

Abstract
Roles of cyclin dependent kinase inhibitors, p21/Cip1 (p21) and p27/Kip1 (p27) in prostate cancer (PCa) progression is still not clear. Lower p27 protein expression in PCa tissues is often associated with poor prognosis, but prognostic significance of p21 is still controversial. Herein, we investigated the role of these molecules in determining PCa growth characteristics. We generated human PCa DU145 cell variants with knocked down levels of p21 (DU-p21) or p27 (DU-p27), or both (DU-p21+p27) via retroviral transduction of respective shRNAs and compared their various characteristics with empty vector-transduced DU145 (DU-EV) cells in vitro as well as in vivo. Knocking down either p21 or p27 did not show any significant change in doubling time, clonogenicity and cell cycle progression in DU145 cells, but simultaneous knock-down of both p21 and p27 significantly enhanced these parameters. In athymic mice, DU-p21+p27 tumors showed higher growth rate than the comparable growth of DU-EV, DU-p21 and DU-p27 tumors. Concurrently, DU-p21+p27 tumors had significantly higher proliferation rate, showing 54% and 48% increase in proliferating cell nuclear antigen (PCNA) and Ki-67-positive cells, respectively, compared to DU-EV tumors. DU-p21+p27 tumors also showed higher microvessel density and increased expression of vascular endothelial growth factor (VEGF). Proliferation and angiogenic status of DU-p21 and DU-p27 tumors was comparable to DU-EV tumors. Both in vitro and in vivo results implicate that p21 and p27 have compensatory roles in advanced prostate cancer cells, and ablation or down-modulation of both these molecules essentially enhances the aggressive prostate carcinoma phenotype.
Keywords: prostate cancer, p21/Cip1, p27/Kip1, tumorigenicity
Introduction
Prostate cancer (PCa) is the most frequently diagnosed cancer in the United States male population.1 In the early stages, it is androgen-dependent and responds to androgen-ablation therapy. However, the disease almost invariably progresses to androgen-independent state which is more malignant and apoptosis-resistant.2 Digital rectal examination and detection of prostate specific antigen levels have been beneficial to screen and diagnose the disease only at its early stages. Thus better biomarkers are needed to identify and predict the tumorigenic potential of the cancer, disease outcome and recurrence rate and to provide more non-invasive treatment options of the malignancy.
The sequence of events in cell cycle progression is highly orchestrated and depends on the cyclic activation and inactivation of cyclin dependent kinases (CDK) which govern the progression of the cells from one phase to another.3 In the event of tumorigenesis, constitutive mitogenic signaling as well as mutations in tumor suppressor genes and proto-oncogenes leads to cell cycle deregulation and uncontrolled proliferation.4 p21 and p27 belong to the Cip/Kip family of cyclin dependent kinase inhibitors (CDKIs), which play a determining role in G1-S transition by regulating cyclin dependent kinase (CDK) activity. p27 is considered to be happloinsufficient due to increased predisposition of both p27 homozygous and heterozygous mice to tumor formation at multiple sites on exposure to carcinogen.5 No such conclusive reports are available about p21. However, the incidence of polymorphism or mutation of their genes in cancer is very rare.6 Their altered protein expression level, on the other hand, is considered important to predict disease outcome in various cancer types. In case of prostate cancer, reduced p27 expression has been consistently associated with more aggressive form of cancer and poor survival.7, 8 However, the significance of p21 protein expression in disease prognosis has remained controversial. Upregulation of p21 by adenoviral infection or various chemotherapeutic agents have shown to induce growth arrest in several androgen-dependent and -independent prostate cancer cell lines.9-11 But paradoxically, elevated p21 level in human prostate cancer tissues has in many cases been correlated with higher Gleason score, poor survival and increased recurrence rate of the disease.12 Some studies suggest the inverse relation of p21 level with survivability to be dependent on ethnicity.13 However, some reports have also shown p21 to be associated with favorable prognosis. Cheng et al reported high p21 and p27 levels are associated with metastasis-free survival in patients treated by salvage prostatectomy after radiation therapy. Thus these mixed observations warrant further studies to examine the roles of these molecules in PCa progression and justify their use as biomarker in prostate cancer patients.
In the present study we aimed to identify the importance of p21 and p27 in advanced prostate cancer cells, in terms of their growth, cell cycle progression, tumorigenicity and angiogenic potential. We generated advanced human PCa DU145 cells with stable knocked-down levels of p21 and/or p27, and observed that diminished expression of either p21 or p27 alone does not significantly alter the proliferation rate or tumorigenicity of DU145 cells, both in vivo and in vitro. However, simultaneous reduction of p21 and p27 protein levels renders these cells more aggressive and angiogenic.
Results
Effect of knocking down of p21 and/or p27 levels on proliferation rate and clonogenic potential of DU145 cells
Stable knockdown of p21 and/or p27 levels were achieved by retroviral transduction of p21 and/or p27 shRNAs in DU145 cells. About 70% and 50% reduction in p21 level was obtained in DU-p21 and DU-p21+p27 cells respectively, compared to empty vector infected cells (DU-EV) (Fig. 1A). About 50 % reduction in p27 levels was achieved in both DU-p27 and DU-p21+p27 cells compared to DU-EV cells (Fig. 1A). Transduction of p21 shRNA did not affect the protein expression of p27 levels and vice versa which proves the target-specificity of the respective shRNAs. Even though we did not obtain a complete knockdown in the levels of p21 and p27 levels in these cell lines, we evaluated the impact of reduction of these critical negative regulators of cell cycle on different phenotypic characteristics of DU145 cells. First we compared the growth characteristics/proliferation rate of these different cell variants. We observed that DU-EV, DU-p21 and DU-p27 cells followed similar growth pattern from 24-96 h after plating. However, DU-p21+p27 cells showed comparatively a much faster proliferation rate (Fig. 1B). The doubling time of the cells was calculated using the formula: ‘h*ln(2)/ln(c2/c1)’ where h is the time difference; c1 is the initial cell number; c2 is the final cell number after h hours. The time span 48-96 h after plating, were selected for calculating the doubling time. We observed that DU-p21+27 cells has a significantly shorter doubling time (28 h) compared to DU-EV cells (34 h), while that of DU-p21 (32 h) and DU-p27 (33 h) cells were very close to DU-EV cells. Moreover, only DU-p21+p27 cells had 30% higher clonogenicity (P<0.001) than DU-EV cells, while the clonogenic potential of DU-p21 and DU-p27 was not different from that of DU-EV (Fig. 1C).
