

Abstract
CD11b+Gr-1+-expressing cells, termed myeloid-derived suppressor cells (MDSC), can mediate immunosuppression and tumor progression. However, the intrinsic molecular events that drive their pro-tumorigenic behavior remain to be elucidated. Although CD11b+Gr-1+ cells exist at low frequencies in normal mice, it also remains unresolved whether they are biologically distinct from those of tumor-bearing hosts. These objectives were investigated using CD11b+Gr-1+ cells from both implantable (4T1) and autochthonous (MMTV-PyMT) mouse models of mammary carcinoma. Limited variation was observed in the expression of markers associated with immunoregulation between CD11b+Gr-1+ cells of both tumor models, as well as with their respective controls. Despite limited differences in phenotype, tumor-induced CD11b+Gr-1+ cells were found to produce a more immunosuppressive cytokine profile than that observed by control CD11b+Gr-1+ cells. Furthermore, when admixed with tumor cells, CD11b+Gr-1+ cells from tumor-bearing mice significantly enhanced neoplastic growth compared to counterpart cells from control mice. However, the pro-tumorigenic behavior of these tumor-induced CD11b+Gr-1+ cells was significantly diminished when the expression of IRF-8, a key myeloid-associated transcription factor, was enhanced. The loss of this pro-tumorigenic effect occurred independently of the host immune system, and correlated with a CD11b+Gr-1+ cytokine/chemokine production pattern that resembled cells from non-tumor-bearing control mice. Overall, our data indicate that (a) tumor-induced CD11b+Gr-1+ cells from both cancer models were phenotypically similar, but biologically distinct from their non-tumor-bearing counterparts; and (b) modulation of IRF-8 levels in tumor-induced CD11b+Gr-1+ cells can significantly abrogate their pro-tumorigenic behavior, which may have important implications for cancer therapy.
Keywords: rodents, monocytes/macrophages, transcription factors, host-tumor interactions, transgenic models
INTRODUCTION
Myeloid-derived suppressor cells (MDSC) (1) have been shown to accumulate during a range of pathologies, including cancer (2, 3), graft-versus-host disease (4), stress (5) and infections (e.g., viral, bacterial and parasitic) (6, 7). There is increasing interest in MDSC, particularly in neoplastic disease, where these cells have been shown to play a role in both immune suppression and angiogenesis (3, 8-10). In mouse models, MDSC are characterized as a heterogeneous mixture of myeloid cells that express both the CD11b and Gr-1 differentiation markers. MDSC-like subsets have also been shown to accumulate in patients with a variety of tumor types, including carcinoma of the head and neck, non-small cell lung carcinoma, renal carcinoma, melanoma and adenocarcinomas of the colon, breast and pancreas (11-17).
Both in vitro and in vivo immune responses can be suppressed by MDSC through a number of mechanisms including, the production of reactive oxygen species, the activation of arginase-1 resulting in arginine depletion and altered TCR signaling (18-20). A range of tumor-derived factors (TDF), including IL-10 (21), CSF-1 (22), VEGF (23-25), GM-CSF (26), IL-1β (27-30), PGE2 (15, 31), IL-6 (28) and G-CSF (32) have been implicated in the development, regulation and/or function of MDSC. CD11b+Gr-1+ cells can be found at low frequencies in the periphery of normal hosts; however, it remains to be clarified whether these cells are phenotypically, functionally and biologically distinct from those of tumor-bearing hosts. We therefore chose to study aspects of MDSC biology in two distinct mouse mammary tumor models; one involved an orthotopic implant of the well-characterized 4T1 tumor cell line (20, 28, 33), while the other utilized a transgenic model of spontaneously arising autochthonous mammary carcinoma that demonstrates longer-term disease progression (34-36).
Interferon regulatory factor-8 (IRF-8), an IFN-γ-inducible transcription factor, has been shown to play an essential role in ensuring normal myelopoiesis (37-39) and the production of certain pro-inflammatory type-1 cytokines, such as IL-12p40 and RANTES (38, 40, 41). In both 4T1 and spontaneous tumor models,we recently observed a strong inverse correlation between IRF-8 expression levels and the emergence of these CD11b+Gr-1+ cells. Whereas CD11b+Gr-1+ cells of non-tumor-bearing control mice constitutively expressed IRF-8 as expected (37, 42), similarly isolated myeloid populations from tumor-bearing mice of both models expressed significantly lower levels (43). Moreover, IRF-8 null mice possess significantly increased frequencies of various myeloid populations, including neutrophils, macrophages and CD11b+Gr-1+ myeloid cells (37-39). Ultimately, IRF-8-deficient mice develop a CML-like syndrome (37-39). Thus, IRF-8 exerts a multi-functional role in both normal and aberrant stages of myeloid cell biology. Using these two mammary carcinoma models, we therefore addressed the following questions: 1) Are CD11b+Gr-1+ cells that arise in an implantable tumor model distinct from those that arise from a slower growing autochthonous tumor model in terms of phenotype and function? 2) Are CD11b+Gr-1+ cells from these tumor-bearing mice distinct from those of non-tumor-bearing control mice in terms of these same features? and 3) What molecular events impact the biological function of these cells, with an emphasis on the role of IRF-8?
MATERIALS AND METHODS
Mice
Female C57BL/6, BALB/c and bg-nu-XID mice were obtained from the National Cancer Institute-Frederick Cancer Research Animal Facility (Frederick, MD). Hybrid CB6F1/J (C57BL × BALB/c) mice were obtained from The Jackson Laboratories (Bar Harbor, ME).
The MMTV-PyMT/B6 transgenic mouse, termed MTAG, expresses the polyomavirus middle T antigen controlled through the MMTV-LTR promoter (34). These transgenic mice were originally derived in FVB mice (34) and were backcrossed > 10 times onto a C57BL/6 (H-2b) background (44) and kindly provided by S. Gendler (Mayo Clinic, Scottsdale, AZ). MTAG mice develop multifocal mammary carcinomas with progression to pulmonary metastases. Only female MTAG mice were used in these experiments and were obtained by breeding transgenic male MTAG mice with wild-type C57BL/6 female mice. Progeny were monitored for transgene expression by PCR, with 100% of transgene-positive (Tg+) female mice developing mammary carcinoma. Transgene-negative (Tg-) littermates were used as age/gender-matched controls. Mice were housed in a specific-pathogen free environment, and the experiments were conducted according to institutional regulations for animal care and use. In accordance with these regulations, no single tumor mass was allowed to exceed 2 cm3, although MTAG mice could develop up to 10 discrete mammary gland tumors. Therefore, total tumor volume in female MTAG mice was determined as the sum of each distinct tumor volume within an individual mouse. Tumor-bearing MTAG mice used in experiments for the isolation of CD11b+Gr-1+ cells harbored total tumor loads > 6 cm3 and were older than 160 days of age. MTAG mice were euthanized when any single tumor mass reached 2 cm3 in size or for other health considerations specified by institutional regulations. Tumor growth was measured weekly in two dimensions, using a digital caliper, and tumor volume was calculated using the formula (w2 × l)/2, whereby w = width and l = length of tumor mass, measured in millimeters
Production of IRF-8 Tg mice
Complementary DNA encoding the mouse IRF-8 coding sequence was cloned into the pcDNA3.1(+) plasmid (Invitrogen, Carlsbad, CA) under the control of the CMV promoter (plasmid kindly provided by K. Ozato, NIH, Bethesda, MD). A 4.3 kb fragment, which contained the transcriptional unit (i.e., promoter, gene, poly A sites) was then generated from the construct by cleavage at an FspI restriction site, and isolation by sucrose gradient purification. This purified material was then microinjected into the pronuclei of one cell embryos of fertilized eggs from B6 mice to obtain several founder mice (Laboratory Animal Sciences Program, Frederick, MD). Transgene integration in progeny was verified by Southern blot analysis using the 4.3 kb fragment as the probe and PCR genotyping of tail snips. The founder line, E5, was selected for this study based on the highest IRF-8 copy number and mRNA levels. Furthermore, no atypical effects of IRF-8 transgene expression were observed on various hematologic parameters, as determined by phenotypic analysis of the spleen and complete blood count (CBC) on peripheral whole blood. F1 progeny were produced by mating transgenic male mice with either wild-type female B6 or BALB/c mice as indicated in the Results. Transgene expression was verified by PCR analysis of tail DNA using the PCR/RT-PCR conditions described below.
Tumor cell lines and culture conditions
The 4T1 tumor cell line (ATCC, Manassas, VA) was originally derived as a spontaneously arising mammary carcinoma from BALB/c mice (33) and maintained in vitro in an RPMI-based culture medium (36). A tumor cell line, termed AT-3, was established from a primary mammary gland carcinoma of an MTAG mouse (36) and propagated in a DMEM-based culture medium (36). All in vitro assays were performed in RPMI-based culture medium. The Mtg2 cell line was derived from a primary mammary gland carcinoma (approximately 1 cm3) of an MTAG mouse in a fashion similar to that of AT-3 (36).
Isolation of CD11b+Gr-1+ Cells
CD11b+Gr-1+ cells were purified from single cell preparations of mouse splenocytes from non-tumor-bearing control mice, 4T1 tumor-bearing mice (1 – 2 cm3) or MTAG tumor-bearing mice (as defined above), using similarly established protocols (26, 45, 46). Selections were performed using CD11b+ magnetic beads (#130-049-801) from Miltenyi Biotec (Auburn, CA) and double column separation protocols on an AutoMACs system. Splenocytes were incubated with the Ab-coated magnetic beads for 30 min on ice prior to separation. The purity of the enriched cells was determined by flow cytometry following each separation, and the percentage of CD11b+ cells was routinely > 95%. Of these cells, > 90% expressed both CD11b and Gr-1 cell surface markers from both control and tumor (4T1 or MTAG)-bearing mice. CD11b+Gr-1+ cells were stimulated by culturing for 24 hr with or without lipopolysaccharide (LPS/L-6143; 1 μg/ml; Sigma-Aldrich, St. Louis, MO)and recombinant mouse IFN-γ (100 U/ml; R&D Systems, Minneapolis, MN). Cell-free supernatants were then collected for cytokine analysis.
Flow cytometry
Cells were incubated with directly conjugated mAb for 30 – 60 min, as described (36). At least 10,000 gated events were collected on a FACSCalibur flow cytometer (Becton-Dickinson, San Jose, CA) and analyzed by CellQuest software (Becton-Dickinson). mAbs reactive against the following cell surface markers were assessed: PE-conjugated CD4, CD8, NK1.1, Gr-1, H-2Kb, H-2Db, H-2Kd, H-2Dd, CD11a, CD11c, CD11b, CD14, CD40, CD48, CD80, CD95 (Fas), CD95L (FasL), CD154; PE-Cy5 conjugated CD3; FITC-conjugated I-Ab, H-2Ld, I-Ad, I-Ed, CD45R, CD4, CD8, CD19, CD54, CD86, Mac-3, Gr-1; and allophycocyanin (APC)-conjugated CD31 (all mAbs listed above were obtained from BD Biosciences/Pharmingen, San Diego, CA). FITC-CD11b (Southern Biotech, Birmingham, AL), PE-conjugated-CD115 (Serotec, Raleigh, NC) and APC-conjugated F4/80 (eBioscience, San Diego, CA) were also used. All samples were run with appropriate isotype controls.
