

Abstract
Chronic obstructive pulmonary disease (COPD) is associated with high incidence of morbidity and mortality. Oxidative stress is intimately associated with the progression and exacerbation of COPD and therefore targeting oxidative stress with antioxidants or boosting the endogenous levels of antioxidants is likely to have beneficial outcome in the treatment of COPD. Among the various antioxidants tried so far, thiol antioxidants and mucolytic agents, such as glutathione, N-acetyl-L-cysteine, N-acystelyn, erdosteine, fudosteine, and carbocysteine; Nrf2 activators, and dietary polyphenols (curcumin, resveratrol, green tea, and catechins/quercetin) have been reported to increase intracellular thiol status alongwith induction of GSH biosynthesis. Such an elevation in the thiol status in turn leads to detoxification of free radicals and oxidants as well as inhibition of ongoing inflammatory responses. In addition, specific spin traps, such as a-phenyl-N-tert-butyl nitrone, a catalytic antioxidant (ECSOD mimetic), porphyrins (AEOL 10150 and AEOL 10113), and a SOD mimetic M40419 have also been reported to inhibit cigarette smoke-induced inflammatory responses in vivo in the lung. Since a variety of oxidants, free radicals and aldehydes are implicated in the pathogenesis of COPD; it is possible that therapeutic administration of multiple antioxidants and mucolytics will be effective in management of COPD. However, a successful outcome will critically depend upon the choice of antioxidant therapy for a particular clinical phenotype of COPD, whose pathophysiology should be first properly understood. This article will review the various approaches adopted to enhance lung antioxidant levels, antioxidant therapeutic advances and recent past clinical trials of antioxidant compounds in COPD.
Keywords: COPD, Smokers, Oxidants, Thiol, Glutathione, Antioxidants, Lungs
Oxidative stress in COPD
COPD and inhaled oxidants
The lung is exposed to a high-oxygen environment and exogenous pollutants/toxicants that come along with the inhaled breath. Considering its large surface area and associated blood supply, the lung is therefore highly susceptible to injury mediated by oxidative stress. Oxidative stress is a consequence of an inability of resident antioxidant mechanisms to neutralize prooxidant factors generated endo- or exogenously. Reactive oxygen and nitrogen species (ROS and RNS) are the most prominent manifestations of oxidative stress and are associated with remodeling of extracellular matrix and blood vessels, elevated mucus secretion, inactivation of antiproteases, apoptosis, autophagy and regulate cell proliferation. Increased levels of ROS have been further implicated in initiating inflammatory responses in the lungs through the activation of transcription factors [nuclear factor-kappaB (NF-κB) and activator protein-1 (AP-1)], signal transduction, chromatin remodeling and gene expression of pro-inflammatory mediators [Janssen-Heininger et al. 2002; Rahman and MacNee 1998; Rahman et al. 2004] (Figure 1). Since a variety of oxidants, free radicals and aldehydes are implicated in the pathogenesis of chronic obstructive pulmonary disease (COPD) it is possible that therapeutic administration of appropriate antioxidants will be effective in the treatment of COPD [Rahman 2002; Rahman et al. 2006]. Given the existence of various clinical phenotypes of COPD [Patel et al. 2006; Makita et al. 2007], it becomes important to understand, which particular type of oxidant(s) is associated with the given COPD phenotype and during exacerbations of COPD and accordingly select an antioxidant variety and regimen.
Figure 1.

