter of the slices, see26, 27). C. The clusters observed by testis dissection, shown here as orange dots, could be explained if a few rare mutations each provide the affected self-renewing spermatogonial cells (SrAp) with a proliferative selective advantage.
To study the two Apert mutations (755C>G and 758C>G), testes from older normal men were dissected into ~200 pieces while retaining information on their three-dimensional relationships to one another. A modified PCR assay28 was used to estimate the mutation frequency in each piece. In each of four older donors (aged 44-62 years) the mutant cells were not found uniformly distributed in the testis FIGURE 2B. Instead, a few pieces (often adjacent ones) contained most of the mutant cells; some pieces had frequencies 103-104 fold greater than the remaining ones26, 27. As another measure of clustering, in general 95% of the mutations were found in just 5% of the testis pieces. In order to address the possibility that these clusters result from mutations early in testis development a mathematical model for germline mutation was proposed26, 27. According to this model, the clustering observed in the testis is incredibly unlikely to be due to early “jackpot”-producing mutations; therefore the hotspot model was rejected. Further data supported the conclusion that the mutation clusters were not initiated early in testis development: for two younger donors (aged 19 and 23 years) there were either no clusters or the clusters had much lower frequencies26; also, a presumably neutral transversion mutation on another chromosome was spread uniformly throughout the testis of an older donor27.
An alternative approach to test the hot spot model, using only sperm, was developed for the Apert syndrome 755C>G mutation24. This approach requires a highly informative SNP extremely close to this site (so that both can be amplified on the same DNA fragment). For each individual heterozygous for the SNP, the frequency of the 755C>G mutation is measured. The same is done for the 755C>T mutation. The researchers assumed that if both substitutions are neutral (in the testis) then the mutation with the higher observed frequency is the result of more mutation events. For each individual (and each mutation separately) the authors also examined how the mutant molecules were partitioned between the two SNP allele types (A or a). Each mutation is expected to have arisen with equal probability on a chromosome with SNP allele A and a chromosome with SNP allele a. Therefore the fraction of mutant molecules with SNP allele A, for example, should be closer to 50% for the higher frequency substitution than for the lower frequency substitution. The opposite of this expectation was reported, and it was concluded that the higher 755C>G substitution frequency was not caused by more mutation events (i.e. rejecting the hotspot model)24. However, whether this sperm data is inconsistent with the hot spot model has been questioned (Text S1 and Figure S1 in27). Finally, other features of Apert syndrome have led to questioning the validity of the hot spot model23, 43-45.
Selection can alter the frequency of germline mutation
An early idea to explain the unexpectedly high frequency of individuals born with Apert syndrome (and achondroplasia) suggested that pre-meiotic testis cells carrying these mutations had a selective advantage23, 45. The same hypothesis can explain the Apert testis dissection data26, 27 FIGURE 2C, and the Apert sperm analysis data24. More specifically, to explain the mutation clusters observed for both 755C>G and 785C>G in the testis dissection data27, the authors proposed that SrAp cells that harbor either of the two mutations had a selective advantage over wild type SrAp. This advantage was suggested to result because mutant SrAp cells occasionally divide symmetrically FIGURE 3 to produce two SrAp cells, instead of only replacing themselves as wild type SrAp cells do26, 27, 44. Thus, even if only 1% of mutant SrAp divisions are symmetrical, the observed high mutation frequency and mutation clusters can be explained even when the nucleotide substitution rate per cell division is similar to the genome average rate inferred for neutral C to G transversions29, 33. The molecular mechanism that might be responsible for such a selective advantage is unknown. However, information on the molecular pathways involved in asymmetric and symmetric cell divisions in model organisms could be especially pertinent46, 47.
Figure 3. Mutation hot spot and selection models of germline cell divisions.
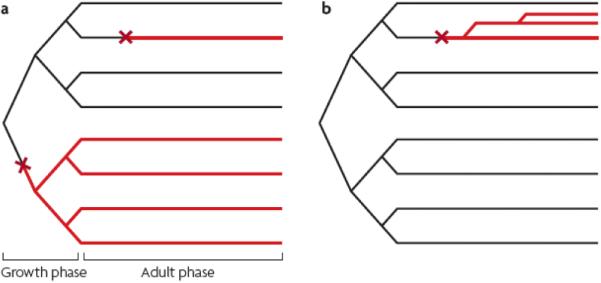
The mutation hot spot (a) and selection (b) models of germline cell division. In each case there is a growth phase and an adult phase. Two independent mutation events (red X) are shown in (a). The mutation in the adult phase produces one mutated SrAp cell lineage (red line), whereas the mutation event in the growth phase produces four mutated SrAp cell lineages. In the selection model (b) one mutation event in the adult phase produces three mutated SrAp cell lineages (from two symmetric rather than asymmetric cell divisions) that would form a cluster within the testis.
Positive germline selection may explain mutations in other genes
In addition to Apert syndrome, other disease mutations may also be subject to positive selection in the germline. In Table 1 we compare the features of Apert syndrome with three other diseases and find the similarities to be highly suggestive of positive selection. For achondroplasia, although complete testis dissection accompanied by quantitative analysis is lacking, analysis of two biopsies from each of four testes48 revealed that for very old individuals, the two biopsies from the same testis had very different mutation frequencies, whereas for the testes from middle-aged donors the two biopsies from the same testis had similar mutation frequencies.
Table 1. Are mutations in other genes subject to germline selection?
Relevant references are given in the main text. FGFR2 and FGFR3-members of the fibroblast growth factor receptor family. RET-rearranged during transfection protooncogene. MEN2B-Multiple Endocrine Neoplasia type 2B. MECP2-member of the methyl-CpG-binding protein family.
| Disease | Recurrent sporadic nucleotide substitution | Mode of transmission | Gene | Unexpected “high” mutation frequency | Virtually exclusive paternal origin | Paternal age effect | Direct testis analysis |
|---|---|---|---|---|---|---|---|
| Apert Syndrome | 755C>G 758C>G |
Dominant | FGFR2 | Yes | Yes | Yes | Yes |
| Achondro plasia | 1138G>A | Dominant | FGFR3 | Yes | Yes | Yes | Partial |
| MEN2B | 2943T>C | Dominant | RET (protooncogene) | Possibly | Yes | Yes | No |
| Rett Syndrome | Recurrent mutations at 8 CpG sites | X-linked (only females affected) | MECP2 | Possibly | Yes | Weak | No |
Like Apert syndrome, mutations leading to achondroplasia and MEN2B occur in receptor tyrosine kinases that are implicated in cancer and have a proliferative role during normal development (see49-51). MEN2B appears to share all the characteristics of Apert syndrome, except that the available data make it difficult to calculate whether the frequency of sporadic cases significantly exceeds that expected for a T>C transition mutation52-55. Interestingly, data suggest that the receptor tyrosine kinase mutated in MEN2B (RET) is required for spermatogonial self-renewal in mice (reviewed in56).
The last and most speculative possibility listed in Table 2 is Rett syndrome (see57-60). Eight different recurrent (usually inactivating) nucleotide substitutions at CpG sites account for 61% of all mutations; the remaining include large and small deletions, insertions and truncations. The Rett gene encodes a member of the methyl-CpG-binding protein family (MECP2) that specifically binds m5CpG sequences (CpG dinucleotides with the cytosine methylated at the 5-position). Alteration or inactivation of MECP2 could have wide ranging (possibly advantageous?) effects on spermatogonia lacking this activity since the protein acts as a transcriptional repressor. However, the absence of a striking paternal age effect (PAE; see the section “Age effects on mutation frequencies”) in Rett syndrome would place limits on the magnitude of any selective advantage provided to spermatogonia by a Rett gene mutation.
Table 2. Effect on male bias (α) of other factors (see text) that could influence the mutation frequency at cytosine in different contexts in males (M) vs. females (F).