Cytokine analysis
Cell-free supernatants were collected and stored at −20°C until assayed. Cytokine analysis was performed initially using a Cytometric Bead Array assay (BD Biosciences). The Th1/Th2 array kit detects IL-2, IL-4, IL-5, IFN-γ and TNF-α. Assays were performed with dual laser acquisition per manufacturer’s protocols. A more comprehensive analysis of cytokines and chemokines was performed using Quansys multipex ELISA services (Quansys Biosciences, Logan, UT), and included IL-1α, IL-1β, IL-2, IL-3, IL-4, IL-5, IL-6, IL-9, IL-10, IL-12p70, MCP-1, TNF-α, GM-CSF, MIP-1α, RANTES, MDC and IP-10. A similarly extensive cytokine/chemokine analysis was conducted on cell culture supernatants of the 4T1 and MTAG tumor cell lines. A mouse “20 plex” Cytometric Bead Array (CBA) system (BD Biosciences) was used, as per manufacturer’s instructions. Supernatants were collected from incubating tumor cells (1×106 cells/ml) for 24 hr in 24 well plates. A Quantikine mouse VEGF immunoassay (R&D Systems, Minneapolis, MN) was used to determine tumor-derived VEGF levels. Serum cytokine levels were analyzed using the CBA system, as above. Sera were collected from terminal blood draws of 4T1 or AT-3 tumor-bearing mice (with average tumor volumes between 1. 5 and 2 cm3), and frozen at -20°C until analysis.
Proliferation/Suppression Assay
Flat-bottomed 96-well plates were coated with anti-CD3 mAb (1 μg/well) by incubation at 4°C overnight. Unbound anti-CD3 mAb was removed by washing. CD4+ or CD8+ T cells were purified from the spleens of naïve CB6F1/J hybrid mice using T cell subset-specific MicroBeads and the AutoMACS separator, and plated at 5×104 cells/well. Splenic CD11b+Gr-1+ cells were purified from the different groups of mice, as described above and added to the plate at a CD11b+Gr-1+ cell:T cell ratio of 2:1. Plates were incubated for 48 hr at 37°C, after which 3H-thymidine (1 μCi/well) was added for an additional 24 hr of culture. Cells were recovered using an automated plate harvester, and proliferation determined by measuring 3H-thymidine uptake.
In vivo tumor growth studies
Mice were injected orthotopically in the abdominal mammary gland (#4) with 5×104 4T1 tumor cells and tumor growth monitored thrice weekly. In the admix experiments, 5×104 4T1 or 5×105 AT-3 tumor cells were mixed with MACS-purified, MHC-matched CD11b+Gr-1+ cells at a 2:1 ratio just prior to orthotopic implantation. Tumors were measured thrice weekly until masses reached ethical limits or until mice were euthanized for other health considerations.
In vivo cell depletions
Mice were injected ip with anti-Gr-1 Ab (clone RB6-8C5; Cedarlane, Ontario, Canada) to deplete Gr-1+ cells or an isotope control Ab. Antibodies were administered at 1 μg/mouse in a 200 μl volume on days 5, 6, 10 and 13 after tumor implantation using a modified protocol (47). CTLA-4 mAb-based immunotherapy was initiated by the administration of purified anti-mouse CTLA-4 mAb (clone 9H10) ip at 100 μg/mouse in 200 μl, on days 7, 11, and 14, where day 0 represented the day of tumor implantation, using a protocol similar to that previously described (48, 49). The anti-CTLA-4 hybridoma was kindly provided by J. Allison (Memorial Sloan-Kettering, NY) via J. Schlom (NCI, NIH), and was purified at the NCI-Protein Expression Laboratory (Frederick, MD). A combination therapy strategy utilized the combined protocols for Gr-1 depletion and CTLA-4 mAb administration as outlined above.
RT-PCR and PCR analyses
For RT-PCR, total RNA was isolated from CD11b+Gr-1+ cells using RNeasy Mini kits from Qiagen (Valencia, CA) according to manufacturer’s instructions. The RNeasy kit includes a silica-membrane technology that efficiently removes genomic DNA without DNase treatment. Total RNA was used for first strand cDNA synthesis using the ThermoScript RT-PCR system (Invitrogen). The cDNA was then used as the template for PCR amplification of mouse GAPDH and mouse IRF-8. PCR reactions were carried out in a PTC-200 thermal cycler (MJ Research, Waltham, MA) under the following conditions: 94°C for 2 min, 30 cycles (94°C for 30 sec, 60°C for 30 sec and 72°C for 1 min) and 72°C for 10 min. The PCR primers for GAPDH were as follows: forward primer: 5’-CATCACCATCTTCCAGGAGCG-3’; reverse primer: 5’-ACGGACACATTGGGGGTAGG-3’. The PCR primers for IRF-8 were as follows: forward primer: 5’-CGTGGAAGACGAGGTTACGCTG-3’; reverse primer: 5’- GCTGAATGGTGTGTGTCATAGGC-3’. For PCR analysis, various cycle numbers were previously tested, and 30 cycles was found to be optimal for effectively distinguishing differences in IRF-8 levels between control and tumor-induced CD11b+Gr-1+ cells. For genotyping transgenic offspring, PCR analysis was performed using these IRF-8 primers (30 cycles), which effectively distinguished transgenic from endogenous genomic IRF-8 levels (the latter of which were undetectable under these conditions), while the PCR primers (40 cycles) for PyMT were as follows: forward primer: 5’-AGTCACTGCTACTGCACCCAG-3’; reverse primer: 5’-CTCTCCTCAGTTCCTCGCTCC-3’. PCR products were separated on 1 or 2% agarose gels, and images captured with the BioDocIt imaging system (UVP, Upland, CA).
Statistical analysis
Analysis of data (see Figs. 4 and 6A) was performed on the slopes to preclude any time effect from the analysis and to determine a global treatment effect. For each data set, a separate linear regression analysis was performed on cubed root transformed data for each mouse to give a single estimated slope value. The estimated slope represents the average growth rate over the entire period. Since the distribution of the slope estimates were not normally distributed, the Wilcoxon rank sum test was used to compare distributions of slope estimates. For Fig. 5, some of the data were not linear after the transformation, thus, O’Brien’s method (50) was used to make overall pair-wise comparisons. For Fig. 6B, Anova on the estimated slopes was performed (after a cubed root transformation). Statistical analysis of the cytokine data was done using the exact Wilcoxon rank sum test (see Figs. 1B and 3A) or the Wilcoxon signed rank test (see Fig. 3B) after establishing ratios between tumor-bearing and control cells. The Hochberg method was used to adjust p-values for multiple comparisons (see Figs. 1B, 4 and 6B). In Fig. 7, paired differences for each of the log10 cytokine values were obtained from each tumor-bearing non-transgenic and IRF-8 transgenic mouse analyzed in the same experiment. Then, ANOVA was used on these values to determine if the differences were equal to zero. In all analyses, a p-value less than 0.05 was considered significant.
Figure 4. CD11b+Gr-1+ cells from tumor-bearing mice are pro-tumorigenic.
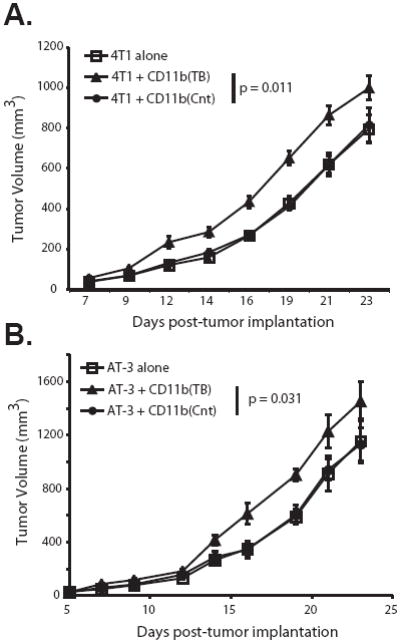
CD11b+Gr-1+ cells were isolated from the spleens of either 4T1- (A) or MTAG (B) tumor-bearing mice (TB) or age/gender-matched non-tumor-bearing control (Cnt) mice, as in Fig. 2. These CD11b+Gr-1+ cells were admixed with 4T1 or AT-3 tumor cells, respectively, just prior to orthotopic tumor challenge in the abdominal mammary gland, as described in the Materials and Methods. Tumor growth was monitored thrice weekly. Error bars represent the standard error of 10 mice per group. P-values are for the overall significance in the tumor growth rate difference between the groups.
Figure 6. Increased expression of IRF-8 in tumor-induced CD11b+Gr-1+ cells exerts an antitumor, rather than pro-tumorigenic effect.
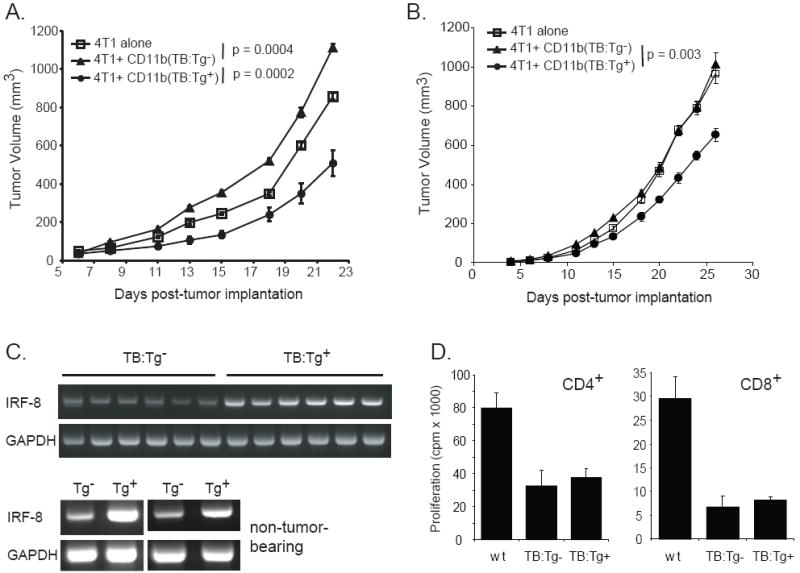
CD11b+Gr-1+ cells were purified from the spleens of 4T1 tumor-bearing hybrid (Tg-) or 4T1 tumor-bearing IRF-8 transgenic hybrid (Tg+) mice. These CD11b+Gr-1+ cells were then admixed with fresh 4T1 tumor cells (at a 1:2 ratio) just prior to tumor cell implantation. The admixed cell preparation was injected orthotopically into the abdominal mammary gland of (A) wild-type BALB/c mice or (B) immunodeficient bg-nu-XID mice. Tumor growth was monitored thrice weekly. Error bars represent the standard error of 7 mice in the group given 4T1 alone, and 10 mice each for the other two groups. P-values for overall significance in the tumor growth rate between groups are shown. (C) Top: IRF-8 expression in splenic CD11b+Gr-1+ cells of representative individual 4T1 tumor-bearing IRF-8 Tg- and IRF-8 Tg+ hybrid mice, as determined by RT-PCR analysis; Bottom: IRF-8 expression in splenic CD11b+Gr-1+ cells of non-tumor-bearing Tg- or non-tumor-bearing IRF-8 Tg+ hybrid mice from two separate experiments. (left) CD11b+Gr-1+ cells pooled from four Tg- and three Tg+ mice; (right) CD11b+Gr-1+ cells pooled from four Tg- and five Tg+ mice. (D) Splenic CD4+ or CD8+ T cells were purified from non-tumor-bearing wild-type (wt) CB6F1/J mice. T cells were mixed with CD11b+Gr-1+ cells (1:2 ratio) purified from the spleens of either wt or 4T1 tumor-bearing IRF-8 transgene-positive (TB:Tg+) or transgene-negative (TB:Tg-) CB6F1/J mice. Proliferation was measured by 3H-thymidine uptake, with the results representing the mean ± SD of triplicate wells.