Inflammatory response is mediated by inhaled and/or cellular oxidants. These oxidants activate alveolar macrophages, neutrophils, eosinophils, and epithelial cells, leading to ROS/RNS generation, redox imbalance, and activation of redox-sensitive transcription factors. They may act as signaling mediators and further involve in the inflammatory responses in patients with COPD. Antioxidants inhibit the oxidative stress by induction of GSH biosynthesis and change of redox status. It is possible that therapeutic administration of antioxidants will be effective in the treatment of COPD. ROS: reactive oxygen species; RNS: reactive nitrogen species
Most prominent source among the environmentally derived ROS involved in driving the pathogenesis of COPD is cigarette smoke (CS) [Snider 1989; Rahman and Adcock, 2006]. CS is an admixture of inorganic and organic oxidants, highly reactive carbonyls and metal ions, such as iron which can through an amplification loop increase ROS formation through Fenton and Haber-Weiss chemistry. COPD is a slow progressive condition characterized by airflow limitation, which is largely irreversible [ATS 1995; Pauwels et al. 2001]. About 90% of patients with COPD are smokers, yet only 15–20% of these show a rapid decline in forced expiratory volume in one second (FEV1) and develop the disease [Snider 1989]. Since CS contains more than 1015 oxidant/free radical molecules per puff and over 4,700 chemicals [Church and Pryor 1985], this adds to the oxidant burden in smokers. Of much recent importance is the report that CS irreversibly modifies glutathione (GSH) levels in airway epithelial cells; a basic mechanism which may underpin CS mediated inflammatory lung damage [Reddy et al. 2002; Van der Toorn et al. 2007].
Cell-derived ROS
Cellular-derived ROS is produced by all cells as a result of metabolism within the mitochondria or by inflammatory cells (macrophages and neutrophils) as part of an inflammatory-immune response towards a pathogen or irritant. The principle ROS-generating enzyme in inflammatory cells is NADPH (nicotinamide adenine dinucleotide phosphate) oxidase, a complex enzyme system that is present in phagocytes and epithelial cells. However, other enzyme systems are also used, such as the xanthine/xanthine oxidase system and the heme peroxidases, levels of which are elevated in COPD patients [Rahman et al. 1996; Pinamonti et al. 1998; Aaron et al. 2001; Gompertz et al. 2001]. Similarly, RNS in the form of nitric oxide production is genereated by nitric oxide sythase. Nitric oxide when in the presence of superoxide anion will form the more powerful and damaging peroxynitrite radical [Janssen-Heininger et al. 2002]. Although, oxidants generated in a cell may be from several sources, NADPH oxidases and mitochondria however, are the major sources. It is being increasingly recognized now that oxidants from different sources may have different physiological consequences as is evident from the observation that while, oxidants derived from NADPH oxidase are involved mainly in signaling processes, those produced by mitochondria induce cell death and preceding cell damage [Bindoli et al. 2008]. In this light it therefore becomes imminent that before chosing an antioxidant therapy, due importance be given in identifying the source of oxidants in a given COPD phenotype and during exacerbations of COPD.
Undoubtedly, ROS generation is now known to be directly associated with oxidative modification of proteins, lipids, carbohydrates and DNA. Markers of this ROS-mediated damage, such as carbonyl-modified or tyrosine-nitrosylated proteins are elevated in many chronic respiratory diseases [Lenz et al. 1999; Balint et al. 2001; Rahman et al. 2002a; Barreiro et al. 2005; Adam 2007]. Such alterations can impact a variety of both, intra- and extracellular physiological processes [Richter et al. 1995; Gutteridge et al. 2000; Kirkham et al. 2003; Kirkham et al. 2004; Marwick et al. 2004] and may be instrumental in cell and tissue damage associated with these chronic inflammatory lung diseases [Hatch 1995; Rahman et al. 1996; Rahman and MacNee 1999; Rahman et al. 2000; Dworski 2000]. ROS/RNS can initiate inflammatory responses in the lungs through the activation of redox sensitive signaling mechanisms and transcription factors [Guyton et al. 1996; Rahman and MacNee 1998], targets being signaling molecules such as G-proteins ras/rac and the heterotrimeric Gi complex, the mitogen-activated protein (MAP) kinases JNK, p38 and Erk, protein phosphatases [Lander et al. 1997; Nishida et al. 2000] and numerous transcription factors, for example; NF-κB, AP-1, Sp-1, c-Myb, p53, c-myc, EGR-1, MIF-1, GR and CREB [Poli et al. 2004]. Similarly, ROS itself can act as a physiological second messenger for activated receptors against tumour necrosis factor α (TNFα) and lipopolysaccharide (LPS) [Rochelle et al. 1998]. Consequently, oxidative stress and the subsequent changes in intracellular redox status could increase the sensitivity and heighten the response to these proinflammatory stimuli [Keatings et al. 1996].