In the case of C at non-CpG sites, the difference in total cell divisions is assumed to be the most important factor in explaining the high α value; no sex difference in deamination or DNA repair is assumed. For C at a CpG site, all the listed factors are also assumed equal in both sexes except for the number of cell divisions (always greater in males). If, however, an inequality (shown in blue) did exist (for example enzymatic deamination was greater in females than males) and was of sufficient magnitude, it might lead to a low α value. Repair is taken to mean either base excision repair or DNA mismatch repair (or both). A is the case where a sex-difference is assumed in methylation; B a difference in enzymatic deamination; and C a difference in repair. CpG (CpG dinucleotide), m5C (cytosine methylated at the 5-position).
| C at non-CpG site | C at CpG site | |||
|---|---|---|---|---|
| Factors | A | B | C | |
| Methylation to m5C | - | F>M | M=F | M=F |
| Deamination of m5C | - | M=F | F>M | M=F |
| Repair of G:T | - | M=F | M=F | M>F |
| Deamination of C | M=F | M=F | F>M | M=F |
| Repair of G:U | M=F | M=F | M=F | M>F |
| # Cell divisions | M>F | M>F | M>F | M>F |
| Expected α (M/F) | HIGH | LOW | LOW | LOW |
Undoubtedly, genes responsible for other autosomal dominant or X-linked genetic conditions that match several of the criteria shown in Table 1 may also be candidates for providing mutant male germline cells with a selective advantage.
Germline selection may not be restricted to nucleotide substitutions
Well documented examples exist for trinucleotide repeat expansion mutations61-68. In Fragile X syndrome, for example, affected adult males with disease alleles in their somatic cells do not produce sperm with the disease allele; instead sperm carry significantly contracted alleles. Examination of gonads of affected fetuses of different ages suggested that germ cells with smaller alleles increase in frequency as testis development proceeds, possibly because they have an advantage over cells with a disease allele.
Also, germline selection may not be limited to males. One very recent study proposes that selection of trisomy 21-carrying ovarian germline cells during fetal and postnatal life may contribute to the well known Down syndrome maternal age effect69.
Finally, new gain-of-function mutations with a germline selective advantage are more likely to be transmitted to the next generation because the effective mutation frequency is elevated beyond what the underlying rate of mutation alone can achieve. When such mutations decrease the fitness in the offspring the genetic burden on the population would increase70.
VARIATION IN SUBSTITUTION FREQUENCY
The direct method, discussed above, measures the mutation frequency at individual nucleotide sites, and has found that in the male germline, at least some sites have frequencies much greater than the genome average. The indirect method infers average frequencies in sequence blocks much wider than a single nucleotide, and has found that these averages vary with the chromosomal position of the blocks. Indeed, sequence divergence varies significantly between blocks within the same chromosome30, 71-73, between different autosomes30, 31, 73, and between autosomes and sex chromosomes30, 31, 73. This last observation will be further discussed in the “Sex differences in mutation” section. Possible explanations for all this variation include: local heterogeneity in the mutation rate or DNA repair efficiency, or selection on specific mutations (positive or negative). We would like to emphasize that here we are discussing selection on the organisms phenotype, whereas in the previous sections we were discussing selection only in the germline. The possibility of selection, and the recent explosion of genome data, has spawned many papers attempting to find genomic factors that vary with divergence and/or diversity, which we will discuss next.
Coding and non-coding comparisons
It has long been appreciated that, due to purifying selection, both diversity and divergence are greater in non-coding regions than in coding regions, and greater at synonymous sites than non-synonymous sites within coding regions. In order to estimate the neutral mutation rate, researchers have traditionally focused on non-coding regions, synonymous sites, or ancestral repeats73. It is, however, difficult to know which regions of the genome are truly neutral (for example73), since numerous non-coding regions have been found to be conserved, presumably due to selection, and there is evidence that synonymous sites are subject to selection. A more subtle difference involving mutation frequency and coding regions has also been observed: there is an asymmetry favoring G and T nucleotides over A and C nucleotides on the coding strand of genes in mammals74(see also75), possibly due to the bias introduced by transcription-coupled repair76. A later study with far more data confirmed this bias in general, but also found the opposite bias confined to the first 1-2 kb downstream of the 5' end of genes77, which requires a distinct explanation. The transcription-coupled repair idea predicts a stronger bias in genes which are highly expressed in the germline; this was tested for a set of ubiquitously expressed housekeeping genes and it was found that the average expression level across a broad range of tissue types was correlated with the strength of the mutational bias78(see also79 ). We are not aware of a study that tests this prediction with expression values measured directly in germline cells, but this would appear to be a worthwhile study.
Base identities
It has also long been appreciated that transitions are more likely than transversions, and that CpGs have higher mutation rates than other sequence contexts (see BOX 3). Relative to their expected frequency in the genome, a large proportion of new mutation events responsible for genome-wide nucleotide polymorphisms and human disease occur at 5' CpG 3' sequences29, 80-82. CpGs are further discussed in the “Sex differences in mutation” section. Researchers have considered other local contexts, and they have found that the identities of these bases also affect mutation rates, though not as strongly as the CpG context (for example,83-86). A very recent paper has further suggested that non-local contexts may also play a role87. Moreover, while not introducing new mutations, biased gene conversion is believed to complicate the analysis discussed in BOX 2 by favoring the transmission of G or C alleles at the expense of A or T alleles at already polymorphic sites, somewhat similar to a biased form of recurrent mutation (see for example 88, 89).
BOX 3. Mutation of methylated CpGs.
The base cytosine (C) is chemically less stable than the other three bases and can spontaneously undergo deamination, converting it to uracil (U) and forming a G:U mismatch. This premutagenic lesion must be repaired in order that a daughter strand is not converted to a A:U bp during the next round of DNA replication, thereby fixing the G:C to A:T mutation. DNA base excision repair (BER) may remove the U at the G:U mismatch using the enzyme UNG (uracil DNA glycosylase) and subsequently replace it with the original C (reviewed in150).
A common epigenetic modification to DNA that occurs virtually exclusively at the palindromic 5' CpG 3' site (CpG dinucleotide) involves methylation of cytosine at the 5-position (m5C) by members of the DNA methyltransferase (DMT) enzyme family151. Deamination of m5C produces a T and results in a G:T mismatch. Base excision repair (BER) involving the enzymes TDG (thymine DNA glycosylase) or MDB4 (methyl-CpG-binding domain protein) can remove the T and reinsert the C that subsequently can be remethylated (reviewed in150). If the premutagenic lesion is not repaired before the next round of DNA replication, a G:C to A:T transition mutation results.
Other factors
Divergence and/or diversity have also been found to vary with G+C content30, 71, 72, 90, CpG density (even for non-CpG mutations91), proximity to the ends of chromosomes30, 72, broad meiotic recombination fractions inferred from pedigrees72, 90, 92, fine-scale recombination hotspots inferred from population genetic data93, 94, and the intensity of Giemsa staining in cytogenetically defined chromosome bands30. Interestingly, both diversity and divergence are positively correlated with broad recombination fractions, but only diversity is correlated with fine-scale hotspots, presumably due to these recombination hotspots being short-lived relative to human-chimpanzee divergence times (reviewed in95).
Of course, many of these genomic factors are confounded with each other. The Chimpanzee Consortium30 has observed, “multiple mechanisms are at work, and that no single factor, such as G+C content or recombination rate, is an adequate predictor of regional variation in the mammalian genome by itself.” Likewise in an earlier review73, Ellegren had written: it is “highly unlikely that there is a single factor that can explain mutation rate variation.” Some researchers have attempted to fit models using many of these factors as dependent variables72, 90, 96.
A further factor that was not included in the above models, and which has since been shown to be correlated with divergence, is the proximity to small (<101 bp) insertions or deletions97 (also called indels). It has been proposed that heterozygosity for an indel, or indel creation itself, could be mutagenic to the surrounding sequences. Although no evidence was found that indel-associated mutation depends on meiotic recombination rate, DNA double strand break (DSB) formation in premeiotic germ cells is still a possible cause of these mutations since DSB repair by non-homologous end joining is potentially error prone98, as is repair by homologous recombination99.
In addition to genomic sequence data, epigenomic data is also being collected. One publication100 observed that, as measured by indirect means, the rate of mutation tends to be less in the open chromatin regions of the genome than the closed regions. These authors argued that this difference was due to greater accessibility of repair mechanisms in the open regions. However, one caveat to this work is that the open and closed chromatin structure was determined in transformed lymphoblastoid cell lines, and these structures may be different in germline cells.
SEX DIFFERENCES IN MUTATION
In this section we first discuss how paternal/maternal mutation bias is assessed (using direct and indirect methods), and then examine a number of possible explanations for this bias (i.e. influence of the type of mutation, DNA methylation and DNA repair). The first hint that a male mutation bias existed came from studies on mutation frequency using disease prevalence data and population genetic reasoning regarding the disease's effect on fitness101. Since then many additional cases have been found in which more de novo disease mutations come either from the father or mother, depending on the type of mutation event (reviewed in20, 44, 102-105).