Figure 5. Depletion of Gr-1-expressing cells enhances T cell-based immunotherapy.
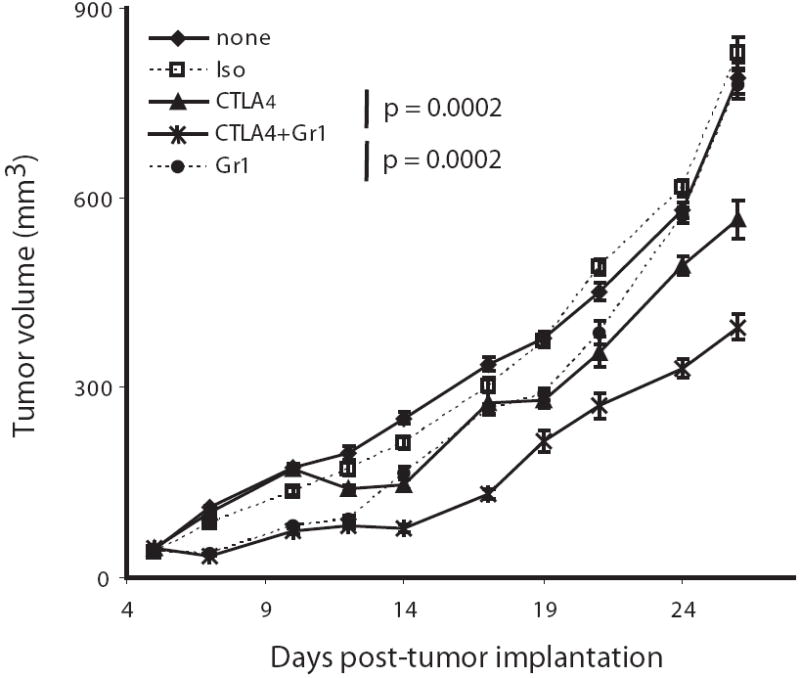
4T1-challenged BALB/c mice were depleted of Gr-1-expressing cells using anti-Gr-1 mAb administered i.p. on days 5, 6, 10, and 13 after tumor implantation. In other groups, mice were treated with anti-CTLA-4 mAb i.p. on days 7, 11, and 14 relative to tumor implantation or a combination of protocols for both Gr-1 depletion and anti-CTLA-4 administration. Tumor growth was monitored thrice weekly. Error bars represent the standard error of 10 mice per group. P-values were calculated by O’Brien’s method, which tested for overall differences in tumor volumes across all time points.
Figure 1. Tumor model-specific differences in the generation of CD11b+Gr-1+ MDSC.
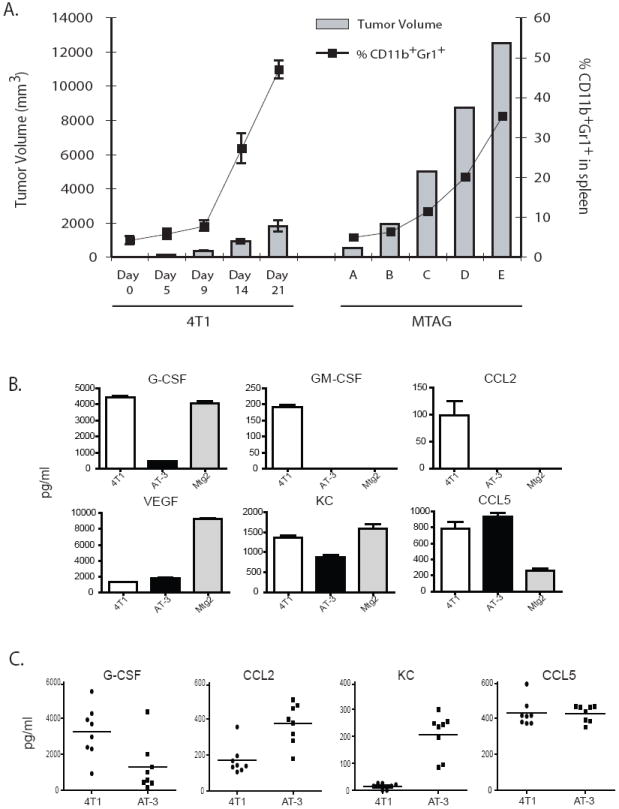
(A) Using both implantable and autochthonous mouse models of mammary tumor growth, spleens of mice with increasing tumor loads were recovered and the percentage of CD11b+Gr-1+ cells determined by flow cytometry. For the 4T1 model, syngeneic BALB/c mice were injected orthotopically with 4T1 tumor cells, and analyzed at various time points post-tumor implantation (n=5 mice per timepoint). Error bars represent standard deviations. For the MTAG model, results for individual MTAG mice with increasing tumor burdens are shown. (B) Cell-free supernatants from 4T1, AT-3 and Mtg2 tumor cells were collected in vitro and analyzed for the indicated cytokines/chemokines. The sample size varied between 5 – 8 determinations per cytokine/chemokine. Data represent mean ± SEM. (C) Sera were collected from 4T1 or AT-3 tumor-bearing mice and analyzed for the indicated cytokine/chemokines. GM-CSF levels were undetectable. Each data point represents the results from an individual mouse (n = 8), while the horizontal bar in the graph indicates the mean.
Figure 3. Cytokine production profiles of CD11b+Gr-1+ cells from 4T1 and MTAG tumor models.
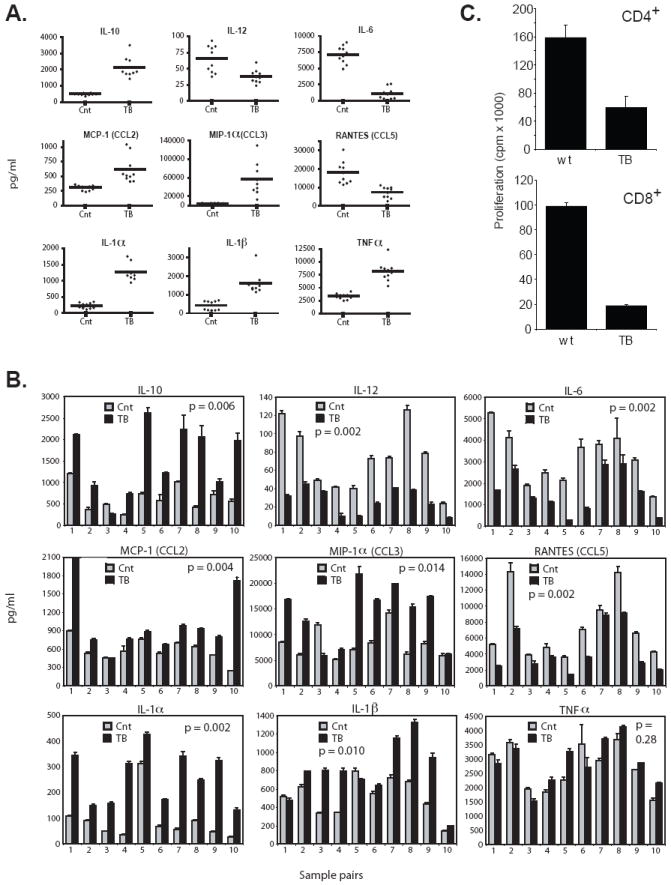
CD11b+Gr-1+ cells were isolated from the spleens of either 4T1 or MTAG tumor-bearing (TB) mice or age/gender-matched non-tumor-bearing control (Cnt) mice, as in Fig. 2. Cells were incubated for 24 hr with or without LPS (1 μg/ml) and IFN-γ (100 U/ml) stimulation. Supernatants were harvested and analyzed for a range of cytokines/chemokines. Cytokines, including IL-2, IL-3, IL-4, IL-5, IL-9 and GM-CSF were also measured, but are not shown because they were undetectable. (A) Supernatants from the culture of CD11b+Gr-1+ cells isolated from either BALB/c Cnt or TB mice are shown. Each individual sample was run in triplicate and the average value plotted. The horizontal line represents the mean of the values for 10 individual mice. Data shown are representative of two separate experiments (n=5 in the first experiment, which is not shown). For all the cytokines/chemokines shown, the differences between cells from TB vs. control mice were highly significant (p < 0.005). (B) Individual TB MTAG mice were directly compared with an age/gender-matched control mouse (usually littermates). Error bars represent the standard deviations of triplicate values. Total tumor volumes for the ten individual mice covered a range from 7.3 cm3 to 12.5 cm3. (C) Splenic CD4+ or CD8+ T cells were purified from non-tumor-bearing wild-type (wt) CB6F1/J mice. T cells were mixed with CD11b+Gr-1+ cells (1:2 ratio) purified from the spleens of either wt (n=4) or one of several 4T1 tumor-bearing (TB) CB6F1/J mice. Cultures were then incubated in the absence or presence of immobilized anti-CD3 mAb for 48 hr. Proliferation was measured by 3H-thymidine uptake after an additional 24 hr of incubation. Results represent the mean ± SD of triplicate wells.
Figure 7. Cytokine production patterns of CD11b+Gr-1+ cells from tumor-bearing IRF-8 transgenic versus non-transgenic mice.
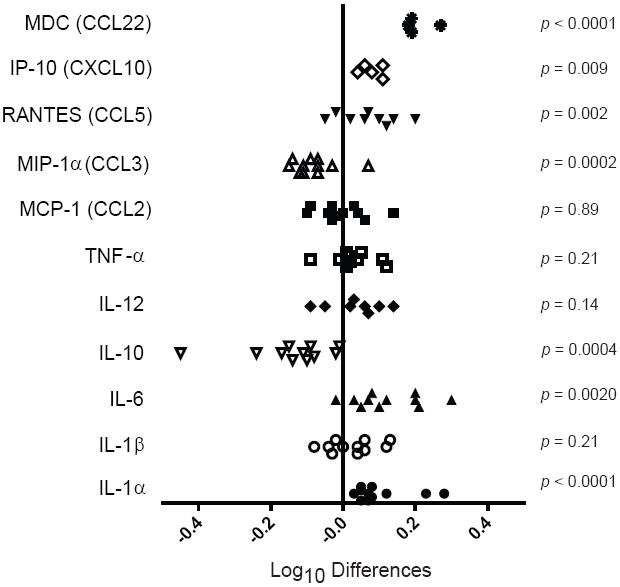
CD11b+Gr-1+ cells were isolated from the spleens of either 4T1 tumor-bearing IRF-8 transgenic or non-transgenic mice, as in Fig. 6. Cells were incubated for 24 hr with or without LPS (1 μg/ml) and IFN-γ (100 U/ml). Supernatants were harvested and analyzed for a range of cytokines/chemokines. The sample size varied between 5 and 11 determinations per group. Prior to this analysis, the cytokine data was transformed (log10(y)), and then the log10 differences between the transgenic and non-transgenic groups were calculated according to sample number. The values of the log differences [i.e., log10 (Tg+ group) minus log10 (Tg- group)] are shown for each cytokine tested, along with the associated two-tailed p-value as determined by ANOVA. In this figure, a positive difference indicates that the Tg+ value is greater than that of the paired Tg- mouse. Seven cytokines had a mean difference that was significantly different from zero.