Oxidative stress biomarkers in exhaled breath condensate
The outcome of any clinical trial/supplementation studies could potentially be monitored by assessing the levels of a representative marker(s) of a said disease. Such a marker should have a typically non-invasive accessibility and easily measured. Recent investigations regarding biomarkers of COPD have focused on applying non-invasive techniques including exhaled breath condensate (EBC) to evaluate oxidative stress markers in COPD subjects [Rahman and Kelly 2003; Rahman and Biswas 2004]. Smokers and patients with COPD have higher levels of exhaled H2O2 than non-smokers, and levels are even higher during exacerbations of COPD [Kharitonov and Barnes 2001; Montuschi and Barnes 2002]. The levels of 8-iso-prostglandin F2α (8-isoprostane) in EBC are elevated in healthy smokers and more markedly in patients with COPD reflecting the degree of oxidative stress [Rahman and MacNee 1998; Montuschi et al. 2000]. Increased levels of pro-inflammatory cytokines and decreased GSH levels were observed in patient with COPD than in healthy non-smokers [Drost et al. 2005]. Lipid peroxidation products, such as thiobarbituric acid reactive substances (TBARS) have also been shown to be elevated in breath condensate and in lungs of patients with stable COPD [Pratico et al.1998; Nowak et al. 1999]. Other specific products of lipid peroxidation such as malondialdehyde (MDA) and 4-hydroxy-2-nonenal (4-HNE) have also been shown to be increased in EBC and lungs of patients with COPD [Rahman et al. 2002a; Aoshiba et al. 2003; Rahman et al. 2000]. As evident from the above, a host of biomarkers are available for monitoring the status of COPD. It would be however, prudent to add a note of caution, that many markers of COPD vary with the stage of the disease and in smokers [Rahman and Biswas 2004] and therefore, only the most consistent marker should be considered and assessed for long-term longtitudinal studies.
Therapeutic intervention with antioxidants
Oxidative stress is implicitly involved with the pathogenesis of chronic airway diseases. A rational approach for the treatment of COPD would therefore be to consider antioxidant intervention not only, to neutralize the increased oxidative stress and the subsequent inflammatory response, but also to identify the source of oxidants and overwhelm their generation. This can be achieved through two approaches, either by increasing the endogenous antioxidant enzyme defenses or by enhancing the non-enzymatic defenses through dietary or pharmacological means. Several small molecular weight compounds that target oxidant signaling, or quench cigarette smoke derived oxidants and reactive aldehydes are currently being tested clinically [Rahman and MacNee 2000a]. Antioxidant agents, such as thiol donors and analogues (GSH and mucolytic drugs, such as N-acetyl-L-cysteine, nacystelyn, erdosteine and carbocysteine lysine salt), dietary polyphenols and flavonoids (curcumin, resveratrol, green tea, quercetin), all have been reported to scavenge/detoxify free radicals/oxidants, increase intracellular thiol levels, and control NF-κB activation and consequently inhibit inflammatory gene expression. A quick glance through the available literature testifies that various approaches have been undertaken to study the benefits of antioxidants in obstructive lung disease (Table 1, Table 2). These range from dietary antioxidants and thiols through to superoxide dismutase (SOD) mimetics. In this review, the various antioxidant strategies discussed have been divided into various chemical and functional categories such as: thiols, spin traps, redox sensor inhibitors, enzyme mimetics, polyphenols, modulators of redox sensitive transcription factors, and antioxidants-mediated reversing of steroid resistance.
Table 1.
Antioxidant therapeutic interventions in COPD
| Antioxidant compounds |
|---|
| Thiol compounds: N-acetyl-L-Cysteine, N-acystelyn, N-isobutyrylcysteine, glutathione |
| esters, thioredoxin, erdosteine, fudosteine, carbocysteine |
| Inducers of glutathione biosynthesis, Nrf2 activators |
| Antioxidant vitamins (vitamin A, E, C), β-carotene, CoQ10 |
| Polyphenols (curcumin, resveratrol, quercetin and green tea-catechins) |
| Nitrone spin traps |
| Superoxide dismutase and glutathione peroxidase mimetics |
| Ebselen |
| Porphyrins |
Table 2.