Figure 1.
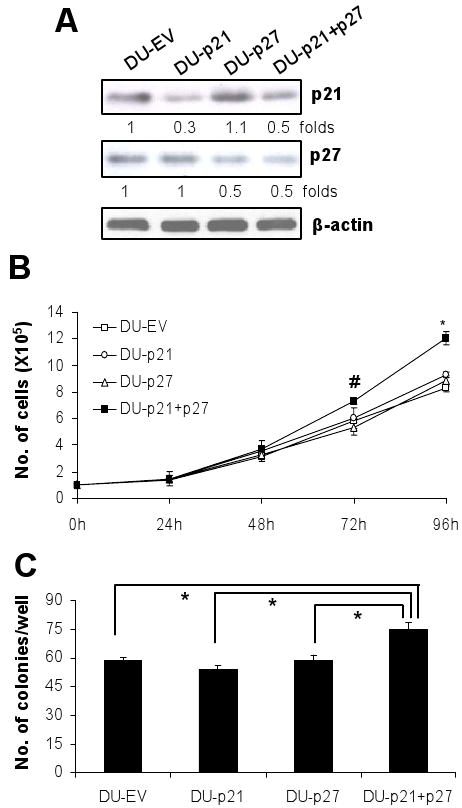
Effect of p21 and/or p27 knockdown on proliferative and clonogenic potential of DU145 cells. A, protein expression levels of p21 and p27 were estimated by western blot analysis in DU145 cells transduced with empty vector, p21, p27 or both p21 and p27 shRNA retroviruses as detailed in Materials and Methods. B, the in vitro proliferation rate of asynchronous DU-EV, DU-p21, DU-p27 and DU-p21+p27 cells were measured by counting the cells after 24, 48, 72 and 96 h of plating. C, the clonogenic potential of the four cell lines were assessed by counting the number of colonies formed after 10 days. Data are represented as mean ± SEM (n=3). #, P<0.05 and *, P<0.001. If not otherwise indicated, statistical analysis is performed versus DU-EV.
Effect of knocking down of p21 and/or p27 levels on cell cycle progression in DU145 cells
Under asynchronous condition, all the four cell lines show similar cell cycle distribution (data not shown). Given the fact that they are all immortalized tumor cells, any discernible difference in their cell cycle progression is not expected under asynchronous condition. However, p21 and p27 are crucial regulators of G1 checkpoint and, therefore, determine the G1 exit of cell population. Thus, if cells are brought to G0-G1 and then allowed to reenter into cell cycle, they might respond differently. Hence, we synchronized the cells in G0-G1 by serum-starvation and released them in serum-containing media and examined the pattern of G1-S transit in all the four cell lines (Fig. 2A). At 0 h of release, all the cell lines showed ∼70% cell population in G0-G1 phase. They started coming out of G1 from 12 h (data not shown), and by 16 h, all the cell lines had a significant increase in population (from 20-22% at 0 h to 47-52% at 16 h) in S phase (Fig. 2B). However, only the DU-p21+p27 cells showed a much higher percentage of S phase population compared to DU-EV cells (52% in DU-p21+p27 versus 47% in DU-EV; P<0.001). The S phase population of both DU-p21 (49%) and DU-p27 (49%) cells after 16 h of release were not significantly different from that of DU-EV cells. The increased S phase population of DU-p21+p27 cells was also evident after 20 h (56% in DU-p21+p27 versus 48% in DU-EV; P<0.05) of release (Fig. 2B). From 16 h to 20 h after release, although cells progressed from S to G2M, however, there was no considerable difference among the cell lines (Fig. 2B).
Figure 2.
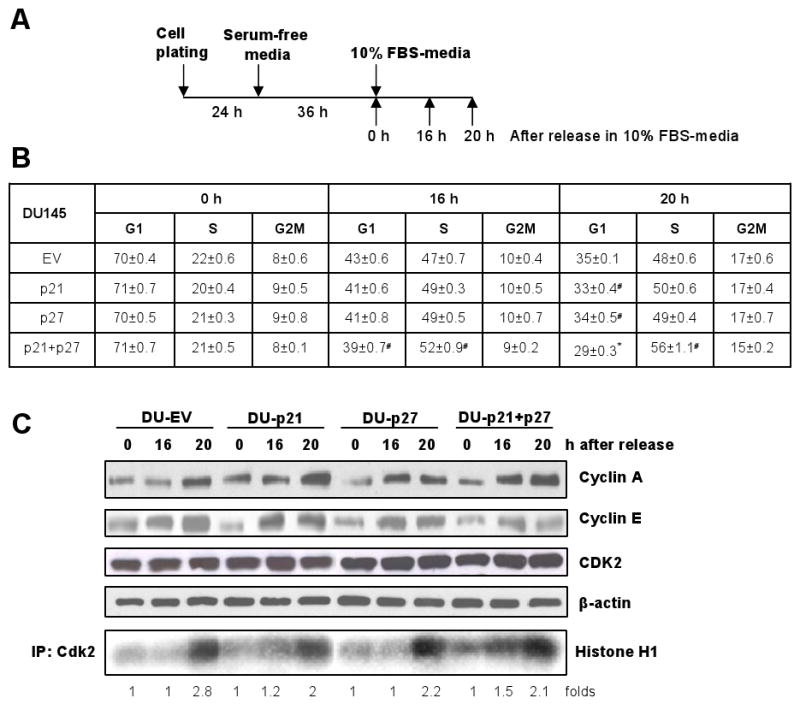
Effect of p21 and/or p27 knockdown on cell cycle progression in DU145 cells. A, DU-EV, DU-p21, DU-p27 and DU-p21+p27 cells were arrested in G0-G1 phase by serum-starvation for 36 h and then released in 10% serum containing media and harvested at different time points as indicated. B, cell cycle progression of all the four cell lines, after release from G0-G1 block, was analyzed by flow cytometry as detailed in Materials and Methods. C, protein expression of cyclin A, cyclin E and CDK2 as well as CDK2 kinase activity were assessed in all the four cell lines at different time points after release from G0-G1 block as detailed in Materials and Methods. The quantitative data shown is mean ± SEM of two independent experiments done in triplicates. #, P<0.05 and *, P<0.001 versus DU-EV. The immunoblots are representative of two independent experiments.