RESULTS
Accumulation of CD11b+Gr-1+ cells in mouse models of implantable and autochthonous mammary tumor growth
The vast majority of studies on these myeloid populations have been conducted using implantable tumor models, where the MDSC usually accumulate rapidly and may not recapitulate the chronic nature of human disease. It remains unclear whether MDSC from such implantable tumor models are distinct from those produced in an autochthonous disease setting where protracted host-tumor interactions may influence MDSC development or biology in a fundamentally distinct manner. We therefore chose to study aspects of MDSC biology in two distinct mouse mammary tumor models; one involved the orthotopic implant of the 4T1 tumor cell line, while the other utilized a transgenic model of spontaneously arising autochthonous mammary carcinoma, which demonstrates longer-term disease progression.
The 4T1 tumor model is a well-characterized model that exhibits aggressive primary tumor growth resulting in spontaneous metastasis to the lymph nodes, lungs, liver, bone, and brain (20, 28, 33). In contrast, the transgenic mouse tumor model engineered to express the polyomavirus middle T antigen (PyMT) under the control of the mouse mammary tumor virus (MMTV) promoter, henceforth termed MTAG, develops autochthonous mammary carcinomas over an approximate 6-month lifespan, with eventual metastatic spread to the lungs (34). This MTAG mouse has also been shown to mimic human breast cancer disease progression at morphological, molecular (35, 51) and immunological levels (36, 52).
We have previously demonstrated that as tumor load increased, there was a concomitant increase in the absolute number and percentage of splenic CD11b+Gr-1+ cells in MTAG mice (36, 52). Here, we compared the onset of development/accumulation of CD11b+Gr-1+ cells in implantable versus autochthonous mouse tumor models. We observed a profound difference in the timing and quantity of CD11b+Gr-1+ cells that arise in the transgenic MTAG model compared to the 4T1 implantable model (Fig. 1A). CD11b+Gr-1+ cells in 4T1 tumor-bearing mice accumulated much more rapidly and in greater numbers than in MTAG mice. By the time total tumor volume approached 2 cm3, the splenic compartment of 4T1 tumor-bearing mice consisted of 30 – 50% CD11b+Gr-1+ cells (Fig. 1A). In contrast, the splenic compartment of MTAG mice contained > 20% CD11b+Gr-1+ cells only when the total tumor load was greater than 6 cm3, which encompassed a period of tumor growth of several months. These CD11b+Gr-1+ myeloid populations therefore developed in both types of models, albeit in a fundamentally distinct dynamic and temporal manner.
The observation that these CD11b+Gr-1+ myeloid populations accumulated in both tumor models via distinct kinetics suggested that this temporal disparity may be attributed to the types or quantities of factors produced by the different tumors. It is now clear that different malignancies secrete a variety of tumor-derived factors (TDF) that modulate the accumulation, differentiation, mobilization and/or function of CD11b+Gr-1+ myeloid cells, notably VEGF, GM-CSF, G-CSF, IL-10, PGE2, IL-1β and IL-6 (21, 23-26, 28, 31, 32). Therefore, we measured TDF produced by 4T1 cells in vitro, along with two distinct autologous MTAG-derived tumor cell lines, termed AT-3 (36) and Mtg2, as surrogates for the spontaneous primary mammary tumors in MTAG mice (Fig. 1B). 4T1 cells were found to produce demonstrable levels of G-CSF, GM-CSF, VEGF as well as two myeloid-associated chemokines, CCL2 (MCP-1) and KC (keratinocyte-derived chemokine; CXCL1) and a lymphoid-associated chemokine, CCL5 (RANTES) (Fig. 1B), but negligible amounts of IL-1β, IL-6 and IL-10 (data not shown). Whereas, AT-3 and Mtg2 cells also produced G-CSF, VEGF, KC and CCL5 at varying amounts, they did not secrete detectable levels of GM-CSF and CCL2 (Fig. 1B). Thus, all three tumor cell lines were shown to produce several TDF that have been implicated in MDSC accumulation. In terms of comparing the two MTAG-derived tumor cell lines to each other, we found that the profiles for GM-CSF and CCL2 profiles were similar, but the profiles for G-CSF, VEGF, KC and CCL5 were different (p < 0.002 for these latter cytokines/chemokines), indicating tumor heterogeneity within this MTAG model. Such tumor heterogeneity may contribute to the variability in CD11b+Gr-1+ myeloid cell accumulation that has been observed between individual MTAG mice (36, 52).
We then analyzed serum cytokine levels from 4T1 and AT-3 tumor-bearing mice to determine correspondence with these in vitro results (Fig. 1B vs. 1C). We assayed sera for several of the cytokines shown in figure 1B, including: G-CSF, CCL2 (MCP-1), KC, CCL5 (RANTES) and GM-CSF (Fig. 1C). We found that the cytokine patterns for G-CSF and RANTES mirrored what was observed in vitro. In the case of G-CSF, higher amounts of G-CSF were detected in 4T1 compared to AT-3 tumor-bearing mice, while comparable amounts of RANTES were observed between both groups of tumor-bearing mice. However, the patterns for CCL2 and KC showed somewhat less correspondence with the in vitro data. In the case of 4T1 tumor-bearing mice, we still detected CCL2, but now only minor amounts of KC. Interestingly, in the case of AT-3 tumor-bearing mice, we detected not only KC, but now also CCL2 which was previously not observed in vitro. GM-CSF was undetectable in both groups of tumor-bearing mice. The reasons for these differences may reflect a number of complex biological factors. Several factors that may influence detection in vivo include: the exact cytokine source(s) (i.e., tumor vs. host response), cytokine utilization (which affects availability) and/or the sites of cytokine action (i.e., local vs. systemic). Overall, the differences in TDF types and levels observed between the models (implantable vs. autochthonous) may account, at least in part, for the distinct kinetics in CD11b+Gr-1+ myeloid cell accumulation.
Phenotype of the various CD11b+Gr-1+ cell populations
To better characterize the CD11b+Gr-1+ cells arising in either the implantable or autochthonous tumor models, we isolated these cells from both control and tumor-bearing mice and examined them for the expression of a range of immunologically relevant cell surface markers. Enrichment for this population was achieved by positive selection of CD11b+ cells (> 95% purity), with at least 90% of these cells being double-positive for the expression of both the CD11b+ and Gr-1+ cell surface markers (Fig. 2B). Differences in the pattern of CD11b and Gr-1 expression were evident between cells isolated from 4T1 tumor-bearing mice versus those of tumor-bearing MTAG mice. The CD11b+ cells from MTAG mice showed greater heterogeneity in Gr-1 staining than CD11b+ cells from 4T1 tumor-bearing mice (Fig. 2B). Heterogeneity in the expression levels of both CD11b and Gr-1 markers were similarly observed in myeloid cells isolated from non-tumor-bearing control BALB/c or C57BL/6 mice (Fig. 2B).
Figure 2. Phenotypic analysis of purified CD11b+Gr-1+ cells from 4T1 and MTAG tumor models.
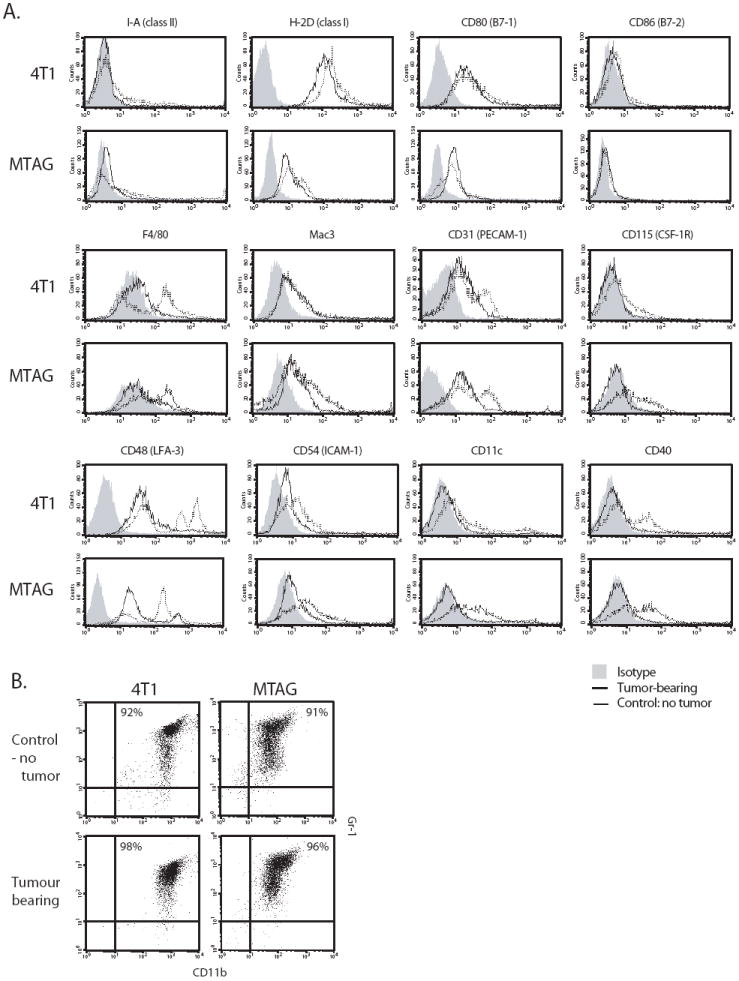
CD11b+Gr-1+ cells were isolated from the spleens of either 4T1 or MTAG tumor-bearing mice, as described in the Materials and Methods, and compared with similarly purified cells from age/gender-matched non-tumor-bearing BALB/c or C57BL/6 control mice, respectively. (A) Cells were stained for a range of cell surface markers. Solid line, cells of tumor-bearing mice; dotted line, cells of control mice; gray-filled histogram, isotype control Ab. Results are representative of at least 2 similar experiments. (B) Purity of the cells analyzed in panel A and their expression of the CD11b and Gr-1 markers.
Despite these observations, and with the exception of Mac-3 staining, there appeared to be no striking differences in the staining profiles between CD11b+Gr-1+ cells from 4T1 versus MTAG tumor-bearing mice (Fig. 2A). However, when comparing CD11b+Gr-1+ cells of tumor-bearing mice of either model with their respective control cells, some differences were noted. Reduced levels of staining were observed on CD11b+Gr-1+ cells from tumor-bearing mice for F4/80, CD31, CD115, CD48, CD54 and CD40 when compared with cells from non-tumor-bearing control mice (Fig. 2A). There were also elevated numbers of CD11c+ cells in the CD11b+Gr-1+-enriched preparations of control mice compared to similarly purified cells from tumor-bearing mice. In contrast, there was no obvious difference between the staining profiles of CD11b+Gr-1+ cells from control and tumor-bearing mice for MHC class I and II molecules, nor for the costimulatory molecules CD80 and CD86 (Fig. 2A). Similar levels of additional cell surface markers, including CD95, CD11a and CD14, were expressed on cells from control and tumor-bearing mice (data not shown). Markers that were not expressed in any of the populations studied, included CD154, CD19, CD45R (B220) and CD4.