A list of antioxidant compounds currently in clinical trials for COPD treatment
| Antioxidant | Structure | Indications | Company | Phase (condition) |
|---|---|---|---|---|
| N-acetyl-L-cysteine |  |
Bronchiesctasis, COPD, cystic fibrosis, pulmonary fibrosis | Zambon | Launched (bronchial hypersecretion) Phase II (idiopathic pulmonary fibrosis) |
| Nacystelyn | 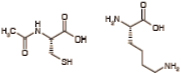 |
COPD, cystic fibrosis | Galephar, Cystic Fibrosis Foundation, SMB Laboratories | Phase II (COPD, Cystic fibrosis) |
| Erdosteine |  |
Bronchitis, cough, cystic fibrosis | Negma-Lerads,Galen, Adams Respiratory Therapeutics | Launched (bronchial hypersecretion and acute exacerbation of chronic bronchitis) Phase II (chronic bronchitis associated with COPD) |
| Fudosteine |  |
Bronchitis, cough | Mitsubishi Tanabe Pharma Corporation | Phase II (chronic bronchitis associated with COPD) |
| Carbocysteine |  |
Bronchitis, cough | Beacon Pharmaceuticals | Phase II (chronic bronchitis associated with COPD) |
| Ebselen |  |
Asthma, atherosclerosis, myocardinal ischemia | Daiich Pharmaceuticals, Sound Pharmaceuticals | Phase II (hearing loss) |
| Recombinant thioredoxin | Lung injury, respiratory distress syndrome, COPD | Redox Bio Science | Phase I/II (respiratory syndrome) | |
| BXT-51072 (ALT-2074) |  |
Asthma, COPD, respiratory distress syndrome, septic shock, coronary artery restenosis | Oxix International Synvista Therapeutics | Phase II (coronary artery disease and percutaneous coronary intervention) |
| Curcumin | COPD, cancer | Sami Labs, M.D. Anderson Cancer Center, National Institute on Aging, University of Pennsylvania | Phase II (Alzheimer’s dementia) | |
| Resveratrol |  |
COPD, Infection | Royalmount Pharma INC | Phase II (chronic bronchitis associated with COPD) |
COPD, chronic obstructive pulmonary dusease
Systemic antioxidant capacity and antioxidant vitamins
The decrease in antioxidant capacity in smokers and in patients with COPD as reflected by, decreased plasma antioxidant capacity and protein sulphydryls [Rahman et al. 1996; Rahman et al. 1997; Corradi et al. 2003], occurs transiently during smoking and during exacerbations of COPD and resolves rapidly after smoking cessation. The depletion of antioxidant capacity could in part be explained by the increased release of ROS from peripheral blood neutrophils, as shown by a significant negative correlation between neutrophil superoxide anion release and plasma antioxidant capacity [Rahman et al. 1996] implying that oxidants in cigarette smoke markedly decrease plasma antioxidants and play an important role in the pathogenesis of COPD. Furthermore, it is possible that inter-individual differences in antioxidant capacity may contribute to the differences in susceptibility to cigarette-smoke induced COPD. Thus, it is imperative to propose the rationale for antioxidant therapy ameliorating the increased oxidative stress and consequently the inflammatory response in COPD.