CDK2 is the primary kinase at late G1 and early S phase and its kinase activity determines the G1-S transit.3 Therefore, we analyzed the CDK2 kinase activity in the synchronized cells and observed that at 0 h of release, all the cell lines had a very low CDK2 basal activity (Fig. 2C). However, keeping with the cell cycle progression results, we observed that only DU-p21+27 cells showed higher kinase activity at 16 h of release (1.5 fold higher than 0 h kinase activity). After 20 h of release, however, all the cell lines showed similar kinase activity (Fig. 2C). The protein levels of S phase-regulating cyclins were also estimated under these conditions, and a gradual induction in their levels, following release, was observed in all the four cell lines with no observable difference in their extent of induction among the four cell lines (Fig. 2C). Overall, these findings indicate that the increased G1 to S phase progression of DU-p21+p27 cells with higher proliferation index compared to the other cell variants was primarily due to increased CDK2 kinase activity which in turn occurs due to simultaneous knockdown of Cip/Kip proteins.
Effect of knocking down of p21 and/or p27 levels on DU145 tumor growth in athymic nude mice
To confirm that the results obtained in vitro can be extrapolated to in vivo conditions, all the DU145 cell variants were implanted s.c. in athymic nude mice and grown for 6 weeks. As anticipated, DU-p21+p27 cells showed higher rate of tumor growth as compared to the DU-EV cells, with significant difference (P<0.001) observable after 4 weeks of cell inoculation. After 6 weeks, the tumor volumes for DU-EV, DU-p21, DU-p27 and DU-p21+p27 cell variants were 276 mm3, 414 mm3, 355 mm3 and 787 mm3 per tumor, respectively (Fig. 3A). For these cell variants, the excised tumor weights were 0.128 g, 0.214 g, 0.177 g and 0.39 g per tumor, respectively, (Fig. 3B). Both tumor volume (2.8-fold) as well as tumor weight (3-fold) for DU-p21+p27 tumors were significantly (P<0.001) higher than that of DU-EV tumors after 6 weeks of inoculation (Fig. 3A-B). The tumor volume and tumor weight for DU-p21 and DU-p27 were slightly higher but insignificant when compared with DU-EV. Even after 6 weeks of inoculation, the tumors from the DU-p21, DU-p27 and DU-p21+p27 cells had attenuated levels of p21 and/or p27 as examined by the western blot analysis of tumor samples (Fig. 3C). Densitometric analysis showed that compared to DU-EV tumors, the mean p21 levels was 60% lower in DU-p21 and DU-p21+p27 tumors, while mean p27 levels was about 50 % lower in DU-p27 and DU-p21+p27 tumors (Fig 3D). These results indicate that knocking down of both p21 and p27 proteins have a pronounced impact on tumor growth potential of DU145 cells in in vivo condition.
Figure 3.
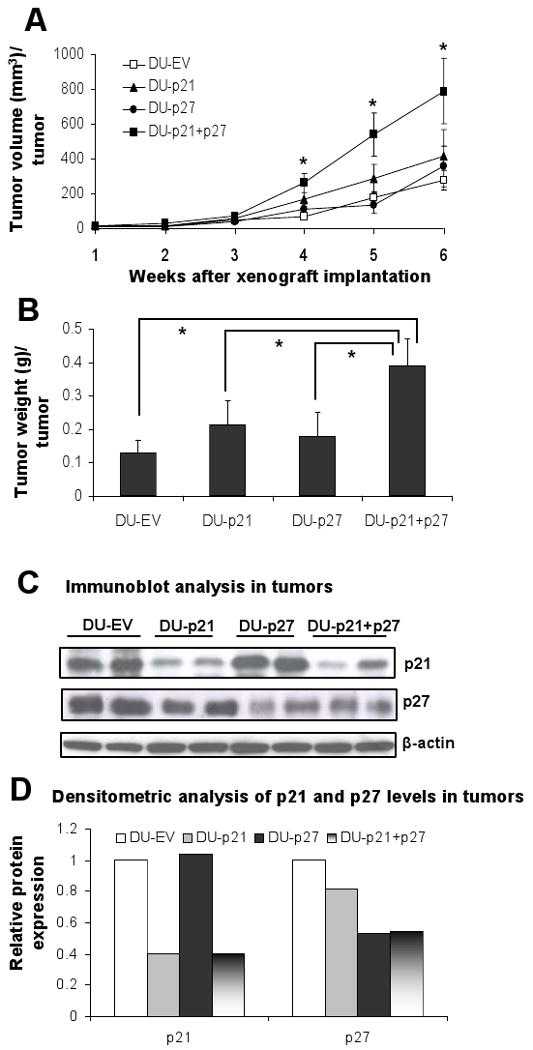
Effect of p21 and/or p27 knockdown on the DU145 tumor growth in athymic nude mice. Mice (n=5 per group) were subcutaneously injected with 3.5 × 106 DU-EV, DU-p21, DU-p27 or DU-p21+p27 cells mixed with matrigel and animals were euthanized after 6 weeks and tumors were collected. A, tumor growth was monitored and tumor volume is represented as average (mean ± SEM) of 5 animals in each group. B, excised tumor weight is represented as average (mean ± SEM) of 5 animals in each group, at the end of 6 weeks. C, two randomly selected tumor samples from each group was subjected to immunoblotting for p21 and p27 protein expression levels as detailed in Materials and Methods. D, densitometry data are shown for p21 and p27 protein levels in tumors from Fig. 3C. *, P<0.001. If not otherwise indicated statistical analysis is performed versus DU-EV.