Tumor-induced CD11b+Gr-1+ cells produce an altered cytokine profile
It has been established that CD11b+Gr-1+ cells from tumor-bearing mice are immunosuppressive, and inhibit T cell proliferation in vitro through a number of mechanisms (3, 8). However, less is known regarding the cytokine production profiles of tumor-induced CD11b+Gr-1+ cells, which may also impact the outcome of an adaptive immune response. The approach taken was to trigger cytokine release in response to LPS and IFN-γ treatment, similarly as described (38, 41, 53). It is important to note that these activation experiments were performed in the absence of any myelopoietic growth factor known to affect differentiation (i.e., GM-CSF, G-CSF or M-CSF). Although little-to-no cytokines were detectable in the absence of stimulation, CD11b+Gr-1+ cells of 4T1 tumor-bearing mice, following LPS and IFN-γ exposure, demonstrated an altered cytokine production profile when compared with similarly purified cells from control mice (Fig. 3A). Tumor-induced CD11b+Gr-1+ cells produced significantly higher levels of IL-10, MCP-1, MIP-1α, IL-1α, IL-1β, TNF-α and significantly lower amounts of IL-12, IL-6 and RANTES. For all cytokine differences shown, the p-values were < 0.005.
The cytokine patterns of CD11b+Gr-1+ cells from MTAG mice were similar to that of 4T1 tumor-bearing mice (Fig. 3B). To account for potential differences in age and tumor burden or inter-assay variability, CD11b+Gr-1+ cells of MTAG mice were directly compared to age- and gender-matched control mice. In most cases, CD11b+Gr-1+ cells of tumor-bearing MTAG mice produced significantly higher amounts of IL-10, MCP-1, MIP-1α, IL-1α and IL-1β and lower levels of IL-6, IL-12 and RANTES. For the ten pairs of mice tested, the overall differences between control and tumor-bearing MTAG-derived CD11b+Gr-1+ cells were significant (p < 0.015) for all the indicated cytokines, except TNF-α (p = 0.28). Screening was also performed for IL-2, IL-3, IL-4, IL-5, IL-9 and GM-CSF; however, these cytokines were undetectable from the cells of both tumor models. Lastly, using the 4T1 model, we verified that the CD11b+Gr-1+ cells from tumor-bearing mice were immunosuppressive based on their ability to inhibit anti-TCR-induced CD4+ or CD8+ T cell proliferation (Fig. 3C). We had previously shown that CD11b+Gr-1+ cells from tumor-bearing MTAG mice also possessed immunosuppressive capabilities (52).
Tumor-induced CD11b+Gr-1+ cells are pro-tumorigenic in vivo
Next, we evaluated whether the tumor-induced CD11b+Gr-1+ cells from these two models demonstrated distinct biological activities compared to their respective non-tumor-bearing controls. In addition to promoting immunosuppression, tumor-induced CD11b+Gr-1+ cells have been described to enhance tumor growth when admixed with syngeneic tumor cells and reinjected into naïve mice (9, 10). We therefore investigated whether CD11b+Gr-1+ cells obtained from either the 4T1 or the MTAG model were similarly pro-tumorigenic, but functionally distinct from CD11b+Gr-1+ cells of control mice.
We found that when 4T1 tumor-induced CD11b+Gr-1+ cells were admixed with fresh 4T1 tumor cells and injected orthotopically into new groups of syngeneic mice, there was an enhanced rate of tumor growth compared to admixtures of CD11b+Gr-1+ cells derived from the spleens of non-tumor-bearing control mice (p = 0.011) (Fig. 4A). In contrast, CD11b+Gr-1+ cells derived from non-tumor-bearing control mice, when admixed with fresh 4T1 tumor cells, had a comparable rate of tumor growth compared to 4T1 tumor cells injected alone (Cnt vs. alone, p = 0.57), revealing that control CD11b+Gr-1+ cells had no pro-tumorigenic effect (Fig. 4A). Similarly, in the MTAG model, when CD11b+Gr-1+ cells generated from tumor-bearing or control mice were admixed with AT-3, an autologous MTAG-derived tumor cell line, a significantly enhanced tumor growth rate was observed in recipient B6 mice using tumor-induced, but not control CD11b+Gr-1+ cells (p = 0.031, when compared to each other) (Fig. 4B). As with the 4T1 model, there was no difference in the rate of AT-3 growth alone when compared to the rate of growth of AT-3 cells admixed with CD11b+Gr-1+ cells from control mice (Cnt vs. alone, p = 0.85). Thus, tumor-induced CD11b+Gr-1+ cells isolated from both implantable and autochthonous models were similarly pro-tumorigenic, but biologically distinct from CD11b+Gr-1+ cells of their non-tumor-bearing counterparts, as determined by this admixture analysis.
Depletion of tumor-induced CD11b+Gr-1+ cells enhance a T cell-based immunotherapy
In addition to using an “admix” approach to determine whether tumor-induced CD11b+Gr-1+ cells augment tumor growth (Fig. 4), we made use of a reciprocal approach to examine whether the elimination of these myeloid populations would reduce tumor growth and, perhaps, further improve response to extrinsic immunotherapy. In the 4T1 model, an Ab-based approach was employed to deplete Gr-1-expressing cells in vivo (47) alone or in combination with CTLA-4-blockade (Fig. 5), which has previously been used successfully to potentiate tumor immunity (48, 49). Depletion of Gr-1+ cells alone retarded 4T1 tumor progression (p = 0.0002), and appeared to be most effective in the early phases of tumor growth. Treatment with anti-CTLA-4 mAb alone also had an overall significant inhibitory effect on tumor growth (p = 0.001), although becoming evident later during tumor progression, as would be expected from the induction of an improved antitumor T cell response. Interestingly, a synergistic enhanced antitumor response was observed when the two treatments were combined, relative to Gr-1 depletion alone (p = 0.0002) or CTLA-4 blockade alone (p = 0.0002).
Therefore, as evident in both the admix (Fig. 4) and depletion experiments (Fig. 5), our findings are consistent with the hypothesis that CD11b+Gr-1+ cells generated in these models favor tumor progression, and that the elimination of potentially immunosuppressive and pro-tumorigenic (Gr-1-expressing) myeloid populations can augment certain T cell-based immunotherapies. These findings also extend other studies, revealing that biological- or immune-based therapies mediate stronger antitumor effects when combined with strategies that eliminate MDSC or inhibit their suppressive function (54-57).
Increased IRF-8 expression decreases the pro-tumorigenic behavior of tumor-induced CD11b+Gr-1+ cells
Recently, we found that IRF-8 expression, an essential myeloid-associated developmental/differentiation transcription factor (37-39) is inversely related to, and a potential negative regulator of, CD11b+Gr-1+ cell accumulation in tumor-bearing MTAG mice (43). IRF-8 has also been shown to play an important role in myeloid cell function, including the regulation of IL-12p40 and RANTES production (38, 40, 41). Given the importance of IRF-8 in myeloid cell biology and function, including ensuring normal myelopoiesis and governing the production of certain type-1 pro-inflammatory cytokines, we investigated whether IRF-8 may be an important molecular determinant of the pro-tumorigenic behavior of CD11b+Gr-1+ cells. We therefore examined whether IRF-8 over-expression could modify the pro-tumorigenic behavior of tumor-induced CD11b+Gr-1+ cells. Based on the earlier observations that revealed that tumor-induced CD11b+Gr-1+ myeloid cells were similarly pro-tumorigenic in both 4T1 and AT-3 tumor models (Fig. 4), we therefore focused on the 4T1 model for the following experiments.
To test this hypothesis, we purified CD11b+Gr-1+ cells from 4T1 tumor-bearing wild-type and IRF-8 transgenic mice and compared their pro-tumorigenic behavior in the tumor cell-myeloid cell admix model. Since the IRF-8 transgenic mouse was originally developed on a B6 background (H-2b), we used semi-syngeneic (hybrid) mice to examine the impact of IRF-8 in the 4T1 tumor model (Fig. 6A). These mice were produced by breeding IRF-8 transgenic male mice with wild-type female BALB/c mice to generate F1 progeny (H-2b/d), which will accept the H-2d-expressing 4T1 tumor allograft. Although in this model we observed no significant impact of the IRF-8 transgene on the percentage of splenic CD11b+Gr-1+ cells or their expression of the CD11b and Gr-1 markers (data not shown), we did however observe a significant difference in the biology of the CD11b+Gr-1+ cells based on their reduced tumorigenic behavior in the tumor cell-myeloid cell admix experiments (Fig. 6).
Similar to what we previously demonstrated in fully syngeneic mice (Fig. 4A), CD11b+Gr-1+ cells of tumor-bearing wild-type mice exerted a pro-tumorigenic effect on 4T1 tumor growth in this hybrid model (p = 0.0004) (Fig. 6A). However, when 4T1 cells were admixed with CD11b+Gr-1+ cells of tumor-bearing IRF-8 transgenic mice, this resulted in a significant decrease in the size and tumor growth rate compared to 4T1 cells injected either alone (p = 0.001) or with CD11b+Gr-1+ cells of tumor-bearing wild-type mice (p = 0.0002) (Fig. 6A). Thus, increased expression of IRF-8 in tumor-induced CD11b+Gr-1+ cells significantly impacted the biology of these cells and led to a reduction in their pro-tumorigenic capacity. The increase in IRF-8 expression in CD11b+Gr-1+ cells from individual 4T1 tumor-bearing IRF-8 transgenic mice compared with similarly isolated cells of 4T1 tumor-bearing wild-type mice, which were used in these admix experiments, was verified by RT-PCR analysis (Fig. 6C; upper panels). An analysis of IRF-8 expression levels revealed that the higher levels of IRF-8 observed in non-tumor-bearing IRF-8 transgenic (Tg+) mice, compared to the non-tumor-bearing wild-type (Tg-) mice, were maintained during tumor growth (Fig. 6C).
To examine whether host immune responses were involved in the mechanism of the observed CD11b+Gr-1+-mediated pro- or antitumor effect, we repeated these experiments in immunodeficient bg-nude-XID mice, which are functionally deficient in T, B and NK cells (Fig. 6B). Interestingly, the pro-tumorigenic effect of CD11b+Gr-1+ cells from tumor-bearing wild-type mice on 4T1 tumor growth was abrogated (p = 0.21). These data indicated that eliminating the host immune system in recipient mice also eliminated the functional capacity of the CD11b+Gr-1+ cells to enhance tumor growth. In contrast, CD11b+Gr-1+ cells of tumor-bearing IRF-8 transgenic mice maintained their ability to significantly inhibit the overall rate of tumor growth when compared to 4T1 cells injected either alone (p < 0.0001) or coinjected with CD11b+Gr-1+ cells of tumor-bearing wild-type mice (p = 0.003). When these cells were examined in vitro for their ability to suppress T cell proliferation, we observed that tumor-induced CD11b+Gr-1+ cells from both IRF-8 transgenic and non-transgenic mice were equally effective in their ability to inhibit anti-TCR-induced CD4+ or CD8+ T cell proliferation (Fig. 6D).