Direct analysis of patients
Discovering the parent of origin of a mutation using SNP data does not require knowledge of the type of mutation but is difficult to interpret. Data on the tendencies for different types of mutation events in the two sexes can provide a better understanding of the factors underlying mutation bias. In many genes the vast majority of new disease mutations are nucleotide substitutions (as in achondroplasia and Apert syndrome) whereas different events (such as deletions and duplications) contribute to other diseases. In achondroplasia and Apert syndrome, the causal base substitutions are virtually always of paternal origin21, 44. However in X-linked Duchenne Muscular Dystrophy (DMD) deletion events show a female bias while base substitutions exhibit a male bias106. In hemophilia A (also X-linked), the majority of mutations are either missense/nonsense substitutions (48%) or large meiotic inversions involving intron 22 (36%). Both the base substitutions107 and inversion events108 show a male bias (arising in the maternal grandfather; fathers do not transmit X-chromosomes to their son) whereas deletions, as in DMD, are female biased107. Data on other disorders is also available (examples in20, 44, 102-105, 109, 110). The situation is clearly highly variable among diseases and must reflect the tendencies of the male and female germlines to undergo different kinds of mutation events.
In general, the most accurate calculation of male/female spontaneous mutation biases requires both the ability to detect a wide variety of mutation types, a large sample of mutations and the absence of ascertainment bias in selecting affected individuals for study since mutations causing severe disease are more likely to be noticed than those causing very mild disease. Also, what appears to be a new germline mutation originating in an unaffected parent might instead result from a postzygotic mutation early in development of the affected individual leading to mosaicism111-118. Such cases are important to ascertain since mutations in the embryo may not arise under the same constraints as in the male or female germlines. Finally, work in mice and fish119-121 raise the possibility that some mosaicism may result from “delayed” or “transgenerational” mutations” where a premutagenic lesion originates in the germline of the unaffected parent but is not fixed as a mutation until after fertilization.
Indirect analysis of bias
Sex-based nucleotide substitution mutation bias, but with a genome wide perspective, can also be inferred from DNA sequence divergence between species104, 105, 122, 123. By adjusting for the fact that over evolutionary time an autosome is equally likely to reside in a male or female germline cell whereas an X chromosome experiences a female cell environment 66% of the time (33% in a male environment), a strong male bias (α) for neutral nucleotide substitutions is detected. For example, the sequence divergence between the X chromosomes of human and chimp can be compared to estimate the autosomal divergence (X/A ratio) (α ~3-8, reviewed in124).
The above “male-driven” evolution (MDE) is usually explained by noting that cell divisions of self-renewing spermatogonia (SrAp) occur throughout a man's life, whereas oogonia cease replication during fetal life after ~30 cell generations125. Thus, sperm from twenty year old men (approximate time between generations during human-chimp evolution29) come from SrAp cells with a history of ~195 cell generations. When compared to a female gamete's cell generation history this amounts to a ~6 fold male bias and is remarkably consistent with the α values based on the DNA divergence studies. In general, such considerations have been taken as evidence that cell replication-dependent events are more important than replication-independent processes in dictating nucleotide substitution frequencies but of course this is not true for all mutation types even when only considering base substitutions (see below).
Do different kinds of nucleotide substitutions show the same male bias?
The recent estimate of α124 based on the human-chimp sequence comparisons also contrasted mutations at CpG sites where the cytosines are often methylated to CpG sites in CpG “islands” where they are rarely methylated. The results were striking: CpG sites in CpG islands show the strong male bias typical of non-CpG sites (α~7-8), but CpG sites elsewhere in the genome show a highly significant reduction in bias (α~2). The authors conclude, as have others based on less extensive data sets (reviewed in124), that G:C to A:T transitions at CpG sites may be less dependent on an excess of male germline cell generations than other nucleotide substitution types because a spontaneous chemical event (deamination) initiates the mutation rather than some other event that is specifically associated with DNA replication. This recent work is important because it points out the advantage of accumulating mutation information from sequence data parsed according to the exact nature and context of the molecular event and also because it influences how we interpret the role of age in new mutation formation (see below).
Alternative explanations for the low male bias of CpG mutations
There is a hidden assumption in the argument that the small male sex bias for CpG mutations that exist outside of CpG islands results from the fact that the mutation event is a spontaneous chemical reaction. This assumption is that the rate limiting step for a transition mutation at an m5CpG site is the spontaneous deamination event itself (see BOX 3). Mechanistically, the situation may be more complex. The chance that a sperm or egg contains a CpG to TpG transition at a particular site actually depends on whether the cytosine was methylated, the relative deamination rates of m5C compared to C (m5C > C126), and the likelihood that the pre-mutagenic lesion (a G:T or G:U mismatch, respectively) is restored to a G:C by DNA repair before the next replication event. It turns out that we know very little about these three processes in human germline tissues. Below we discuss what we know about these factors in humans and speculate (Table 2) on how they might interact with one another to result in a low male bias at CpG sites.
With regard to the methylation status of CpG sites in males vs females, data on mice indicate the time course of methylation and demethylation in the male and female germlines but the past focus has been on retrotransposons, X-inactivation or imprinted genes (reviewed in127, 128) rather than the majority of single copy coding sequences that would be major targets of disease nucleotide substitutions. In humans, a large and more inclusive set of single copy targets have recently been studied by bisulfite sequencing129. However, only sperm, (rather than self-renewing spermatogonia or female germline tissues) were examined. A very small number of human CpG sites in disease gene coding sequences have been studied in the germline130: analysis of a total of 33 CpG sites at the Factor VIII and FGFR3 genes130 showed little difference in CpG methylation levels in mature oocytes relative to mature sperm.
Regarding m5C and C deamination, it is usually assumed to be spontaneous. However, enzymatic deamination pathways are known to exist (see131-133). Data on male and female germline tissues is limited regarding the possible roles of enzymatic deamination, but does suggest that levels of one cytosine deaminase is higher in oocytes than spermatogonia131, 132.
Knowledge of germline DNA repair capabilities in humans, especially of G:T and G:U mismatches, is also scarce (see134-136). Data on efficiencies of some repair pathways are available for mouse germ cells (see137, 138). The timing of G:T and G:U pre-mutagenic lesion repair in males and females is also important. If not repaired quickly enough in male embryonic and fetal germ cells or adult male SrAp cells then the mutation will be fixed following DNA replication. The same is true in the female germline until mitotic divisions cease during fetal life. Thereafter, pre-mutagenic lesion repair could occur at anytime through adulthood until the first round of DNA replication is initiated after the mature egg is fertilized. Note that G:T mismatches may also be subject to other repair pathways (to a limited degree, see139) and that the expression levels of repair proteins in different pathways can be differentially cell cycle regulated (see140, 141) perhaps even in a sex-specific way.
The considerations described in this section suggest alternatives that might explain the low α value at CpG sites. Table 2 shows, qualitatively, how the low male mutation bias at CpG sites outside of CpG islands124 could be due to a sex difference in methylation (A), enzymatic deamination (B) or repair (C). Clearly, combinations of such alterations could achieve the same theoretical result; more experimental data is needed to understand the basis for the low male-bias at CpG sites.
AGE EFFECTS ON MUTATION FREQUENCIES
Maternal age effects for chromosomal aneuploidy (eg. Down syndrome) are well known2, 3. Based on the above discussion of sex differences in base substitution mutations and the high ratio of male to female germline cell generations, it is not surprising that for a number of genetic conditions, the chance of a sporadic case being born increases as the father ages (paternal age effect, PAE20, 21, 44, 103, 142, 143, see however144). MDE would predict a linear paternal age-dependent increase (due to the asymmetric divisions of SrAp cells). However, for a number of diseases the increase is exponential143. Direct studies on Apert syndrome and achondroplasia sperm mutation frequencies22-24, 48 reveal an increase with a man's age but have had variable success in establishing that the increase is exponential as suggested by epidemiological studies143. This inconsistency was first explained by proposing that sperm mutation frequencies may increase linearly but that sperm (not premeiotic cells) carrying such mutations have a selective advantage in fertilization leading to an overall exponential increase in sporadic cases22, 23, 48. The sperm data themselves however have been questioned in regards to the sensitivity of the mutation assays, sperm donor sample sizes and other factors (see23, 145). One sperm study on the Apert syndrome (755C>G) mutation argued that its data was consistent with an exponential increase in births but did not test the hypothesis statistically24. A more recent study examining both Apert syndrome mutations in each sperm sample has provided statistical verification for an exponential increase using a larger sample size of sperm donors and a more sensitive Apert mutation assay (unpublished work of the authors; S-R Yoon and collaborators).