Effect of knocking down of p21 and/or p27 levels on tumor cell proliferation
Larger tumors are often a consequence of higher proliferation rate, which can be measured by the immunoreactivity for PCNA and Ki-67 (Fig. 4A, C). Thus, we assessed the percentage of PCNA and Ki-67-positive cells to analyze the correlation between the proliferative potential and tumor growth of the four types of tumors obtained in the study. DU-EV tumors showed 37% PCNA-positive cells, while DU-p21 and DU-p27 tumors showed only a marginal increase in proliferation rate with 40% and 42% PCNA-positive cells, respectively (Fig. 4B). However, DU-p21+p27 tumors had 58% PCNA-positive cells indicating 56% increased (P<0.001) proliferation compared to DU-EV tumors (Fig. 4B). The observed increase in proliferation rate in DU-p21+p27 was reconfirmed by staining for Ki-67, another well known marker for tumor cell proliferation, which also correlates with tumor grade and clinical course.14 In this analysis, DU-p21+p27 tumors showed 49% Ki-67 positive cells (P<0.001 vs. DU-EV) which was significantly higher than that of DU-EV (33%), DU-p21 (36%) and DU-p27 (39%) tumors (Fig 4D). Thus, the higher growth rate of tumor was accompanied by higher tumor cell proliferation in p21+p27 knock-down DU145 cells.
Figure 4.
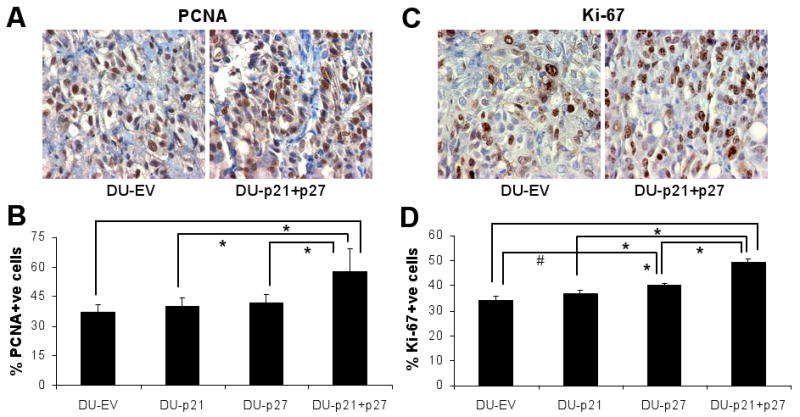
Proliferation status of DU-EV, DU-p21, DU-p27 and DU-p21+p27 tumors. The tumor samples collected at the end of 6 weeks of the study detailed in Figure 3, were processed for immunohistochemical analysis of the proliferation markers (A) PCNA and (C) Ki-67 as detailed in Materials and Methods. Representative images of DU-EV and DU-p21+p27 tumors are shown in each case. Immunostaining was quantified for (B) PCNA and (D) Ki-67 positive cells as detailed in Materials and Methods. #, P<0.05; *, P<0.001.
Effect of knocking down of p21 and/or p27 levels on angiogenic potential of tumors
Angiogenesis allows solid tumors to grow beyond a diameter of 1-2 mm.15 Since we observed a prominent difference in tumor sizes among the different cell line-derived tumors, we investigated whether the larger tumor size of DU-p21+p27 was associated with increased vasculature. The expression of CD31, a marker for endothelial cells, and VEGF was immunohistochemically assessed (Fig. 5A, C). DU-p21+p27 tumors showed 28% increase in CD31-positive endothelial cells over DU-EV tumors (P<0.001), while DU-p21 and DU-p27 tumors had equivalent microvessel density as DU-EV (Fig. 5B). Moreover, VEGF production and secretion by tumors is critical to angiogenesis. Since we observed differences in microvessel density in the different tumor types, we also assessed their intracellular VEGF levels by immunohistochemical analysis (Fig. 5C). Similar to the tumor vasculature data, only DU-p21+p27 tumors showed higher VEGF immunoreactivity (20% higher than DU-EV; P<0.05), while that of DU-EV, DU-p21 and DU-p27 were comparable (Fig. 5D). These results indicate that simultaneous down-regulation of both p21 and p27 makes DU145 tumors more angiogenic to support their growth.
Figure 5.
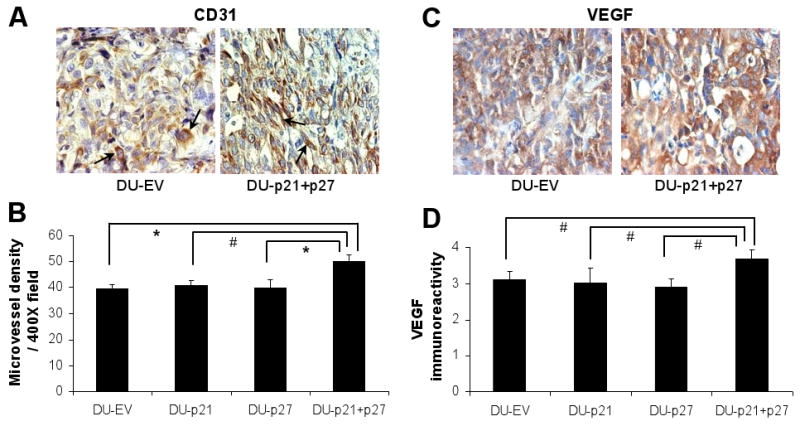
Angiogenic status of DU-EV, DU-p21, DU-p27 and DU-p21+p27 tumors. The tumor samples collected at the end of 6 weeks of the study detailed in Figure 3, were processed for immunohistochemical analysis for (A) CD31 and (C) VEGF staining. Representative images of DU-EV and DU-p21+p27 tumors are shown in each case. Immunostaining was quantified for (B) CD31-positive cells and (D) VEGF immuno-intensity as detailed in Materials and Methods. The arrows in the images for CD31 (panel A) show representative positive staining for CD31. #, P<0.05; *, P<0.001.