Depletion of total antioxidant capacity in smokers is associated with decreased levels of major plasma antioxidants in smokers [Petruzzelli et al. 1990; Duthie et al. 1991; Antwerpen et al. 1993; Bridges et al. 1993; Mezzetti et al. 1995; Rahman and MacNee 1996]. These studies show depletion of ascorbic acid, vitamin E, β-carotene and selenium in the serum of chronic smokers and in patients with COPD [Duthie et al. 1991; Antwerpen et al. 1993; Bridges et al. 1993; Mezzetti et al. 1995; Romieu and Trenga 2001; Couillard et al. 2002; Tug et al. 2004]. Moreover, decreased vitamin E and vitamin C levels were reported in leukocytes and BAL fluids from smokers [Barton and Roath 1976; Bridges et al. 1990; Theron et al. 1990;]. Vitamin C appears to be a particularly important antioxidant in the plasma [Pacht et al. 1988; Lykkesfeldt and Moos 2005]. Reduced levels of vitamin E and a marginal increase in vitamin C in the BAL fluid of smokers, compared to nonsmokers have also been reported [reviewed in Rahman and MacNee 1996]. Similarly, alveolar macrophages from smokers have both increased levels of ascorbic acid and augmented uptake of ascorbate, suggesting that these cells have adaptive response to redress their antioxidant balance [Rahman and MacNee 1996].
Dietary antioxidants supplementation is one of the simplest approaches to boost antioxidant defense systems. Supplementation of vitamin C, vitamin E and β-carotene has been attempted in cigarette smokers and patients with COPD [Cross et al. 1994; Habib et al. 1999; Rautalahti et al. 1997; Allard et al. 1994; Steinberg and Chait 1998; Aghdassi et al. 1999; Lykkesfeldt et al. 2000]. To this extent a positive association between dietary intake of antioxidant vitamins and lung function has been observed in the study population. However, other epidemiological studies have demonstrated negative associations of dietary antioxidant intake with pulmonary function and with obstructive airway disease [Grievink et al. 1998]. Britton and co-workers [Britton et al. 1995] showed a positive association between dietary intake of the antioxidant vitamin E and lung function in a population of 2,633 subjects, supporting the hypothesis that this antioxidant may have a role in protecting against the development of COPD. Another study has suggested that differences in the dietary intake levels of antioxidant could be a possible explanation for differences in COPD mortality in different populations [Sargeant et al. 2000]. Dietary polyunsaturated fatty acids may also protect cigarette smokers against the development of COPD [Shahar et al. 1999].
GSH and its biosynthesis
The major cellular thiol antioxidant and redox recycler, GSH is more concentrated in epithelial lining fluid compared with plasma [Cantin et al. 1987; Pacht et al. 1988] and has an important protective role in the airspaces and intracellularly in epithelial cells. Several studies have suggested that GSH homeostasis may play a central role in the maintenance of the integrity of the lung airspace epithelial barrier, an observation supported by the report that decreased GSH in epithelial cells leads to loss of barrier function and increased permeability [Li et al. 1996; Morrison et al. 1999]. Human studies have shown elevated levels of GSH in epithelial lining fluid (ELF) in chronic cigarette smokers compared with nonsmokers [Cantin et al. 1987; Morrison et al. 1999]. However, this increase is not commenced immediately after acute cigarette smoking [Morrison et al. 1999]. The two-fold increase in ELF GSH in chronic smokers may not be sufficient to deal with the excessive oxidant burden during smoking, when acute depletion of GSH may occur [Harju et al. 2002]. Harju and colleagues have found that the immunoreactivity of glutamate cystine ligase, GCL [the rate limiting enzyme in GSH synthesis-formerly known as γ-GCS], was decreased in the airways of smokers compared to non-smokers, suggesting that cigarette smoke predisposes lung cells to ongoing oxidant stress [Harju et al. 2002]. Neurohr and colleagues recently showed that decreased GSH levels in BALF cells of chronic smokers were associated with a decreased expression of γ-GCS-light subunit without a change in γ-GCS-heavy subunit expression [Neurohr et al. 2003]. Increasing the activity of γ-GCS, would be expected to increase cellular GSH levels. The induction of γ-GCS by molecular means to increase cellular GSH levels or γ-GCS gene therapy also holds great promise in protection against chronic inflammation and oxidant-mediated injury in COPD. In this light it is pertinent to mention that the primary role of GSH is the donation of reducing equivalents within the cell, resulting in its oxidation to the disulphide form, which is recycled back to GSH by glutathione reductase. In turn, GSH is an important component in a pathway for recycling of vitamin C (vit Coxidized to vit Creduced), which in turn is used to recycle vitamin E (vit Eoxidized to vit Ereduced) [Wilson 2002]. Since, vit C, vit E and different GSH analogues and thiol donors have been tested for COPD therapy, it becomes more logical to accurately pinpoint the particular antioxidant that is deficient in a given patient. For example, administration of vit C or Vit E in a patient having GSH or GCL deficiency may not work since the redox coupling between GSH and vit C and therefore between vit C and vit E will remain incomplete in absence or deficiency of GSH.