An exponential increase in mutagenic events or an exponential decrease in DNA repair capacity as the germline ages could also supplement SrAp cell-generation driven linear MDE (α ~3-8, reviewed in124) and provide a basis for an observed exponential increase44, but experimental evidence in humans is lacking for both these possibilities. More importantly, neither factor could explain the observed clustering of Apert mutations in testes26, 27 (described above). Selection on mutant SrAp cells24, 26, 27, 44, however, can explain both the exponential increase observed in the epidemiological data and the mutation clustering in older donors' testes. Likewise, the selection model can explain the lower mutation frequencies and smaller clusters observed in the testes from younger donors26, 27.
A number of diseases exhibit a more linear increase in sporadic cases with the father's age (e.g. neurofibromatosis type 1, see20, 44, 143). MDE alone, in the absence of selection, could theoretically account for such cases. However the magnitude of the linear increase itself could depend on the nature of the mutations causing the disease. As discussed above, nucleotide substitution mutations at non-CpG sites (or CpG sites in CpG islands) have a sufficiently large α value (7-8) that a linear PAE would be expected. For diseases where most mutations arise at CpG sites outside of CpG islands, the expected α value (~2) would make it difficult to detect a PAE (for example see144, 146) other than in an exceptionally large epidemiological study. However, in achondroplasia, where all new mutations arise at the same CpG site (shown to be methylated in mature sperm130), the observed exponential PAE is likely due to selection acting on SrAp cells carrying the mutation. Overall, most diseases arise from a mixture of different genetic events, some of which show a strong male- and others a strong female-bias (see above). Some, but not all of these mutation events may show an age-dependent frequency increase. In such cases, detecting a paternal or maternal age effect would depend on the relative proportion of the different disease-mutation types.
Summary and future prospects
In recent years new experimental approaches have made it possible to estimate the frequencies of new human germline mutations at specific nucleotide sites. Such studies have contributed evidence that some mutations may provide germline cells with a selective advantage thereby increasing the observed mutation frequency beyond the actual number of mutation events that occur each generation. Equally exciting is the possibility that other types of mutation events besides base changes may also contribute disproportionately to the human mutational load because of germline selection. Clearly, examination of the diseases listed as possible candidates in this review, as well as others, might provide new insights into the role that germline selection may play in affecting mutation frequencies.
Recent computational approaches to studying germline mutations using the ever increasing amount of genomic data have also revealed new complexities about the factors that could influence mutation processes. These studies have the advantage of a genome-wide perspective and the large amounts of data have provided convincing evidence for their conclusions. The accumulation of more DNA sequence data as well as new analytic tools should help to further our understanding. We would like to emphasize that the experimental and computational approaches are complementary. The indirect approach could never have found the elevated frequencies at the Apert and achondroplasia disease sites, nor the paternal age effect. Since mutation rates are relatively low, the indirect method of comparing species or individuals within the same species has provided far more mutation data than is currently feasible with the direct method. However, as sequencing technology becomes less expensive, more direct studies may be possible by sequencing pedigrees.
A number of long standing assumptions about the role different factors may play in human germline mutation have little direct experimental support primarily because of the difficulty of studying human germline tissues. Methods for culturing mouse germline stem cells under in vitro conditions are developing and procedures for transplanting such cells into the mouse germline or, in some cases, even carrying out spermatogenesis in vitro have been described147-149. As such systems mature it could provide an ideal starting point for studying patterns of DNA methylation and DNA repair on homogeneous populations of relevant germline cells that someday may even extend to human tissues.
GLOSSARY DEFINITIONS
- Achondroplasia
A common form of dwarfism, inherited in an autosomal dominant fashion. OMIN #100800
- Apert syndrome
an autosomal disorder characterized by premature closing of cranial sutures, fused fingers and toes and that is transmitted in a dominant.fashion OMIN #101200
- Recombination fraction
Estimate of the proportion of all gametes that were derived from meiotic crossing over events in a chosen interval.
- Epigenomics
Analysis of epigenetic marks (DNA and protein modifications) on a genome-wide scale.
- DMD Duchenne muscular dystrophy (OMIN #310200)
A disorder caused by mutations in the X-linked dystrophin gene and characterized by rapidly-worsening muscle weakness.
- Hemophilia A
A blood clotting disease resulting from mutations in the X-linked Factor VIII gene (OMIN +306700).
- Bisulfite sequencing
Chemical treatment of genomic DNA before sequencing allowing identification of those cytosines that were methylated in the DNA from a particular tissue source. Unmethylated Cs are converted to Us, whereas methylated Cs remain unmodified.
- MEN2B Multiple Endocrine Neoplasia type 2B (OMIN #162300)
Mutation in the rearranged during transfection (RET) proto-oncogene. The mutation is inherited in an autosomal dominant fashion and leads to early childhood thyroid cancer.
- Rett syndrome
An X-linked neurodevelomental disorder associated with mental retardation (OMIN #312750). It is found sporadically almost exclusively in females who inherit a new mutation in the methyl-CpG-binding protein 2 gene (MECP2) from their father.
REFERENCES
- 1.Weatherall DJ. The global problem of genetic disease. Ann Hum Biol. 2005;32:117–22. doi: 10.1080/03014460500075480. [DOI] [PubMed] [Google Scholar]
- 2.Hassold T, Hall H, Hunt P. The origin of human aneuploidy: where we have been, where we are going. Hum Mol Genet. 2007;16(Spec No 2):R203–8. doi: 10.1093/hmg/ddm243. [DOI] [PubMed] [Google Scholar]
- 3.Pacchierotti F, Adler ID, Eichenlaub-Ritter U, Mailhes JB. Gender effects on the incidence of aneuploidy in mammalian germ cells. Environ Res. 2007;104:46–69. doi: 10.1016/j.envres.2006.12.001. [DOI] [PubMed] [Google Scholar]
- 4.Rosenbusch BE. Mechanisms giving rise to triploid zygotes during assisted reproduction. Fertil Steril. 2008;90:49–55. doi: 10.1016/j.fertnstert.2007.06.031. [DOI] [PubMed] [Google Scholar]
- 5.Ellegren H. Microsatellites: simple sequences with complex evolution. Nat Rev Genet. 2004;5:435–45. doi: 10.1038/nrg1348. [DOI] [PubMed] [Google Scholar]
- 6.Kelkar YD, Tyekucheva S, Chiaromonte F, Makova KD. The genome-wide determinants of human and chimpanzee microsatellite evolution. Genome Res. 2008;18:30–8. doi: 10.1101/gr.7113408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bois P, Jeffreys AJ. Minisatellite instability and germline mutation. Cell Mol Life Sci. 1999;55:1636–48. doi: 10.1007/s000180050402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lupski JR, Stankiewicz P. Genomic disorders: molecular mechanisms for rearrangements and conveyed phenotypes. PLoS Genet. 2005;1:e49. doi: 10.1371/journal.pgen.0010049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Inoue K, Lupski JR. Molecular mechanisms for genomic disorders. Annu Rev Genomics Hum Genet. 2002;3:199–242. doi: 10.1146/annurev.genom.3.032802.120023. [DOI] [PubMed] [Google Scholar]