However, the cytokine/chemokine production profiles of tumor-induced CD11b+Gr-1+ cells from IRF-8 transgenic mice were different from those of the corresponding non-transgenic mice (Fig. 7). CD11b+Gr-1+ cells from tumor-bearing IRF-8 transgenic mice produced significantly higher levels of RANTES, IL-6 and IL-1α, as well as significantly lower levels of IL-10 and MIP-1α. Although we did not observe a significant change in IL-12 levels, there was a tendency toward the production of higher amounts by CD11b+Gr-1+ myeloid cells of the IRF-8 transgenic group. Furthermore, we observed significant increases in two other pro-inflammatory chemokines, MDC (macrophage-derived chemokine) and IP-10. Thus, enhanced IRF-8 expression within the tumor-induced CD11b+Gr-1+ population resulted in a cytokine production pattern that had reverted to a more “normal”, non-tumor-bearing phenotype (Figs. 3 & 7). Together, these data indicated that IRF-8 expression directly affected the pro-tumorigenic behavior of these cells, and rendered tumor-induced CD11b+Gr-1+ cells less tumorigenic, presumably due to cell contact-dependent or –independent (cytokine-driven) interactions with the tumor and/or other non-immune host cells within the tumor microenvironment.
DISCUSSION
In this study, we investigated whether tumor-induced CD11b+Gr-1+ cells were biologically distinct from those of non-tumor-bearing hosts and whether IRF-8 regulated molecular events influenced their pro-tumorigenic behavior. To do so, we made use of both implantable and autochthonous mouse models of mammary carcinoma. We found that the accumulation of CD11b+Gr-1+ cells in the 4T1 model occurred more rapidly and at much lower tumor loads than that of MTAG mice. This disparity may reflect the acute nature of the implant model, or the aggressiveness of 4T1 tumor growth, which may override host homeostatic mechanisms that restrict CD11b+Gr-1+ cell expansion. In contrast, in the autochthonous MTAG model where host-tumor interactions occur over a prolonged period of time, these homeostatic mechanisms may have the opportunity to elicit some control.
The differential expansion of CD11b+Gr-1+ cells in the 4T1 versus MTAG models may also reflect the types or levels of TDF produced. For instance, several studies have reported that different tumors secrete various TDF that modulate the development, differentiation and/or function of CD11b+Gr-1+ cells, namely VEGF, GM-CSF, G-CSF, IL-10, PGE2, IL-1β and IL-6 (21, 23-32). Indeed, this is supported by our analysis of TDF produced by 4T1 and MTAG-derived (AT-3, Mtg2) tumor cells. These tumor cell lines produced varying levels of G-CSF, GM-CSF, MCP-1 and/or VEGF, which may influence the tumor growth rates and govern, at least in part, the rate and magnitude of MDSC accumulation in tumor-bearing hosts. Another TDF that was expressed by these tumor cell lines was KC, which is functionally equivalent to GROα (58). Although the production of KC by tumor cells has not previously been shown to be related to MDSC accumulation, trafficking or function, it has the potential to affect MDSC biology due to its ability to chemoattract certain myeloid subsets. Future studies are needed to evaluate the contribution of these and/or other TDF that affect MDSC biology, and the physiological significance of the different time-courses of CD11b+Gr-1+ cell generation on host immunosuppression and tumor growth.
We also demonstrated that other than Mac3 staining, there appeared to be no overt differences in the expression of a range of cell surface markers associated with immunoregulation between CD11b+Gr-1+ cells derived from either the 4T1 (BALB/c) or MTAG (C57BL/6) models. However, some interesting phenotypic differences were observed between tumor-induced CD11b+Gr-1+ cells of either model when compared to similarly isolated cells from corresponding non-tumor-bearing control mice. For example, reduced levels of F4/80, CD31, CD48, CD54 and CD40 staining were detected on tumor-induced CD11b+Gr-1+ cells when compared with control cells of either model. In contrast, no differences in MHC class I or II staining or staining for the costimulatory molecules, CD80 and CD86, were observed. Although it remains to be further investigated, it is conceivable that the reduction in markers, such as the adhesion molecules CD31, CD48 and CD54 and/or the costimulatory molecule CD40, may contribute to the immunosuppressive phenotype of the tumor-induced CD11b+Gr-1+ cells and may be used to further define specific functional subsets of MDSC. Despite the marked differences in the rate of CD11b+Gr-1+ cell accumulation between the two mammary tumor models, these tumor-induced myeloid populations were essentially phenotypically and functionally similar.
To evaluate cytokine phenotype, we made use of IFN-γ and LPS stimulation. This rationale was based on the notion that if tumor growth intrinsically altered the biology of these myeloid cells, then their cytokine response under these same activation conditions would be distinct from that observed with myeloid cells from non-tumor-bearing control mice. Our observations supported that hypothesis, indicating that the cytokine phenotype of CD11b+Gr-1+ myeloid cells from tumor-bearing mice were indeed intrinsically altered. Thus, despite the use of an activation cocktail that is thought to elicit a M1 phenotype in “naïve” (control) CD11b+Gr-1+ myeloid cells, this same stimuli combination led to a distinct cytokine pattern in those cells from tumor-bearing mice. Overall, this profile revealed not only a higher production of certain inflammatory cytokines/chemokines, such as IL-1α, IL-1β, MCP-1, MIP-1α and IL-10, but also a lower production of IL-12, RANTES and IL-6. The elevated levels of IL-1α, IL-1β, MCP-1 and MIP-1α may contribute to diverse aspects of MDSC function, such as tissue or tumor infiltration, as well as how they affect the biology of other infiltrating myeloid populations, such as tumor-associated macrophages (59). IL-10 is a major determinant of a type-2 response (60), and can enhance the immunosuppressive behavior of MDSC (21). Additionally, elevated IL-10 production from these cells may promote T cell anergy (61), block DC-mediated priming of naïve T cells (61) and, when detected in sera, serve as a poor prognostic indicator in a variety of cancers (62, 63). IL-1β has also been shown to promote tumor progression by enhancing the accumulation of MDSC (27-30).
Additional studies have shown that MDSC can impair tumor immunity by increasing macrophage production of IL-10 and decreasing IL-12, thus leading to diminished T cell activation (64). Therefore, along with a reduction in RANTES, which may lessen the extent of T cell migration to the tumor microenvironment, decreased IL-12 levels could impair an adaptive immune response. In terms of IL-6, it was somewhat surprising that tumor-induced CD11b+Gr-1+ cells produced significantly less IL-6 than control cells, since elevated IL-6 levels has been associated with poor prognosis in cancer patients (65) and MDSC accumulation (21). However, because IL-6 may be utilized by MDSC as shown in vivo, (28) reduced IL-6 levels of tumor-induced CD11b+Gr-1+ cells in vitro may reflect in part cytokine consumption, which remains to be elucidated. Collectively, these data identify previously unrecognized cytokine/chemokine patterns of tumor-induced CD11b+Gr-1+ cells, notably elevated levels of MCP-1 and MIP-1α and diminished amounts of RANTES, which may also contribute to their pro-tumorigenic phenotype.
Lastly, we investigated the role of IRF-8 in the modulation of the pro-tumorigenic capabilities of these CD11b+Gr-1+ cells. Because MDSC represent an aberrantly expanded heterogeneous population of CD11b+Gr-1+ myeloid cells, we postulated that this altered phenotype may reflect altered IRF-8 levels that ordinarily control key aspects of normal myeloid cell differentiation and biology (37-39, 42). Therefore, we further reasoned that modulation of IRF-8 levels in tumor-induced CD11b+Gr-1+ cells may restore, at least to some extent, normal myeloid cell functions. Using the tumor cell-myeloid cell admix approach, we found that CD11b+Gr-1+ cells that accumulated in tumor-bearing IRF-8 transgenic mice were significantly less pro-tumorigenic, although not immunosuppressive (at least in vitro), than similar cells produced in tumor-bearing non-transgenic mice. These observations were made in both immune-competent and immune-deficient mice, indicating that such IRF-8-mediated changes also directly affected myeloid cell-tumor cell interactions and/or myeloid-associated interactions with other non-immune host cells within the tumor microenvironment. Future studies are warranted to investigate these possibilities in detail, as well as to examine further the roles played by IRF-8.
In addition to IRF-8 modulation of the in vivo biology of these tumor-induced CD11b+Gr-1+ cells, our data indicate that the cytokine/chemokine patterns of these cells can be partially restored to a more normal cytokine profile, particularly in terms of IL-6 and RANTES levels, which were significantly elevated, as well as IL-10 and MIP-1α levels, which were significantly reduced relative to tumor-induced CD11b+Gr-1+ cells from non-transgenic mice. Moreover, it is interesting to note that IP-10 (CXCL10) and MDC (CCL22) levels produced by CD11b+Gr-1+ cells from tumor-bearing IRF-8 transgenic mice were significantly higher compared with similarly isolated cells from tumor-bearing non-transgenic mice. IP-10 has been shown to inhibit tumor growth by blocking tumor angiogenesis, enhancing apoptosis and facilitating CTL development (66, 67). Ectopic expression of the gene encoding MDC in a murine lung tumor model has been shown to promote tumor rejection in vivo, and both T cells and DC have been shown to play important roles in this MDC-mediated mechanism of tumor rejection (68). Thus, tumor-induced changes in the IRF-8 levels of CD11b+Gr-1+ myeloid cells may have important pathological as well as therapeutic implications, and represent an integral determinant for the regulation of these myeloid populations.
Acknowledgments
This research was supported [in part] by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research. SIA was also supported in part by RPCI Institutional Funding. We thank Drs. Keiko Ozato (NICHD, NIH) for providing the IRF-8 plasmid, James Allison (Memorial Sloan-Kettering) for the anti-CTLA-4 hybridoma, Sandra Gendler (Mayo Clinic) for originally providing the B6-MTAG mouse model and Lionel Feigenbaum (SAIC/NCI-Frederick) for assistance with the production of the IRF-8 transgenic mouse.
Grant Support: This research was supported [in part] by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research. SIA was also supported in part by RPCI Institutional Funding.
Abbreviations used in this paper
- IRF-8
interferon regulatory factor-8
- LPS
lipopolysaccharide
- MTAG
Middle T AntiGen
- MDSC
myeloid-derived suppressor cells
- MMTV-PyMT
mouse mammary tumor virus-polyomavirus middle T antigen
- TDF
tumor-derived factors
- Tg
transgenic
Footnotes
Disclosures: All authors declare no competing financial interests.