Discussion
G1 to S phase transition plays a crucial role in maintaining the genomic integrity since it commits the cells to DNA replication and subsequent mitosis. The G1 checkpoint is often compromised in the event of carcinogenesis and thus tumor cells undergo limitless replication with accumulating genomic damage. p21 and p27 physically interact with CDK and inhibit its kinase activity.16 However, recent studies have revealed diverse aspects of their physiological roles as well as their aberrant functions in various pathogenesis, including cancer. Though termed as Cdk inhibitors, Cip/Kip proteins are shown to be required for the activation of Cdk4-cyclin complexes in early G1 phase.16, 17 More intriguingly, aberrant accumulation of p21 has being observed in specific cancer types including PCa.12, 13 In various cancer cell lines, cells lacking p21 show increased sensitivity to apoptosis induced by cytotoxic agents.18 Moreover, the sub-cellular compartmentalization of p21 and p27 also seems important to determine cell survival or cell growth. Cytoplasmic sequestration of Cip/Kip proteins which prevents them from inhibiting nuclear cyclin-Cdk complex, has been shown to facilitate cell proliferation.19-21 Moreover, cytoplasmic p21 interacts with Ask1 and make the cells more resistant to apoptosis.22 Li et al reported cytoplasmic p27 correlated with worse recurrence-free survival of PCa patients.23 Thus the importance of these molecules in cancer needs to be validated more cautiously. This provided us with the rationale to investigate the impact of the lack of either or both of these proteins on PCa growth characteristics which was investigated in both in vitro and in vivo conditions.
DU145 cells represent an advanced stage of PCa malignancy, since it is not only androgen-independent but also the crucial cell cycle molecules e.g. p53, retinoblastoma (Rb) are mutated.24, 25 In this case, the role of Cip/Kip proteins could be critical in regulating the proliferation of DU145 cells. Also, in many clinical cases of advanced PCa, their lower levels have been reported.7 Therefore, it is likely that modulation in their expression level can lead to a difference in the cellular phenotypic characteristics. To explore this possibility, we knocked-down the expression of p21 and p27, either individually or simultaneously in DU145 cells. Surprisingly, reducing the expression of either of these CDKIs did not show any considerable change in the growth and clonogenicity characteristics of the DU145 cells. These findings indicated that both these CDKIs are closely related in regulating the biological activities including cell proliferation and clonogenicity in DU145 cells. This implies that p21 and p27 proteins play a compensatory role in advanced PCa, at least in DU145 cells. When both of these CDKIs were ablated, an enhanced characteristic for cell growth and clonogenicity were observed, indicating that simultaneous down-modulation of p21 and p27 could be a mechanism leading to aggressive phenotype in PCa.
While exploring the underlying mechanisms for aggressive PCa phenotype, first we observed that it is only the simultaneous knock-down of both p21 and p27 that shorten the first gap phase of the cell cycle (G1) to make the cells readily available for DNA synthesis in S phase. The accelerated transition through G1-S check-point was through the early increase in CDK2 kinase activity that happened only with the simultaneous absence of both p21 and p27. These observations are supported by the fact that both p21 and p27 can physically interact with CDK2 to inhibit its kinase activity. Therefore, the absence of either of them at a time could be taken care by the presence of the other to regulate the CDK2 kinase activity as observed in DU-EV, DU-p21 and DU-p27 cell variants.
Next, we studied the in vivo tumor growth characteristics of DU145 cells having different levels of p21 and/or p27 in stable transfectants in nude mice. Similar to the results from in vitro studies, enhanced tumor growth rate was observed in cell variant knocked-down for both p21 and p27 as compared to DU-EV, DU-p21 and DU-p27 cell variants. The immunohistochemical analysis of tumors for PCNA also showed comparatively higher number of proliferating cells in tumors developed from the DU145 cells with lowered expression of both p21 and p27. Further, we studied the angiogenic status of these tumors and observed increased vasculature of DU-p21+p27 tumors compared to DU-EV tumors. This is an interesting observation that p21 and p27 could modulate angiogenesis, although the increased angiogenesis observed in DU-p21+p27 tumors could also be an indirect consequence of increased proliferation rate in tumors. Therefore, further studies are needed to examine the precise role, specifically direct or indirect effect, of Cip/Kip proteins in regulation of tumor angiogenesis. Nevertheless, our findings support that simultaneous down-regulation of both p21 and p27 could be a potential mechanism to acquire aggressive tumor growth with enhanced tumor vasculature in PCa.
Overall, studies on prostatectomy samples from PCa patients have implicated ethnicity to be an important context for correlation of Cip/Kip levels with disease outcome. p21 has been shown to predict disease free survival in Caucasians, but not in Afro-Americans, and in this regard, DU145 cells are from a Caucasian patient.13 Moreover, DU145 cells are already aggressive cancer cells, and even in these cells, loss of p21 and p27 proteins induced a further aggressive phenotype, especially in vivo. Therefore, the rationalized implication of present study would be to investigate the roles of p21 and p27 in PCa growth and progression, in PCa cell lines from different ethnic origin as well as with weak and aggressive tumorigenic phenotypes.26 The impact of Cip/Kip protein knockdown in these cells will help to clarify the importance of these molecules at different stages of the malignancy in different ethnic population. In recent studies, p21 and p27 have often been implicated to induce growth arrest or apoptosis by various chemopreventive agents.11, 27, 28 Thus, other implication of the present study is that the generation of stable cell lines with knockdown levels of p21 and p27 could be an effective tool to dissect out the importance of these molecules in mediating the pharmacologic antitumor responses of chemopreventive agents in both in vitro and in vivo conditions. Finally, our present study underscored the role of Cip/Kip (p21/p27) as tumor suppressor molecules whose expression levels have a significant impact on tumor phenotype of even advanced PCa cells.