- 10.Sharp AJ, Cheng Z, Eichler EE. Structural variation of the human genome. Annu Rev Genomics Hum Genet. 2006;7:407–42. doi: 10.1146/annurev.genom.7.080505.115618. [DOI] [PubMed] [Google Scholar]
- 11.Sen SK, et al. Human genomic deletions mediated by recombination between Alu elements. Am J Hum Genet. 2006;79:41–53. doi: 10.1086/504600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Scherer SW, et al. Challenges and standards in integrating surveys of structural variation. 2007 doi: 10.1038/ng2093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Turner DJ, et al. Germline rates of de novo meiotic deletions and duplications causing several genomic disorders. Nat Genet. 2008;40:90–5. doi: 10.1038/ng.2007.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Babushok DV, Kazazian HH., Jr. Progress in understanding the biology of the human mutagen LINE-1. Hum Mutat. 2007;28:527–39. doi: 10.1002/humu.20486. [DOI] [PubMed] [Google Scholar]
- 15.Chen JM, Stenson PD, Cooper DN, Ferec C. A systematic analysis of LINE-1 endonuclease-dependent retrotranspositional events causing human genetic disease. Hum Genet. 2005;117:411–27. doi: 10.1007/s00439-005-1321-0. [DOI] [PubMed] [Google Scholar]
- 16.Cordaux R, Hedges DJ, Herke SW, Batzer MA. Estimating the retrotransposition rate of human Alu elements. Gene. 2006;373:134–7. doi: 10.1016/j.gene.2006.01.019. [DOI] [PubMed] [Google Scholar]
- 17.Jacobs PA, Browne C, Gregson N, Joyce C, White H. Estimates of the frequency of chromosome abnormalities detectable in unselected newborns using moderate levels of banding. J Med Genet. 1992;29:103–8. doi: 10.1136/jmg.29.2.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Baptista J, et al. Breakpoint mapping and array CGH in translocations: comparison of a phenotypically normal and an abnormal cohort. Am J Hum Genet. 2008;82:927–36. doi: 10.1016/j.ajhg.2008.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen J-M, Cooper DN, Chuzhanova N, Ferec C, Patrinos GP. Gene conversion: mechanisms, evolution and human disease. Nat Rev Genet. 2007;8:762–775. doi: 10.1038/nrg2193. [DOI] [PubMed] [Google Scholar]
- 20.Vogel F, Motulsky AG. Human genetics : problems and approaches. Springer, Berlin; New York: 1997. [Google Scholar]
- 21.Crow JF. The origins, patterns and implications of human spontaneous mutation. Nat Rev Genet. 2000;1:40–7. doi: 10.1038/35049558. [DOI] [PubMed] [Google Scholar]
- 22.Glaser RL, et al. The paternal-age effect in Apert syndrome is due, in part, to the increased frequency of mutations in sperm. Am J Hum Genet. 2003;73:939–47. doi: 10.1086/378419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tiemann-Boege I, et al. The observed human sperm mutation frequency cannot explain the achondroplasia paternal age effect. Proc Natl Acad Sci U S A. 2002;99:14952–7. doi: 10.1073/pnas.232568699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Goriely A, McVean GA, Rojmyr M, Ingemarsson B, Wilkie AO. Evidence for selective advantage of pathogenic FGFR2 mutations in the male germ line. Science. 2003;301:643–6. doi: 10.1126/science.1085710. [DOI] [PubMed] [Google Scholar]
- 25.Cole DN, Carlson JA, Wilson VL. Human germline and somatic cells have similar TP53 and Kirsten-RAS gene single base mutation frequencies. Environ Mol Mutagen. 2008;49:417–25. doi: 10.1002/em.20390. [DOI] [PubMed] [Google Scholar]
- 26.Choi SK, Yoon SR, Calabrese P, Arnheim N. A germ-line-selective advantage rather than an increased mutation rate can explain some unexpectedly common human disease mutations. Proc Natl Acad Sci U S A. 2008;105:10143–8. doi: 10.1073/pnas.0801267105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Qin J, et al. The molecular anatomy of spontaneous germline mutations in human testes. PLoS Biol. 2007;5:e224. doi: 10.1371/journal.pbio.0050224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu Q, Sommer SS. Detection of extremely rare alleles by bidirectional pyrophosphorolysis-activated polymerization allele-specific amplification (Bi-PAP-A): measurement of mutation load in mammalian tissues. Biotechniques. 2004;36:156–66. doi: 10.2144/04361DD03. [DOI] [PubMed] [Google Scholar]
- 29.Nachman MW, Crowell SL. Estimate of the mutation rate per nucleotide in humans. Genetics. 2000;156:297–304. doi: 10.1093/genetics/156.1.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Initial sequence of the chimpanzee genome and comparison with the human genome. Nature. 2005;437:69–87. doi: 10.1038/nature04072. [DOI] [PubMed] [Google Scholar]
- 31.Ebersberger I, Metzler D, Schwarz C, Paabo S. Genomewide comparison of DNA sequences between humans and chimpanzees. Am J Hum Genet. 2002;70:1490–7. doi: 10.1086/340787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen FC, Li WH. Genomic divergences between humans and other hominoids and the effective population size of the common ancestor of humans and chimpanzees. Am J Hum Genet. 2001;68:444–56. doi: 10.1086/318206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kondrashov AS. Direct estimates of human per nucleotide mutation rates at 20 loci causing Mendelian diseases. Hum Mutat. 2003;21:12–27. doi: 10.1002/humu.10147. [DOI] [PubMed] [Google Scholar]
- 34.Orioli IM, Castilla EE, Scarano G, Mastroiacovo P. Effect of paternal age in achondroplasia, thanatophoric dysplasia, and osteogenesis imperfecta. Am J Med Genet. 1995;59:209–17. doi: 10.1002/ajmg.1320590218. [DOI] [PubMed] [Google Scholar]
- 35.Horton WA, Hall JG, Hecht JT. Achondroplasia. Lancet. 2007;370:162–72. doi: 10.1016/S0140-6736(07)61090-3. [DOI] [PubMed] [Google Scholar]
- 36.Cohen MM, et al. Birth prevalence study of the Apert syndrome. Am J Med Genet. 1992;42:655–9. doi: 10.1002/ajmg.1320420505. [DOI] [PubMed] [Google Scholar]
- 37.Tolarova MM, Harris JA, Ordway DE, Vargervik K. Birth prevalence, mutation rate, sex ratio, parents' age, and ethnicity in Apert syndrome. Am J Med Genet. 1997;72:394–8. doi: 10.1002/(sici)1096-8628(19971112)72:4<394::aid-ajmg4>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 38.Rousseau F, et al. Mutations in the gene encoding fibroblast growth factor receptor-3 in achondroplasia. Nature. 1994;371:252–4. doi: 10.1038/371252a0. [DOI] [PubMed] [Google Scholar]
- 39.Shiang R, et al. Mutations in the transmembrane domain of FGFR3 cause the most common genetic form of dwarfism, achondroplasia. Cell. 1994;78:335–42. doi: 10.1016/0092-8674(94)90302-6. [DOI] [PubMed] [Google Scholar]
- 40.Bellus GA, et al. Achondroplasia is defined by recurrent G380R mutations of FGFR3. Am J Hum Genet. 1995;56:368–73. [PMC free article] [PubMed] [Google Scholar]
- 41.Park WJ, et al. Analysis of phenotypic features and FGFR2 mutations in Apert syndrome. Am J Hum Genet. 1995;57:321–8. [PMC free article] [PubMed] [Google Scholar]
- 42.Wilkie AO, et al. Apert syndrome results from localized mutations of FGFR2 and is allelic with Crouzon syndrome. Nat Genet. 1995;9:165–72. doi: 10.1038/ng0295-165. [DOI] [PubMed] [Google Scholar]
- 43.Goriely A, et al. Gain-of-function amino acid substitutions drive positive selection of FGFR2 mutations in human spermatogonia. Proc Natl Acad Sci U S A. 2005;102:6051–6. doi: 10.1073/pnas.0500267102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Crow JF. Age and sex effects on human mutation rates: an old problem with new complexities. J Radiat Res (Tokyo) 2006;47(Suppl B):B75–82. doi: 10.1269/jrr.47.b75. [DOI] [PubMed] [Google Scholar]