A direct increase cellular level of GSH in lung appears to be a logical approach to enhance the antioxidant potential in the treatment of COPD. In fact, extracellular augmentation of GSH has been tried through intravenous administration of GSH, oral ingestion of GSH, and aerosol inhalation of nebulized GSH in an attempt to reduce inflammation in various lung diseases [Rahman and MacNee 1999; Rahman and MacNee 2000b]. However, all these route of administration lead to undesirable effects (bronchoconstriction) suggesting that direct GSH therapy may not be an appropriate way of increasing GSH levels in lung ELF and cells in COPD subjects. The bioavailability of GSH, pH and osmalility in the inflammatory micro-environment and the resultant formation of toxic products (thiyl radical, GSSG and GSH-adducts) are further challenges for direct GSH administration. Alternative formulations may address bioavailability, such as liposomal delivery, but at present it seems that direct administration of GSH will not be successful in treating COPD.
Therapeutic Antioxidant Agents
In the following sections we will consider the various antioxidants, which are either used or have the potential to be used as therapeutic agents in the control and amelioration of COPD.
Thiols
N-acetyl-L-cysteine
The free thiol group of N-acetyl-L-cysteine (NAC) is capable of interacting with the electrophilic group of free radicals thereby forming NAC thiol, with NAC disulphide or NAC conjugates as a major end product [Cotgreave 1997]. The availability of cysteine for GSH is a fundamental factor in the regulation of GSH production. NAC, a cysteine-donating reducing compound, acts as a cellular precursor of GSH and becomes deacetylated in the gut to cysteine following oral administration. NAC when orally administered is metabolized into another compound since accompanying increase in non-protein and protein sulphydryl groups were found in plasma [Cotgreave et al. 1987]. The oral availability of NAC varied between 6 and 10% in man [Borgstrom et al. 1984]. It is important to note that increasing the dose also increased NAC bioavailability and time for maximal plasma concentration [Borgstrom and Kagedal 1990], still higher concentration in plasma may be achieved after intravenous administration [Crouch and Rusho 2005]. Ongoing clinical trials with NAC in these patients suggest that this thiol compound is well tolerated and may increase pulmonary levels of GSH.
NAC may also reduce cystine to cysteine, which is an important mechanism for intracellular GSH elevation in vivo in lungs [Burgunder et al. 1989]. Other then reducing disulphide bonds, NAC also interacts directly with oxidants. NAC is also used as a mucolytic agent, to reduce mucus viscosity and to improve mucociliary clearance. A number of systematic reviews have evaluated the effects of treatment with mucolytic agents (NAC, 400–1200 mg/day) in patients with COPD [Grandjean et al. 2000; Dekhuijzen 2004]. Mucolytic treatment was associated with a significant reduction of 0.79 exacerbations per patient per year compared with placebo, a 29% decrease (Table 3) [Grandjean et al. 2000; Decramer et al. 2001; Poole and Black 2003; Dekhuijzen 2004; Decramer et al. 2005]. Although how mucolytic agents work is not clear, the mechanism may include alteration of mucus production via breakdown of sulphydryl group, or through antibacterial and/or immunostimulatory effects [Poole and Black 2001 and 2003]. However, small-scale trials and few meta-analysis have either failed to demonstrate any clear clinical benefits or have demonstrated a small but significant clinical benefit (23% decreases in the number of acute exacerbations compared with placebo) in COPD [Grandjean et al. 2000; Dekhuijzen 2004]. Pharmacological administration of NAC has been used in an attempt to enhance lung GSH in patients with COPD with varying success [Rasmusse and Glennow 1988; Bridgemen et al. 1991 and 1994; Dekhuijzen and Van Beurden 2006]. Zuin and colleague reported that treatment with