Materials and Methods
Generation of p21, p27, p21 + p27 knockdown stable cell lines
For generating DU-p21 or DU-p27 cells, pSUPER-RETRO (OligoEngine, Seattle, WA) was used as the vector to make puromycin resistant retroviruses. For p21, the targeted sequence is GACCATGTGGACCTGTCAC, which is at position 325 to 343 in the p21 ORF. This plasmid was obtained from R. Bernards (The Netherlands Cancer Institute) by way of Andriy Marusky in Dr. James DeGregori's lab (UCHSC). For p27, the targeted sequence is CCTGTGTACATAACTCTGTAA at position 2127-2147 of the 3′UTR of p27. Retroviruses were made by transient transfection of Phoenix-Ampho packaging cells together with pCL-AMPHO helper plasmid using ExGen 500 transfection reagent (Fermentas; Glen Burnie, MD). Transduction of DU145 cells with retroviruses was performed using 3 rounds of infections at 3 h intervals with 50% diluted virus-containing media supplemented with 6 μg/ml polybrene (Sigma, St Louis, MO) to obtain over 95% infection efficiencies. Stable transfectants were selected using puromycin at 2.5 μg/ml. For producing the double p21+p27 shRNA DU145 cells, a G418 version of the p21 shRNA retrovirus was made by subcloning a 240 bp EcoR1-Sal1 fragment from the pSUPER-RETRO construct into pSUPER –RETRONEO (OligoEngine) and then producing virus as described above. DU145 shRNA p27 cells were then used for infection. Stable transfectants were selected using G418 at 0.4 mg/ml in the presence of puromycin to select for both p21 and p27 shRNA.
Cell counting and clonogenic assay
For growth curves, 1 × 105 cells were plated in 60 mm dishes and were counted in triplicates after 24-96 h. To assess the clonogenic potential of the different cell lines, cells were seeded at a density of 1000 cells / well in 6-well plates. After 10 days, cells were fixed in methanol: acetic acid (3:1) for 10 mins and then stained with 0.1% crystal violet for 30 mins. The plates were washed thrice with PBS and the colonies (>50 cells/ colony) were counted under inverted microscope.
Cell synchronization and cell cycle analysis
Cells (1 × 105) were plated in 60 mm dishes and after 24 h, serum-free media was added, which was replaced after every 12 h to minimize autocrine stimulation. After 36 h of serum-deprivation, cells were released in 10% FBS-containing media and harvested after 0-24 h and analyzed for cell cycle distribution by flow cytometry.
Western immunoblotting, immunoprecipitation and kinase assay
Cell lysates, from cell culture and tumor xenograft studies, were prepared in non-denaturing lysis buffer as reported previously, and subjected to sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) on 12% Tris–glycine gel.29 The separated proteins were transferred on to nitrocellulose membrane followed by blocking with 5% non-fat milk powder (w/v) in Tris-buffered saline (10 mM Tris–HCl, pH 7.5, 100 mM NaCl, 0.1% Tween 20) for 1 h at room temperature. Membranes were probed with antibodies for p21 and p27 (Millipore, Billerica, MA), Cyclin A and Cyclin E (Santa Cruz Biotechnology Inc., Santa Cruz, CA) followed by peroxidase-conjugated appropriate secondary antibody and visualized by ECL detection system (GE Healthcare Bioscience, Piscataway, NJ). To ensure equal protein loading, each membrane was stripped and reprobed with anti-β-actin antibody (Sigma, St Louis, MO) to normalize for differences in protein loading. For Cdk2 kinase assay, 200 μg of protein lysates per sample were immunoprecipitated with anti-CDK2 antibody (Santa Cruz Biotechnology). The beads were washed three times with lysis buffer and once with kinase assay buffer (50 mM Tris-HCl, pH 7.4, 10 mM MgCl2 and 1 mM DTT). Phosphorylation of histone H1 substrate (Boehringer Mannheim Corp., Indianapolis, IN) was measured by incubating the beads with 30 μl of reaction solution (0.2 μl (2.5 μg) of histone H1, 0.5 μl (5 μCi) of γ-32P-ATP, 0.5 μl of 0.1 mM ATP and 28.8 μl of kinase buffer) for 30 min at 37°C. Reaction was stopped by boiling the samples in SDS buffer, and samples were resolved on 12% SDS-PAGE and subjected to autoradiography as reported.30
In vivo tumor xenograft study
Cells were detached from the culture dishes by trypsinization and then collected, washed, and resuspended in serum- and antibiotic-free RPMI 1640 media. To establish DU145 tumor xenografts in mice, 6-week-old athymic nu/nu male mice were injected s.c. with 3.5 × 106 cells mixed with Matrigel (1:1) (Collaborative Biomedical Products, Bedford, MA) in the right flank of each mouse. Once xenografts started growing, their sizes were measured weekly with digital caliper. The tumor volume was calculated by the formula “0.5236 L1(L2)2, where L1 is the long axis and L2 is the short axis of the tumor” as reported previously.31 The average tumor volume of each group was calculated as ‘total tumor volume of all the animals in the group, divided by the number of animals in the group’. At the end of experiment, tumors were excised, weighed, and stored at -80°C until additional analysis.
Immunohistochemistry
Tumor samples (one piece of each) were fixed in 10% buffered formalin for 12 h and processed conventionally. The paraffin-embedded tumor sections (5-μm thick) were heat immobilized, deparaffinized using xylene, and then rehydrated in a graded series of ethanol followed by antigen retrieval and blocking of endogenous peroxidase activity as reported.32 Sections were then incubated with specific primary antibodies including, anti-PCNA (Dako, Carpinteria, CA), anti-Ki67 (Dako), anti-CD31 (Abcam, Cambridge, MA) or anti-VEGF antibody (Santa Cruz Biotechnology), and then incubated with biotinylated secondary antibody, streptavidin and 3,3′-diaminobenzidine (Sigma). For PCNA and Ki-67 quantification, cells were counted in five arbitrarily selected fields at 400× magnification and data is represented as the number of positive (brown) cells × 100/total number of cells. For microvessel density quantification, the number of CD31-positive vessels per 400× field were counted in five randomly selected fields and the data is represented as the number of CD31-positive microvessels per 400× microscopic field. For VEGF, the staining intensity was graded as 0, 1, 2, 3, and 4 representing nil, weak, moderate, strong and very strong immunoreactivity, respectively, and the average of five random fields per sample at 400× magnification was quantified.