- 45.Kan SH, et al. Genomic screening of fibroblast growth-factor receptor 2 reveals a wide spectrum of mutations in patients with syndromic craniosynostosis. Am J Hum Genet. 2002;70:472–86. doi: 10.1086/338758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Morrison SJ, Kimble J. Asymmetric and symmetric stem-cell divisions in development and cancer. Nature. 2006;441:1068–74. doi: 10.1038/nature04956. [DOI] [PubMed] [Google Scholar]
- 47.Knoblich JA. Mechanisms of asymmetric stem cell division. Cell. 2008;132:583–97. doi: 10.1016/j.cell.2008.02.007. [DOI] [PubMed] [Google Scholar]
- 48.Dakouane Giudicelli M, et al. Increased achondroplasia mutation frequency with advanced age and evidence for G1138A mosaicism in human testis biopsies. Fertil Steril. 2007 doi: 10.1016/j.fertnstert.2007.04.037. [DOI] [PubMed] [Google Scholar]
- 49.Eswarakumar VP, Lax I, Schlessinger J. Cellular signaling by fibroblast growth factor receptors. Cytokine Growth Factor Rev. 2005;16:139–49. doi: 10.1016/j.cytogfr.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 50.Thisse B, Thisse C. Functions and regulations of fibroblast growth factor signaling during embryonic development. Dev Biol. 2005;287:390–402. doi: 10.1016/j.ydbio.2005.09.011. [DOI] [PubMed] [Google Scholar]
- 51.Runeberg-Roos P, Saarma M. Neurotrophic factor receptor RET: structure, cell biology, and inherited diseases. Ann Med. 2007;39:572–80. doi: 10.1080/07853890701646256. [DOI] [PubMed] [Google Scholar]
- 52.Carlson KM, et al. Single missense mutation in the tyrosine kinase catalytic domain of the RET protooncogene is associated with multiple endocrine neoplasia type 2B. Proc Natl Acad Sci U S A. 1994;91:1579–83. doi: 10.1073/pnas.91.4.1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Carlson KM, et al. Parent-of-origin effects in multiple endocrine neoplasia type 2B. Am J Hum Genet. 1994;55:1076–82. [PMC free article] [PubMed] [Google Scholar]
- 54.Eng C, et al. Point mutation within the tyrosine kinase domain of the RET proto-oncogene in multiple endocrine neoplasia type 2B and related sporadic tumours. Hum Mol Genet. 1994;3:237–41. doi: 10.1093/hmg/3.2.237. [DOI] [PubMed] [Google Scholar]
- 55.Wray CJ, et al. Failure to recognize multiple endocrine neoplasia 2B: more common than we think? Ann Surg Oncol. 2008;15:293–301. doi: 10.1245/s10434-007-9665-4. [DOI] [PubMed] [Google Scholar]
- 56.Oatley JM, Brinster RL. Regulation of spermatogonial stem cell self-renewal in mammals. Annu Rev Cell Dev Biol. 2008;24:263–86. doi: 10.1146/annurev.cellbio.24.110707.175355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Amir RE, et al. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet. 1999;23:185–8. doi: 10.1038/13810. [DOI] [PubMed] [Google Scholar]
- 58.Bird A. The methyl-CpG-binding protein MeCP2 and neurological disease. Biochem Soc Trans. 2008;36:575–83. doi: 10.1042/BST0360575. [DOI] [PubMed] [Google Scholar]
- 59.Percy AK, et al. Rett syndrome: North American database. J Child Neurol. 2007;22:1338–41. doi: 10.1177/0883073807308715. [DOI] [PubMed] [Google Scholar]
- 60.Trappe R, et al. MECP2 mutations in sporadic cases of Rett syndrome are almost exclusively of paternal origin. Am J Hum Genet. 2001;68:1093–101. doi: 10.1086/320109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Malter HE, et al. Characterization of the full fragile-X-syndrome mutation in fetal gametes. Nature Genetics. 1997;15:165–169. doi: 10.1038/ng0297-165. [DOI] [PubMed] [Google Scholar]
- 62.Moutou C, Vincent MC, Biancalana V, Mandel JL. Transition from premutation to full mutation in fragile X syndrome is likely to be prezygotic. Hum Mol Genet. 1997;6:971–9. doi: 10.1093/hmg/6.7.971. [DOI] [PubMed] [Google Scholar]
- 63.Temmerman ND, et al. Intergenerational Instability of the Expanded CTG Repeat in the DMPK Gene: Studies in Human Gametes and Preimplantation Embryos. 2004;75:325–329. doi: 10.1086/422762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Moseley ML, et al. SCA8 CTG repeat: en masse contractions in sperm and intergenerational sequence changes may play a role in reduced penetrance. Hum Mol Genet. 2000;9:2125–30. doi: 10.1093/hmg/9.14.2125. [DOI] [PubMed] [Google Scholar]
- 65.Silveira I, et al. High germinal instability of the (CTG)n at the SCA8 locus of both expanded and normal alleles. Am J Hum Genet. 2000;66:830–40. doi: 10.1086/302827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.De Michele G, et al. Parental gender, age at birth and expansion length influence GAA repeat intergenerational instability in the X25 gene: pedigree studies and analysis of sperm from patients with Friedreich's ataxia. Hum Mol Genet. 1998;7:1901–6. doi: 10.1093/hmg/7.12.1901. [DOI] [PubMed] [Google Scholar]
- 67.Delatycki MB, et al. Sperm DNA analysis in a Friedreich ataxia premutation carrier suggests both meiotic and mitotic expansion in the FRDA gene. J Med Genet. 1998;35:713–6. doi: 10.1136/jmg.35.9.713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Salat U, Bardoni B, Wohrle D, Steinbach P. Increase of FMRP expression, raised levels of FMR1 mRNA, and clonal selection in proliferating cells with unmethylated fragile X repeat expansions: a clue to the sex bias in the transmission of full mutations? J Med Genet. 2000;37:842–50. doi: 10.1136/jmg.37.11.842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hulten MA, et al. On the origin of trisomy 21 Down syndrome. Mol Cytogenet. 2008;1:21. doi: 10.1186/1755-8166-1-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hastings IM. Germline selection: population genetic aspects of the sexual/asexual life cycle. Genetics. 1991;129:1167–76. doi: 10.1093/genetics/129.4.1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Smith NG, Webster MT, Ellegren H. Deterministic mutation rate variation in the human genome. Genome Res. 2002;12:1350–6. doi: 10.1101/gr.220502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hellmann I, et al. Why do human diversity levels vary at a megabase scale? Genome Res. 2005;15:1222–31. doi: 10.1101/gr.3461105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ellegren H, Smith NG, Webster MT. Mutation rate variation in the mammalian genome. Curr Opin Genet Dev. 2003;13:562–8. doi: 10.1016/j.gde.2003.10.008. [DOI] [PubMed] [Google Scholar]
- 74.Green P, Ewing B, Miller W, Thomas PJ, Green ED. Transcription-associated mutational asymmetry in mammalian evolution. Nat Genet. 2003;33:514–7. doi: 10.1038/ng1103. [DOI] [PubMed] [Google Scholar]
- 75.Touchon M, Arneodo A, d'Aubenton-Carafa Y, Thermes C. Transcription-coupled and splicing-coupled strand asymmetries in eukaryotic genomes. Nucleic Acids Res. 2004;32:4969–78. doi: 10.1093/nar/gkh823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hanawalt PC, Spivak G. Transcription-coupled DNA repair: two decades of progress and surprises. 2008;9:958–970. doi: 10.1038/nrm2549. [DOI] [PubMed] [Google Scholar]
- 77.Polak P, Arndt PF. Transcription induces strand-specific mutations at the 5' end of human genes. Genome Res. 2008;18:1216–23. doi: 10.1101/gr.076570.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Majewski J. Dependence of mutational asymmetry on gene-expression levels in the human genome. Am J Hum Genet. 2003;73:688–92. doi: 10.1086/378134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Webster MT, Smith NG, Lercher MJ, Ellegren H. Gene expression, synteny, and local similarity in human noncoding mutation rates. Mol Biol Evol. 2004;21:1820–30. doi: 10.1093/molbev/msh181. [DOI] [PubMed] [Google Scholar]