References
- 1.Gabrilovich DI, Bronte V, Chen SH, Colombo MP, Ochoa A, Ostrand-Rosenberg S, Schreiber H. The terminology issue for myeloid-derived suppressor cells. Cancer Res. 2007;67:425. doi: 10.1158/0008-5472.CAN-06-3037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Serafini P, Borrello I, Bronte V. Myeloid suppressor cells in cancer: recruitment, phenotype, properties, and mechanisms of immune suppression. Semin Cancer Biol. 2006;16:53–65. doi: 10.1016/j.semcancer.2005.07.005. [DOI] [PubMed] [Google Scholar]
- 3.Talmadge JE. Pathways mediating the expansion and immunosuppressive activity of myeloid-derived suppressor cells and their relevance to cancer therapy. Clin Cancer Res. 2007;13:5243–5248. doi: 10.1158/1078-0432.CCR-07-0182. [DOI] [PubMed] [Google Scholar]
- 4.Bobe P, Benihoud K, Grandjon D, Opolon P, Pritchard LL, Huchet R. Nitric oxide mediation of active immunosuppression associated with graft-versus-host reaction. Blood. 1999;94:1028–1037. [PubMed] [Google Scholar]
- 5.Makarenkova VP, Bansal V, Matta BM, Perez LA, Ochoa JB. CD11b+/Gr-1+ myeloid suppressor cells cause T cell dysfunction after traumatic stress. J Immunol. 2006;176:2085–2094. doi: 10.4049/jimmunol.176.4.2085. [DOI] [PubMed] [Google Scholar]
- 6.Bronte V, Wang M, Overwijk WW, Surman DR, Pericle F, Rosenberg SA, Restifo NP. Apoptotic death of CD8+ T lymphocytes after immunization: induction of a suppressive population of Mac-1+/Gr-1+ cells. J Immunol. 1998;161:5313–5320. [PMC free article] [PubMed] [Google Scholar]
- 7.Schleifer KW, Mansfield JM. Suppressor macrophages in African trypanosomiasis inhibit T cell proliferative responses by nitric oxide and prostaglandins. J Immunol. 1993;151:5492–5503. [PubMed] [Google Scholar]
- 8.Serafini P, De Santo C, Marigo I, Cingarlini S, Dolcetti L, Gallina G, Zanovello P, Bronte V. Derangement of immune responses by myeloid suppressor cells. Cancer Immunol Immunother. 2004;53:64–72. doi: 10.1007/s00262-003-0443-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang L, DeBusk LM, Fukuda K, Fingleton B, Green-Jarvis B, Shyr Y, Matrisian LM, Carbone DP, Lin PC. Expansion of myeloid immune suppressor Gr+CD11b+ cells in tumor-bearing host directly promotes tumor angiogenesis. Cancer Cell. 2004;6:409–421. doi: 10.1016/j.ccr.2004.08.031. [DOI] [PubMed] [Google Scholar]
- 10.Yang L, Huang J, Ren X, Gorska AE, Chytil A, Aakre M, Carbone DP, Matrisian LM, Richmond A, Lin PC, Moses HL. Abrogation of TGF beta signaling in mammary carcinomas recruits Gr-1+CD11b+ myeloid cells that promote metastasis. Cancer Cell. 2008;13:23–35. doi: 10.1016/j.ccr.2007.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Almand B, Clark JI, Nikitina E, van Beynen J, English NR, Knight SC, Carbone DP, Gabrilovich DI. Increased production of immature myeloid cells in cancer patients: a mechanism of immunosuppression in cancer. J Immunol. 2001;166:678–689. doi: 10.4049/jimmunol.166.1.678. [DOI] [PubMed] [Google Scholar]
- 12.Schmielau J, Finn OJ. Activated granulocytes and granulocyte-derived hydrogen peroxide are the underlying mechanism of suppression of t-cell function in advanced cancer patients. Cancer Res. 2001;61:4756–4760. [PubMed] [Google Scholar]
- 13.Zea AH, Rodriguez PC, Atkins MB, Hernandez C, Signoretti S, Zabaleta J, McDermott D, Quiceno D, Youmans A, O’Neill A, Mier J, Ochoa AC. Arginase-producing myeloid suppressor cells in renal cell carcinoma patients: a mechanism of tumor evasion. Cancer Res. 2005;65:3044–3048. doi: 10.1158/0008-5472.CAN-04-4505. [DOI] [PubMed] [Google Scholar]
- 14.Filipazzi P, Valenti R, Huber V, Pilla L, Canese P, Iero M, Castelli C, Mariani L, Parmiani G, Rivoltini L. Identification of a new subset of myeloid suppressor cells in peripheral blood of melanoma patients with modulation by a granulocyte-macrophage colony-stimulation factor-based antitumor vaccine. J Clin Oncol. 2007;25:2546–2553. doi: 10.1200/JCO.2006.08.5829. [DOI] [PubMed] [Google Scholar]
- 15.Ochoa AC, Zea AH, Hernandez C, Rodriguez PC. Arginase, prostaglandins, and myeloid-derived suppressor cells in renal cell carcinoma. Clin Cancer Res. 2007;13:721s–726s. doi: 10.1158/1078-0432.CCR-06-2197. [DOI] [PubMed] [Google Scholar]
- 16.Srivastava MK, Bosch JJ, Thompson JA, Ksander BR, Edelman MJ, Ostrand-Rosenberg S. Lung cancer patients’ CD4(+) T cells are activated in vitro by MHC II cell-based vaccines despite the presence of myeloid-derived suppressor cells. Cancer Immunol Immunother. 2008;57:1493–1504. doi: 10.1007/s00262-008-0490-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Diaz-Montero CM, Salem ML, Nishimura MI, Garrett-Mayer E, Cole DJ, Montero AJ. Increased circulating myeloid-derived suppressor cells correlate with clinical cancer stage, metastatic tumor burden, and doxorubicin-cyclophosphamide chemotherapy. Cancer Immunol Immunother. 2009;58:49–59. doi: 10.1007/s00262-008-0523-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu Y, Van Ginderachter JA, Brys L, De Baetselier P, Raes G, Geldhof AB. Nitric oxide-independent CTL suppression during tumor progression: association with arginase-producing (M2) myeloid cells. J Immunol. 2003;170:5064–5074. doi: 10.4049/jimmunol.170.10.5064. [DOI] [PubMed] [Google Scholar]
- 19.Rodriguez PC, Quiceno DG, Zabaleta J, Ortiz B, Zea AH, Piazuelo MB, Delgado A, Correa P, Brayer J, Sotomayor EM, Antonia S, Ochoa JB, Ochoa AC. Arginase I production in the tumor microenvironment by mature myeloid cells inhibits T-cell receptor expression and antigen-specific T-cell responses. Cancer Res. 2004;64:5839–5849. doi: 10.1158/0008-5472.CAN-04-0465. [DOI] [PubMed] [Google Scholar]
- 20.Sinha P, Okoro C, Foell D, Freeze HH, Ostrand-Rosenberg S, Srikrishna G. Proinflammatory s100 proteins regulate the accumulation of myeloid-derived suppressor cells. J Immunol. 2008;181:4666–4675. doi: 10.4049/jimmunol.181.7.4666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Apolloni E, Bronte V, Mazzoni A, Serafini P, Cabrelle A, Segal DM, Young HA, Zanovello P. Immortalized myeloid suppressor cells trigger apoptosis in antigen-activated T lymphocytes. J Immunol. 2000;165:6723–6730. doi: 10.4049/jimmunol.165.12.6723. [DOI] [PubMed] [Google Scholar]
- 22.Lin EY, Gouon-Evans V, Nguyen AV, Pollard JW. The macrophage growth factor CSF-1 in mammary gland development and tumor progression. J Mammary Gland Biol Neoplasia. 2002;7:147–162. doi: 10.1023/a:1020399802795. [DOI] [PubMed] [Google Scholar]
- 23.Gabrilovich D, Ishida T, Oyama T, Ran S, Kravtsov V, Nadaf S, Carbone DP. Vascular endothelial growth factor inhibits the development of dendritic cells and dramatically affects the differentiation of multiple hematopoietic lineages in vivo. Blood. 1998;92:4150–4166. [PubMed] [Google Scholar]
- 24.Gabrilovich D. Mechanisms and functional significance of tumour-induced dendritic-cell defects. Nat Rev Immunol. 2004;4:941–952. doi: 10.1038/nri1498. [DOI] [PubMed] [Google Scholar]
- 25.Melani C, Chiodoni C, Forni G, Colombo MP. Myeloid cell expansion elicited by the progression of spontaneous mammary carcinomas in c-erbB-2 transgenic BALB/c mice suppresses immune reactivity. Blood. 2003;102:2138–2145. doi: 10.1182/blood-2003-01-0190. [DOI] [PubMed] [Google Scholar]
- 26.Serafini P, Carbley R, Noonan KA, Tan G, Bronte V, Borrello I. High-dose granulocyte-macrophage colony-stimulating factor-producing vaccines impair the immune response through the recruitment of myeloid suppressor cells. Cancer Res. 2004;64:6337–6343. doi: 10.1158/0008-5472.CAN-04-0757. [DOI] [PubMed] [Google Scholar]
- 27.Song X, Krelin Y, Dvorkin T, Bjorkdahl O, Segal S, Dinarello CA, Voronov E, Apte RN. CD11b+/Gr-1+ immature myeloid cells mediate suppression of T cells in mice bearing tumors of IL-1beta-secreting cells. J Immunol. 2005;175:8200–8208. doi: 10.4049/jimmunol.175.12.8200. [DOI] [PubMed] [Google Scholar]
- 28.Bunt SK, Yang L, Sinha P, Clements VK, Leips J, Ostrand-Rosenberg S. Reduced inflammation in the tumor microenvironment delays the accumulation of myeloid-derived suppressor cells and limits tumor progression. Cancer Res. 2007;67:10019–10026. doi: 10.1158/0008-5472.CAN-07-2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Marrache F, Tu SP, Bhagat G, Pendyala S, Osterreicher CH, Gordon S, Ramanathan V, Penz-Osterreicher M, Betz KS, Song Z, Wang TC. Overexpression of interleukin-1beta in the murine pancreas results in chronic pancreatitis. Gastroenterology. 2008;135:1277–1287. doi: 10.1053/j.gastro.2008.06.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tu S, Bhagat G, Cui G, Takaishi S, Kurt-Jones EA, Rickman B, Betz KS, Penz-Oesterreicher M, Bjorkdahl O, Fox JG, Wang TC. Overexpression of interleukin-1beta induces gastric inflammation and cancer and mobilizes myeloid-derived suppressor cells in mice. Cancer Cell. 2008;14:408–419. doi: 10.1016/j.ccr.2008.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sinha P, Clements VK, Fulton AM, Ostrand-Rosenberg S. Prostaglandin E2 promotes tumor progression by inducing myeloid-derived suppressor cells. Cancer Res. 2007;67:4507–4513. doi: 10.1158/0008-5472.CAN-06-4174. [DOI] [PubMed] [Google Scholar]
- 32.Shojaei F, Wu X, Zhong C, Yu L, Liang XH, Yao J, Blanchard D, Bais C, Peale FV, van Bruggen N, Ho C, Ross J, Tan M, Carano RA, Meng YG, Ferrara N. Bv8 regulates myeloid-cell-dependent tumour angiogenesis. Nature. 2007;450:825–831. doi: 10.1038/nature06348. [DOI] [PubMed] [Google Scholar]
- 33.Aslakson CJ, Miller FR. Selective events in the metastatic process defined by analysis of the sequential dissemination of subpopulations of a mouse mammary tumor. Cancer Res. 1992;52:1399–1405. [PubMed] [Google Scholar]