Statistical analysis
Statistical significance of difference between DU-EV and all other cell lines was determined by one-way ANOVA followed by Bonferroni t-test using SigmaStat 2.03 software. P<0.05 was considered statistically significant.
Acknowledgments
Grant Support: USPHS grant RO1 CA116636 from the National Cancer Institute, NIH.
Abbreviations
- p21
p21/Cip1
- p27
p27/Kip1
- CDK
cyclin-dependent kinase
- CDKI
cyclin-dependent kinase inhibitor
- PCa
prostate cancer
- PCNA
proliferating cell nuclear antigen
- CD31
cluster of differentiation molecule 31, also called PECAM-1 for platelet endothelial cell adhesion molecule
- VEGF
vascular endothelial growth factor
References
- 1.Jemal A, Siegel R, Ward E, Murray T, Xu J, Thun MJ. Cancer Statistics, 2007. CA Cancer J Clin. 2007;57:43–66. doi: 10.3322/canjclin.57.1.43. [DOI] [PubMed] [Google Scholar]
- 2.Chen CD, Welsbie DS, Tran C, Baek SH, Chen R, Vessella R, Rosenfeld MG, Sawyers CL. Molecular determinants of resistance to antiandrogen therapy. Nat Med. 2004;10:33–9. doi: 10.1038/nm972. [DOI] [PubMed] [Google Scholar]
- 3.Obaya AJ, Sedivy JM. Regulation of cyclin-Cdk activity in mammalian cells. Cell Mol Life Sci. 2002;59:126–42. doi: 10.1007/s00018-002-8410-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.MacLachlan TK, Sang N, Giordano A. Cyclins, cyclin-dependent kinases and cdk inhibitors: implications in cell cycle control and cancer. Crit Rev Eukaryot Gene Expr. 1995;5:127–56. doi: 10.1615/critreveukargeneexpr.v5.i2.20. [DOI] [PubMed] [Google Scholar]
- 5.Fero ML, Randel E, Gurley KE, Roberts JM, Kemp CJ. The murine gene p27Kip1 is haplo-insufficient for tumour suppression. Nature. 1998;396:177–80. doi: 10.1038/24179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shiohara M, el-Deiry WS, Wada M, Nakamaki T, Takeuchi S, Yang R, Chen DL, Vogelstein B, Koeffler HP. Absence of WAF1 mutations in a variety of human malignancies. Blood. 1994;84:3781–4. [PubMed] [Google Scholar]
- 7.Cheng L, Lloyd RV, Weaver AL, Pisansky TM, Cheville JC, Ramnani DM, Leibovich BC, Blute ML, Zincke H, Bostwick DG. The Cell Cycle Inhibitors p21WAF1 and p27KIP1 Are Associated with Survival in Patients Treated by Salvage Prostatectomy after Radiation Therapy. Clin Cancer Res. 2000;6:1896–9. [PubMed] [Google Scholar]
- 8.Doganavsargil B, Simsir A, Boyacioglu H, Cal C, Hekimgil M. A comparison of p21 and p27 immunoexpression in benign glands, prostatic intraepithelial neoplasia and prostate adenocarcinoma. BJU Int. 2006;97:644–8. doi: 10.1111/j.1464-410X.2006.06054.x. [DOI] [PubMed] [Google Scholar]
- 9.Gotoh A, Shirakawa T, Wada Y, Fujisawa M, Okada H, Kamidono S, Hamada K. The growth inhibitory effect of p21 adenovirus on androgen-dependent and -independent human prostate cancer cells. BJU Int. 2003;92:314–8. doi: 10.1046/j.1464-410x.2003.04318.x. [DOI] [PubMed] [Google Scholar]
- 10.De Siervi A, Marinissen M, Diggs J, Wang XF, Pages G, Senderowicz A. Transcriptional activation of p21(waf1/cip1) by alkylphospholipids: role of the mitogen-activated protein kinase pathway in the transactivation of the human p21(waf1/cip1) promoter by Sp1. Cancer Res. 2004;64:743–50. doi: 10.1158/0008-5472.can-03-2505. [DOI] [PubMed] [Google Scholar]
- 11.Roy S, Kaur M, Agarwal C, Tecklenburg M, Sclafani RA, Agarwal R. p21 and p27 induction by silibinin is essential for its cell cycle arrest effect in prostate carcinoma cells. Mol Cancer Ther. 2007;6:2696–707. doi: 10.1158/1535-7163.MCT-07-0104. [DOI] [PubMed] [Google Scholar]
- 12.Rigaud J, Tiguert R, Decobert M, Hovington H, Latulippe E, Laverdiere J, Larue H, Lacombe L, Fradet Y. Expression of p21 cell cycle protein is an independent predictor of response to salvage radiotherapy after radical prostatectomy. Prostate. 2004;58:269–76. doi: 10.1002/pros.10329. [DOI] [PubMed] [Google Scholar]
- 13.Sarkar FH, Li Y, Sakr WA, Grignon DJ, Madan SS, Wood DP, Jr, Adsay V. Relationship of p21(WAF1) expression with disease-free survival and biochemical recurrence in prostate adenocarcinomas (PCa) Prostate. 1999;40:256–60. doi: 10.1002/(sici)1097-0045(19990901)40:4<256::aid-pros7>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 14.Rose DS, Maddox PH, Brown DC. Which proliferation markers for routine immunohistology? A comparison of five antibodies. J Clin Pathol. 1994;47:1010–4. doi: 10.1136/jcp.47.11.1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Folkman J. What Is the Evidence That Tumors Are Angiogenesis Dependent? J Natl Cancer Inst. 1990;82:4–7. doi: 10.1093/jnci/82.1.4. [DOI] [PubMed] [Google Scholar]