- 80.Anagnostopoulos T, Green PM, Rowley G, Lewis CM, Giannelli F. DNA variation in a 5-Mb region of the X chromosome and estimates of sex-specific/type-specific mutation rates. Am J Hum Genet. 1999;64:508–17. doi: 10.1086/302250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Krawczak M, Ball EV, Cooper DN. Neighboring-nucleotide effects on the rates of germ-line single-base-pair substitution in human genes. Am J Hum Genet. 1998;63:474–88. doi: 10.1086/301965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cooper DN, Krawczak M. The mutational spectrum of single base-pair substitutions causing human genetic disease: patterns and predictions. Hum Genet. 1990;85:55–74. doi: 10.1007/BF00276326. [DOI] [PubMed] [Google Scholar]
- 83.Hwang DG, Green P. Bayesian Markov chain Monte Carlo sequence analysis reveals varying neutral substitution patterns in mammalian evolution. Proc Natl Acad Sci U S A. 2004;101:13994–4001. doi: 10.1073/pnas.0404142101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Arndt PF, Hwa T. Identification and measurement of neighbor-dependent nucleotide substitution processes. Bioinformatics. 2005;21:2322–8. doi: 10.1093/bioinformatics/bti376. [DOI] [PubMed] [Google Scholar]
- 85.Hess ST, Blake JD, Blake RD. Wide variations in neighbor-dependent substitution rates. J Mol Biol. 1994;236:1022–33. doi: 10.1016/0022-2836(94)90009-4. [DOI] [PubMed] [Google Scholar]
- 86.Zhao Z, Boerwinkle E. Neighboring-nucleotide effects on single nucleotide polymorphisms: a study of 2.6 million polymorphisms across the human genome. Genome Res. 2002;12:1679–86. doi: 10.1101/gr.287302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hodgkinson A, Ladoukakis E, Eyre-Walker A. Cryptic variation in the human mutation rate. PLoS Biol. 2009;7:e1000027. doi: 10.1371/journal.pbio.1000027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Jeffreys AJ, Neumann R. Reciprocal crossover asymmetry and meiotic drive in a human recombination hot spot. Nat Genet. 2002;31:267–71. doi: 10.1038/ng910. [DOI] [PubMed] [Google Scholar]
- 89.Duret L, Arndt PF. The impact of recombination on nucleotide substitutions in the human genome. PLoS Genet. 2008;4:e1000071. doi: 10.1371/journal.pgen.1000071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hardison RC, et al. Covariation in frequencies of substitution, deletion, transposition, and recombination during eutherian evolution. Genome Res. 2003;13:13–26. doi: 10.1101/gr.844103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Walser JC, Ponger L, Furano AV. CpG dinucleotides and the mutation rate of non-CpG DNA. Genome Res. 2008;18:1403–14. doi: 10.1101/gr.076455.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lercher MJ, Hurst LD. Human SNP variability and mutation rate are higher in regions of high recombination. Trends Genet. 2002;18:337–40. doi: 10.1016/s0168-9525(02)02669-0. [DOI] [PubMed] [Google Scholar]
- 93.Myers S, et al. The distribution and causes of meiotic recombination in the human genome. Biochem Soc Trans. 2006;34:526–30. doi: 10.1042/BST0340526. [DOI] [PubMed] [Google Scholar]
- 94.Spencer CC, et al. The influence of recombination on human genetic diversity. PLoS Genet. 2006;2:e148. doi: 10.1371/journal.pgen.0020148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Arnheim N, Calabrese P, Tiemann-Boege I. Mammalian meiotic recombination hot spots. Annu Rev Genet. 2007;41:369–99. doi: 10.1146/annurev.genet.41.110306.130301. [DOI] [PubMed] [Google Scholar]
- 96.Tyekucheva S, et al. Human-macaque comparisons illuminate variation in neutral substitution rates. Genome Biol. 2008;9:R76. doi: 10.1186/gb-2008-9-4-r76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Tian D, et al. Single-nucleotide mutation rate increases close to insertions/deletions in eukaryotes. Nature. 2008;455:105–8. doi: 10.1038/nature07175. [DOI] [PubMed] [Google Scholar]
- 98.Honma M, et al. Non-homologous end-joining for repairing I-SceI-induced DNA double strand breaks in human cells. DNA Repair. 2007;6:781–788. doi: 10.1016/j.dnarep.2007.01.004. [DOI] [PubMed] [Google Scholar]
- 99.Rattray AJ, Shafer BK, McGill CB, Strathern JN. The roles of REV3 and RAD57 in double-strand-break-repair-induced mutagenesis of Saccharomyces cerevisiae. Genetics. 2002;162:1063–77. doi: 10.1093/genetics/162.3.1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Prendergast JG, et al. Chromatin structure and evolution in the human genome. BMC Evol Biol. 2007;7:72. doi: 10.1186/1471-2148-7-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Haldane JBS. The mutation rate of the gene for haemophilia and its segregation ratios in males and females. Ann. Eugen. 1947;13:262–271. doi: 10.1111/j.1469-1809.1946.tb02367.x. [DOI] [PubMed] [Google Scholar]
- 102.Hurst LD, Ellegren H. Sex biases in the mutation rate. Trends Genet. 1998;14:446–52. doi: 10.1016/s0168-9525(98)01577-7. [DOI] [PubMed] [Google Scholar]
- 103.Glaser RL, Jabs EW. Dear old dad. Sci Aging Knowledge Environ. 2004;2004:re1. doi: 10.1126/sageke.2004.3.re1. [DOI] [PubMed] [Google Scholar]
- 104.Li WH, Yi S, Makova K. Male-driven evolution. Curr Opin Genet Dev. 2002;12:650–6. doi: 10.1016/s0959-437x(02)00354-4. [DOI] [PubMed] [Google Scholar]
- 105.Ellegren H. Characteristics, causes and evolutionary consequences of male-biased mutation. Proc Biol Sci. 2007;274:1–10. doi: 10.1098/rspb.2006.3720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Grimm T, et al. On the origin of deletions and point mutations in Duchenne muscular dystrophy: most deletions arise in oogenesis and most point mutations result from events in spermatogenesis. J Med Genet. 1994;31:183–6. doi: 10.1136/jmg.31.3.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Becker J, et al. Characterization of the factor VIII defect in 147 patients with sporadic hemophilia A: family studies indicate a mutation type-dependent sex ratio of mutation frequencies. Am J Hum Genet. 1996;58:657–70. [PMC free article] [PubMed] [Google Scholar]
- 108.Rossiter JP, et al. Factor VIII gene inversions causing severe hemophilia A originate almost exclusively in male germ cells. Hum Mol Genet. 1994;3:1035–9. doi: 10.1093/hmg/3.7.1035. [DOI] [PubMed] [Google Scholar]
- 109.Kluwe L, et al. The parental origin of new mutations in neurofibromatosis 2. Neurogenetics. 2000;3:17–24. doi: 10.1007/s100480000088. [DOI] [PubMed] [Google Scholar]
- 110.Sommer SS, Scaringe WA, Hill KA. Human germline mutation in the factor IX gene. Mutat Res. 2001;487:1–17. doi: 10.1016/s0921-8777(01)00108-2. [DOI] [PubMed] [Google Scholar]
- 111.Zlotogora J. Germ line mosaicism. Hum Genet. 1998;102:381–6. doi: 10.1007/s004390050708. [DOI] [PubMed] [Google Scholar]
- 112.Youssoufian H, Pyeritz RE. Mechanisms and consequences of somatic mosaicism in humans. Nat Rev Genet. 2002;3:748–58. doi: 10.1038/nrg906. [DOI] [PubMed] [Google Scholar]
- 113.Erickson RP. Somatic gene mutation and human disease other than cancer. Mutat Res. 2003;543:125–36. doi: 10.1016/s1383-5742(03)00010-3. [DOI] [PubMed] [Google Scholar]
- 114.Hall JG. Review and hypotheses: somatic mosaicism: observations related to clinical genetics. Am J Hum Genet. 1988;43:355–63. [PMC free article] [PubMed] [Google Scholar]
- 115.Sippel KC, et al. Frequency of somatic and germ-line mosaicism in retinoblastoma: implications for genetic counseling. Am J Hum Genet. 1998;62:610–9. doi: 10.1086/301766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Leuer M, et al. Somatic mosaicism in hemophilia A: a fairly common event. Am J Hum Genet. 2001;69:75–87. doi: 10.1086/321285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Evans DG, et al. Mosaicism in neurofibromatosis type 2: an update of risk based on uni/bilaterality of vestibular schwannoma at presentation and sensitive mutation analysis including multiple ligation-dependent probe amplification. J Med Genet. 2007;44:424–8. doi: 10.1136/jmg.2006.047753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kehrer-Sawatzki H, Cooper DN. Mosaicism in sporadic neurofibromatosis type 1: variations on a theme common to other hereditary cancer syndromes? J Med Genet. 2008;45:622–31. doi: 10.1136/jmg.2008.059329. [DOI] [PubMed] [Google Scholar]