- 34.Guy CT, Cardiff RD, Muller WJ. Induction of mammary tumors by expression of polyomavirus middle T oncogene: a transgenic mouse model for metastatic disease. Mol Cell Biol. 1992;12:954–961. doi: 10.1128/mcb.12.3.954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lin EY, Jones JG, Li P, Zhu L, Whitney KD, Muller WJ, Pollard JW. Progression to malignancy in the polyoma middle T oncoprotein mouse breast cancer model provides a reliable model for human diseases. Am J Pathol. 2003;163:2113–2126. doi: 10.1016/S0002-9440(10)63568-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stewart TJ, Abrams SI. Altered Immune Function during Long-Term Host-Tumor Interactions Can Be Modulated to Retard Autochthonous Neoplastic Growth. J Immunol. 2007;179:2851–2859. doi: 10.4049/jimmunol.179.5.2851. [DOI] [PubMed] [Google Scholar]
- 37.Holtschke T, Lohler J, Kanno Y, Fehr T, Giese N, Rosenbauer F, Lou J, Knobeloch KP, Gabriele L, Waring JF, Bachmann MF, Zinkernagel RM, Morse HC, 3rd, Ozato K, Horak I. Immunodeficiency and chronic myelogenous leukemia-like syndrome in mice with a targeted mutation of the ICSBP gene. Cell. 1996;87:307–317. doi: 10.1016/s0092-8674(00)81348-3. [DOI] [PubMed] [Google Scholar]
- 38.Masumi A, Tamaoki S, Wang IM, Ozato K, Komuro K. IRF-8/ICSBP and IRF-1 cooperatively stimulate mouse IL-12 promoter activity in macrophages. FEBS Lett. 2002;531:348–353. doi: 10.1016/s0014-5793(02)03556-1. [DOI] [PubMed] [Google Scholar]
- 39.Tamura T, Ozato K. ICSBP/IRF-8: its regulatory roles in the development of myeloid cells. J Interferon Cytokine Res. 2002;22:145–152. doi: 10.1089/107999002753452755. [DOI] [PubMed] [Google Scholar]
- 40.Liu J, Ma X. Interferon regulatory factor 8 regulates RANTES gene transcription in cooperation with interferon regulatory factor-1, NF-kappaB, and PU.1. J Biol Chem. 2006;281:19188–19195. doi: 10.1074/jbc.M602059200. [DOI] [PubMed] [Google Scholar]
- 41.Wang IM, Contursi C, Masumi A, Ma X, Trinchieri G, Ozato K. An IFN-gamma-inducible transcription factor, IFN consensus sequence binding protein (ICSBP), stimulates IL-12 p40 expression in macrophages. J Immunol. 2000;165:271–279. doi: 10.4049/jimmunol.165.1.271. [DOI] [PubMed] [Google Scholar]
- 42.Tamura T, Yanai H, Savitsky D, Taniguchi T. The IRF family transcription factors in immunity and oncogenesis. Annu Rev Immunol. 2008;26:535–584. doi: 10.1146/annurev.immunol.26.021607.090400. [DOI] [PubMed] [Google Scholar]
- 43.Stewart TJ, Greeneltch KM, Reid JE, Liewehr DJ, Steinberg SM, Liu K, Abrams SI. Interferon regulatory factor-8 modulates the development of tumor-induced CD11b+Gr-1+ myeloid cells. J Cell Mol Med. doi: 10.1111/j.1582-4934.2009.00685.x. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen D, Xia J, Tanaka Y, Chen H, Koido S, Wernet O, Mukherjee P, Gendler SJ, Kufe D, Gong J. Immunotherapy of spontaneous mammary carcinoma with fusions of dendritic cells and mucin 1-positive carcinoma cells. Immunology. 2003;109:300–307. doi: 10.1046/j.1365-2567.2003.01656.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gallina G, Dolcetti L, Serafini P, De Santo C, Marigo I, Colombo MP, Basso G, Brombacher F, Borrello I, Zanovello P, Bicciato S, Bronte V. Tumors induce a subset of inflammatory monocytes with immunosuppressive activity on CD8+ T cells. J Clin Invest. 2006;116:2777–2790. doi: 10.1172/JCI28828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Movahedi K, Guilliams M, Van den Bossche J, Van den Bergh R, Gysemans C, Beschin A, De Baetselier P, Van Ginderachter JA. Identification of discrete tumor-induced myeloid-derived suppressor cell subpopulations with distinct T cell-suppressive activity. Blood. 2008;111:4233–4244. doi: 10.1182/blood-2007-07-099226. [DOI] [PubMed] [Google Scholar]
- 47.Terabe M, Matsui S, Park JM, Mamura M, Noben-Trauth N, Donaldson DD, Chen W, Wahl SM, Ledbetter S, Pratt B, Letterio JJ, Paul WE, Berzofsky JA. Transforming growth factor-beta production and myeloid cells are an effector mechanism through which CD1d-restricted T cells block cytotoxic T lymphocyte-mediated tumor immunosurveillance: abrogation prevents tumor recurrence. J Exp Med. 2003;198:1741–1752. doi: 10.1084/jem.20022227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science. 1996;271:1734–1736. doi: 10.1126/science.271.5256.1734. [DOI] [PubMed] [Google Scholar]
- 49.Ryan MH, Bristol JA, McDuffie E, Abrams SI. Regression of extensive pulmonary metastases in mice by adoptive transfer of antigen-specific CD8+ CTL reactive against tumor cells expressing a naturally occurring rejection epitope. J Immunol. 2001;167:4286–4292. doi: 10.4049/jimmunol.167.8.4286. [DOI] [PubMed] [Google Scholar]
- 50.O’Brien PC. Procedures for comparing samples with multiple endpoints. Biometrics. 1984;40:1079–1087. [PubMed] [Google Scholar]
- 51.Maglione JE, Moghanaki D, Young LJ, Manner CK, Ellies LG, Joseph SO, Nicholson B, Cardiff RD, MacLeod CL. Transgenic Polyoma middle-T mice model premalignant mammary disease. Cancer Res. 2001;61:8298–8305. [PubMed] [Google Scholar]
- 52.Stewart TJ, Lutsiak ME, Abrams SI. Immune consequences of protracted host-tumor interactions in a transgenic mouse model of mammary carcinoma. Cancer Invest. 2008;26:237–249. doi: 10.1080/07357900701708419. [DOI] [PubMed] [Google Scholar]
- 53.Delano MJ, Scumpia PO, Weinstein JS, Coco D, Nagaraj S, Kelly-Scumpia KM, O’Malley KA, Wynn JL, Antonenko S, Al-Quran SZ, Swan R, Chung CS, Atkinson MA, Ramphal R, Gabrilovich DI, Reeves WH, Ayala A, Phillips J, Laface D, Heyworth PG, Clare-Salzler M, Moldawer LL. MyD88-dependent expansion of an immature GR-1(+)CD11b(+) population induces T cell suppression and Th2 polarization in sepsis. J Exp Med. 2007;204:1463–1474. doi: 10.1084/jem.20062602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kusmartsev S, Cheng F, Yu B, Nefedova Y, Sotomayor E, Lush R, Gabrilovich D. All-trans-retinoic acid eliminates immature myeloid cells from tumor-bearing mice and improves the effect of vaccination. Cancer Res. 2003;63:4441–4449. [PubMed] [Google Scholar]
- 55.Serafini P, Meckel K, Kelso M, Noonan K, Califano J, Koch W, Dolcetti L, Bronte V, Borrello I. Phosphodiesterase-5 inhibition augments endogenous antitumor immunity by reducing myeloid-derived suppressor cell function. J Exp Med. 2006;203:2691–2702. doi: 10.1084/jem.20061104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Suzuki E, Kapoor V, Jassar AS, Kaiser LR, Albelda SM. Gemcitabine selectively eliminates splenic Gr-1+/CD11b+ myeloid suppressor cells in tumor-bearing animals and enhances antitumor immune activity. Clin Cancer Res. 2005;11:6713–6721. doi: 10.1158/1078-0432.CCR-05-0883. [DOI] [PubMed] [Google Scholar]
- 57.Shojaei F, Wu X, Malik AK, Zhong C, Baldwin ME, Schanz S, Fuh G, Gerber HP, Ferrara N. Tumor refractoriness to anti-VEGF treatment is mediated by CD11b+Gr1+ myeloid cells. Nat Biotechnol. 2007;25:911–920. doi: 10.1038/nbt1323. [DOI] [PubMed] [Google Scholar]
- 58.Kobayashi Y. The role of chemokines in neutrophil biology. Front Biosci. 2008;13:2400–2407. doi: 10.2741/2853. [DOI] [PubMed] [Google Scholar]
- 59.Pollard JW. Tumour-educated macrophages promote tumour progression and metastasis. Nat Rev Cancer. 2004;4:71–78. doi: 10.1038/nrc1256. [DOI] [PubMed] [Google Scholar]
- 60.Mocellin S, Wang E, Marincola FM. Cytokines and immune response in the tumor microenvironment. J Immunother. 2001;24:392–407. [PubMed] [Google Scholar]
- 61.Steinbrink K, Wolfl M, Jonuleit H, Knop J, Enk AH. Induction of tolerance by IL-10-treated dendritic cells. J Immunol. 1997;159:4772–4780. [PubMed] [Google Scholar]
- 62.Wittke F, Hoffmann R, Buer J, Dallmann I, Oevermann K, Sel S, Wandert T, Ganser A, Atzpodien J. Interleukin 10 (IL-10): an immunosuppressive factor and independent predictor in patients with metastatic renal cell carcinoma. Br J Cancer. 1999;79:1182–1184. doi: 10.1038/sj.bjc.6690189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.De Vita F, Orditura M, Galizia G, Romano C, Lieto E, Iodice P, Tuccillo C, Catalano G. Serum interleukin-10 is an independent prognostic factor in advanced solid tumors. Oncol Rep. 2000;7:357–361. doi: 10.3892/or.7.2.357. [DOI] [PubMed] [Google Scholar]
- 64.Sinha P, Clements VK, Bunt SK, Albelda SM, Ostrand-Rosenberg S. Cross-talk between myeloid-derived suppressor cells and macrophages subverts tumor immunity toward a type 2 response. J Immunol. 2007;179:977–983. doi: 10.4049/jimmunol.179.2.977. [DOI] [PubMed] [Google Scholar]
- 65.Trikha M, Corringham R, Klein B, Rossi JF. Targeted anti-interleukin-6 monoclonal antibody therapy for cancer: a review of the rationale and clinical evidence. Clin Cancer Res. 2003;9:4653–4665. [PMC free article] [PubMed] [Google Scholar]
- 66.Pertl U, Luster AD, Varki NM, Homann D, Gaedicke G, Reisfeld RA, Lode HN. IFN-gamma-inducible protein-10 is essential for the generation of a protective tumor-specific CD8 T cell response induced by single-chain IL-12 gene therapy. J Immunol. 2001;166:6944–6951. doi: 10.4049/jimmunol.166.11.6944. [DOI] [PubMed] [Google Scholar]
- 67.Yao L, Pike SE, Pittaluga S, Cherney B, Gupta G, Jaffe ES, Tosato G. Anti-tumor activities of the angiogenesis inhibitors interferon-inducible protein-10 and the calreticulin fragment vasostatin. Cancer Immunol Immunother. 2002;51:358–366. doi: 10.1007/s00262-002-0294-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Guo J, Wang B, Zhang M, Chen T, Yu Y, Regulier E, Homann HE, Qin Z, Ju DW, Cao X. Macrophage-derived chemokine gene transfer results in tumor regression in murine lung carcinoma model through efficient induction of antitumor immunity. Gene Ther. 2002;9:793–803. doi: 10.1038/sj.gt.3301688. [DOI] [PubMed] [Google Scholar]