- 16.Lee MH, Yang HY. Negative regulators of cyclin-dependent kinases and their roles in cancers. Cell Mol Life Sci. 2001;58:1907–22. doi: 10.1007/PL00000826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev. 1999;13:1501–12. doi: 10.1101/gad.13.12.1501. [DOI] [PubMed] [Google Scholar]
- 18.Tian H, Wittmack EK, Jorgensen TJ. p21WAF1/CIP1 Antisense Therapy Radiosensitizes Human Colon Cancer by Converting Growth Arrest to Apoptosis. Cancer Res. 2000;60:679–84. [PubMed] [Google Scholar]
- 19.Li Y, Dowbenko D, Lasky LA. AKT/PKB Phosphorylation of p21Cip/WAF1 Enhances Protein Stability of p21Cip/WAF1 and Promotes Cell Survival. J Biol Chem. 2002;277:11352–61. doi: 10.1074/jbc.M109062200. [DOI] [PubMed] [Google Scholar]
- 20.Denicourt C, Saenz CC, Datnow B, Cui XS, Dowdy SF. Relocalized p27Kip1 Tumor Suppressor Functions as a Cytoplasmic Metastatic Oncogene in Melanoma. Cancer Res. 2007;67:9238–43. doi: 10.1158/0008-5472.CAN-07-1375. [DOI] [PubMed] [Google Scholar]
- 21.Liang J, Zubovitz J, Petrocelli T, Kotchetkov R, Connor MK, Han K, Lee JH, Ciarallo S, Catzavelos C, Beniston R, Franssen E, Slingerland JM. PKB/Akt phosphorylates p27, impairs nuclear import of p27 and opposes p27-mediated G1 arrest. Nat Med. 2002;8:1153–60. doi: 10.1038/nm761. [DOI] [PubMed] [Google Scholar]
- 22.Zhan J, Easton JB, Huang S, Mishra A, Xiao L, Lacy ER, Kriwacki RW, Houghton PJ. Negative regulation of ASK1 by p21Cip1 involves a small domain that includes Serine 98 that is phosphorylated by ASK1 in vivo. Mol Cell Biol. 2007;27:3530–41. doi: 10.1128/MCB.00086-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li R, Wheeler TM, Dai H, Sayeeduddin M, Scardino PT, Frolov A, Ayala GE. Biological correlates of p27 compartmental expression in prostate cancer. J Urol. 2006;175:528–32. doi: 10.1016/S0022-5347(05)00151-5. [DOI] [PubMed] [Google Scholar]
- 24.Carroll AG, Voeller HJ, Sugars L, Gelmann EP. p53 oncogene mutations in three human prostate cancer cell lines. Prostate. 1993;23:123–34. doi: 10.1002/pros.2990230206. [DOI] [PubMed] [Google Scholar]
- 25.Bookstein R, Shew JY, Chen PL, Scully P, Lee WH. Suppression of tumorigenicity of human prostate carcinoma cells by replacing a mutated RB gene. Science. 1990;247:712–5. doi: 10.1126/science.2300823. [DOI] [PubMed] [Google Scholar]
- 26.Gleave M, Hsieh JT, Gao C, von Eschenbach AC, Chung LWK. Acceleration of Human Prostate Cancer Growth in Vivo by Factors Produced by Prostate and Bone Fibroblasts. Cancer Res. 1991;51:3753–61. [PubMed] [Google Scholar]
- 27.Don MJ, Chang YH, Chen KK, Ho LK, Chau YP. Induction of CDK inhibitors (p21(WAF1) and p27(Kip1)) and Bak in the beta-lapachone-induced apoptosis of human prostate cancer cells. Mol Pharmacol. 2001;59:784–94. doi: 10.1124/mol.59.4.784. [DOI] [PubMed] [Google Scholar]
- 28.Yang ES, Burnstein KL. Vitamin D Inhibits G1 to S Progression in LNCaP Prostate Cancer Cells through p27Kip1 Stabilization and Cdk2 Mislocalization to the Cytoplasm. J Biol Chem. 2003;278:46862–8. doi: 10.1074/jbc.M306340200. [DOI] [PubMed] [Google Scholar]
- 29.Deep G, Singh RP, Agarwal C, Kroll DJ, Agarwal R. Silymarin and silibinin cause G1 and G2-M cell cycle arrest via distinct circuitries in human prostate cancer PC3 cells: a comparison of flavanone silibinin with flavanolignan mixture silymarin. Oncogene. 2006;25:1053–69. doi: 10.1038/sj.onc.1209146. [DOI] [PubMed] [Google Scholar]
- 30.Agarwal C, Singh RP, Dhanalakshmi S, Tyagi AK, Tecklenburg M, Sclafani RA, Agarwal R. Silibinin upregulates the expression of cyclin-dependent kinase inhibitors and causes cell cycle arrest and apoptosis in human colon carcinoma HT-29 cells. Oncogene. 2003;22:8271–82. doi: 10.1038/sj.onc.1207158. [DOI] [PubMed] [Google Scholar]
- 31.Singh RP, Dhanalakshmi S, Tyagi AK, Chan DC, Agarwal C, Agarwal R. Dietary feeding of silibinin inhibits advance human prostate carcinoma growth in athymic nude mice and increases plasma insulin-like growth factor-binding protein-3 levels. Cancer Res. 2002;62:3063–9. [PubMed] [Google Scholar]
- 32.Singh RP, Deep G, Blouin MJ, Pollak MN, Agarwal R. Silibinin suppresses in vivo growth of human prostate carcinoma PC-3 tumor xenograft. Carcinogenesis. 2007;28:2567–74. doi: 10.1093/carcin/bgm218. [DOI] [PubMed] [Google Scholar]