- 119.Winn RN, et al. Transgenic lambda medaka as a new model for germ cell mutagenesis. Environ Mol Mutagen. 2008;49:173–84. doi: 10.1002/em.20364. [DOI] [PubMed] [Google Scholar]
- 120.Dubrova YE, Plumb M, Gutierrez B, Boulton E, Jeffreys AJ. Transgenerational mutation by radiation. Nature. 2000;405:37. doi: 10.1038/35011135. [DOI] [PubMed] [Google Scholar]
- 121.Dubrova YE. Radiation-induced transgenerational instability. Oncogene. 2003;22:7087–93. doi: 10.1038/sj.onc.1206993. [DOI] [PubMed] [Google Scholar]
- 122.Makova KD, Li WH. Strong male-driven evolution of DNA sequences in humans and apes. Nature. 2002;416:624–6. doi: 10.1038/416624a. [DOI] [PubMed] [Google Scholar]
- 123.Miyata T, Hayashida H, Kuma K, Mitsuyasu K, Yasunaga T. Male-driven molecular evolution: a model and nucleotide sequence analysis. Cold Spring Harb Symp Quant Biol. 1987;52:863–7. doi: 10.1101/sqb.1987.052.01.094. [DOI] [PubMed] [Google Scholar]
- 124.Taylor J, Tyekucheva S, Zody M, Chiaromonte F, Makova KD. Strong and weak male mutation bias at different sites in the primate genomes: insights from the human-chimpanzee comparison. Mol Biol Evol. 2006;23:565–73. doi: 10.1093/molbev/msj060. [DOI] [PubMed] [Google Scholar]
- 125.Drost JB, Lee WR. Biological basis of germline mutation: comparisons of spontaneous germline mutation rates among drosophila, mouse, and human. Environ Mol Mutagen. 1995;25:48–64. doi: 10.1002/em.2850250609. [DOI] [PubMed] [Google Scholar]
- 126.Shen JC, Rideout WM, 3rd, Jones PA. The rate of hydrolytic deamination of 5-methylcytosine in double-stranded DNA. Nucleic Acids Res. 1994;22:972–6. doi: 10.1093/nar/22.6.972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Trasler JM. Gamete imprinting: setting epigenetic patterns for the next generation. Reprod Fertil Dev. 2006;18:63–9. doi: 10.1071/rd05118. [DOI] [PubMed] [Google Scholar]
- 128.Lees-Murdock DJ, Walsh CP. DNA methylation reprogramming in the germ line. Epigenetics. 2008;3:5–13. doi: 10.4161/epi.3.1.5553. [DOI] [PubMed] [Google Scholar]
- 129.Eckhardt F, et al. DNA methylation profiling of human chromosomes 6, 20 and 22. Nat Genet. 2006;38:1378–85. doi: 10.1038/ng1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.El-Maarri O, et al. Methylation levels at selected CpG sites in the factor VIII and FGFR3 genes, in mature female and male germ cells: implications for male- driven evolution. Am J Hum Genet. 1998;63:1001–8. doi: 10.1086/302065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Morgan HD, Dean W, Coker HA, Reik W, Petersen-Mahrt SK. Activation-induced Cytidine Deaminase Deaminates 5-Methylcytosine in DNA and Is Expressed in Pluripotent Tissues. J. Biol. Chem. 2004;279:52353–52360. doi: 10.1074/jbc.M407695200. [DOI] [PubMed] [Google Scholar]
- 132.Schreck S, et al. Activation-induced cytidine deaminase (AID) is expressed in normal spermatogenesis but only infrequently in testicular germ cell tumours. J Pathol. 2006;210:26–31. doi: 10.1002/path.2014. [DOI] [PubMed] [Google Scholar]
- 133.Bransteitter R, Pham P, Scharff MD, Goodman MF. Activation-induced cytidine deaminase deaminates deoxycytidine on single-stranded DNA but requires the action of RNase. Proc Natl Acad Sci U S A. 2003;100:4102–7. doi: 10.1073/pnas.0730835100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Olsen A-K, Lindeman B, Wiger R, Duale N, Brunborg G. How do male germ cells handle DNA damage?. Toxicology and Applied Pharmacology Living in a Safe Chemical World. Proceedings of the 10th International Congress of Toxicology; Tampere, Finland. 11-15 July, 2004; 2005. pp. 521–531. [DOI] [PubMed] [Google Scholar]
- 135.Jaroudi S, SenGupta S. DNA repair in mammalian embryos. Mutation Research/Reviews in Mutation Research. 2007;635:53–77. doi: 10.1016/j.mrrev.2006.09.002. [DOI] [PubMed] [Google Scholar]
- 136.Menezo Y, Jr., Russo G, Tosti E, El Mouatassim S, Benkhalifa M. Expression profile of genes coding for DNA repair in human oocytes using pangenomic microarrays, with a special focus on ROS linked decays. J Assist Reprod Genet. 2007;24:513–20. doi: 10.1007/s10815-007-9167-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Intano GW, et al. Base Excision Repair Is Limited by Different Proteins in Male Germ Cell Nuclear Extracts Prepared from Young and Old Mice. Mol. Cell. Biol. 2002;22:2410–2418. doi: 10.1128/MCB.22.7.2410-2418.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Xu G, et al. Nucleotide Excision Repair Activity Varies Among Murine Spermatogenic Cell Types. Biol Reprod. 2005;73:123–130. doi: 10.1095/biolreprod.104.039123. [DOI] [PubMed] [Google Scholar]
- 139.Cortazar D, Kunz C, Saito Y, Steinacher R, Schar P. The enigmatic thymine DNA glycosylase. DNA Repair (Amst) 2007;6:489–504. doi: 10.1016/j.dnarep.2006.10.013. [DOI] [PubMed] [Google Scholar]
- 140.Schroering AG, Edelbrock MA, Richards TJ, Williams KJ. The cell cycle and DNA mismatch repair. Experimental Cell Research. 2007;313:292–304. doi: 10.1016/j.yexcr.2006.10.018. [DOI] [PubMed] [Google Scholar]
- 141.Hardeland U, Kunz C, Focke F, Szadkowski M, Schar P. Cell cycle regulation as a mechanism for functional separation of the apparently redundant uracil DNA glycosylases TDG and UNG2. Nucleic Acids Res. 2007;35:3859–67. doi: 10.1093/nar/gkm337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Crow JF. Spontaneous mutation in man. Mutat Res. 1999;437:5–9. doi: 10.1016/s1383-5742(99)00063-0. [DOI] [PubMed] [Google Scholar]
- 143.Risch N, Reich EW, Wishnick MM, McCarthy JG. Spontaneous mutation and parental age in humans. Am J Hum Genet. 1987;41:218–48. [PMC free article] [PubMed] [Google Scholar]
- 144.Ketterling RP, et al. Germline origins in the human F9 gene: frequent G:C-->A:T mosaicism and increased mutations with advanced maternal age. Hum Genet. 1999;105:629–40. doi: 10.1007/s004399900158. [DOI] [PubMed] [Google Scholar]
- 145.Wilkie AO. Bad bones, absent smell, selfish testes: the pleiotropic consequences of human FGF receptor mutations. Cytokine Growth Factor Rev. 2005;16:187–203. doi: 10.1016/j.cytogfr.2005.03.001. [DOI] [PubMed] [Google Scholar]
- 146.Edghill EL, et al. Origin of de novo KCNJ11 mutations and risk of neonatal diabetes for subsequent siblings. J Clin Endocrinol Metab. 2007;92:1773–7. doi: 10.1210/jc.2006-2817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Kubota H, Brinster RL. Culture of rodent spermatogonial stem cells, male germline stem cells of the postnatal animal. Methods Cell Biol. 2008;86:59–84. doi: 10.1016/S0091-679X(08)00004-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.McLean DJ. Spermatogonial stem cell transplantation, testicular function, and restoration of male fertility in mice. Methods Mol Biol. 2008;450:149–62. doi: 10.1007/978-1-60327-214-8_11. [DOI] [PubMed] [Google Scholar]
- 149.Falciatori I, Lillard-Wetherell K, Wu Z, Hamra FK, Garbers DL. Deriving mouse spermatogonial stem cell lines. Methods Mol Biol. 2008;450:181–92. doi: 10.1007/978-1-60327-214-8_13. [DOI] [PubMed] [Google Scholar]
- 150.Walsh CP, Xu GL. Cytosine methylation and DNA repair. Curr Top Microbiol Immunol. 2006;301:283–315. doi: 10.1007/3-540-31390-7_11. [DOI] [PubMed] [Google Scholar]
- 151.Klose RJ, Bird AP. Genomic DNA methylation: the mark and its mediators. Trends in Biochemical Sciences. 2006;31:89–97. doi: 10.1016/j.tibs.2005.12.008. [DOI] [PubMed] [Google Scholar]