NAC (1200 mg/day, 10 days) improved biological markers and clinical outcomes in patients with COPD exacerbations [Zuin et al. 2005]. In a double blind, placebo controlled, phase II randomized trial Van Schooten et al. [Van Schooten et al. 2002] reported that a 6-month oral dose of 600 mg b.i.d. reduced various plasma and BAL fluid oxidative biomarkers in smokers. It has been shown that treatment with NAC 600 mg once daily for 12 months also reduce the concentration of H2O2 in exhaled breath condensate compared with placebo in stable COPD patients [Kasielski and Nowak 2001]. Another clinical trial also proved that oral administration of NAC 600 mg b.i.d. for 2 months rapidly reduces the oxidant burden in airways of stable COPD patients [De Benedetto et al. 2005]. This reduction in oxidative biomarkers results in clinical benefit, such as reduction in bronchial hypersecretion [Aylward et al. 1980], in addition to a decline in FEV1 and in exacerbations [Stey et al. 2000]. Orally dosed NAC has been shown to increase phagocytic activity of BAL macrophages from healthy smokers [Linden et al. 1993], but similar results were not seen in COPD patients, possibly due to active concentrations of NAC not reaching the lung [Vecchiarelli et al. 1994]. Furthermore, it has also been reported that orally dosed NAC increased the quadricep endurance time of severe COPD patients [Koechlin et al. 2004], thus suggesting that NAC administration may have beneficial effects on the systemic oxidative stress associated with COPD. The reason for this effect is not known but presumably NAC acts as a sink for ROS, which are increased in these patients [Rahman 2005]. However, a multicenter study using NAC delivered by metered dose inhalers in patients with chronic cough failed to show a positive effect on wellbeing, sensation of dyspnoea, cough or lung function [Dueholm et al. 1992]. One small trial has shown that there was a significant effect on neutrophil chemoattractant properties after long time (600 mg/day, 10 months) administration of NAC at COPD patients’ sputum [Van Overveld et al. 2000]. A more recent report has suggested that NAC attenuates lung ischemia-reperfusion injury after lung transplantation [Inci et al. 2007]. In addition, on the mechanistic front, NAC has been shown to attenuate allergic lung disease by regulating the activation of NF-κB and hypoxia-inducible factor-1α (HIF-1α) [Lee et al. 2007].
Table 3.
Clinical trials conducted for the efficacy of antioxidants in smokers and COPD
| Trial Name | Antioxidant used | Aim of Study | Disease/ Condition | Outcome | Ref |
|---|---|---|---|---|---|
| BRONCUS | NAC | Effect of NAC on FEV1 | COPD | No effect. A reduction in lung over inflation in patients with severe COPD without inhaled glucocorticoids No change on decline in FEV1. Decreas of exacerbation if NAC-ICS | (Decramer et al. 2001 and 2005) |
| Systematic Cochrane review of 23 randomized, controlled trails | NAC (2 months of oral therapy) | Effect of NAC and antibiotics on number of days of disability | COPD | No difference in lung function. Significant reduction in days of disability (0.65 day per patient per month) and 29% reduction in exacerbations | (Poole and Black 2001 and 2003) |
| Systematic Cochrane review of randomized, controlled trials; 11 of 39 retrieved trials | NAC | Use of validated score to evaluate the quality of each study | COPD | 9 trials showed prevention of exacerbation and 5 of which addressed improvement of symptoms compared with 34.6% of patients receiving placebo | (Stey et al. 2000) |
| Meta-analysis of published trials | NAC | Assess possible prophylactic benefit of prolonged treatment | COPD | 23% decrease in number of acute exacerbations | (Grandjean et al. 2000) |
| - | NAC (600 mg/d, 5 days and 600 mg, 3/d. 5days) | Effect of NAC on GSH and cycteine | COPD | Increase of GSH at day 5 and cycteine (plasma) at day 5 | (Bridgemen et al. 1994) |
| - | NAC (600 mg/d, 7days) | Effect of NAC on FEV1, breathlessness | COPD | No difference compared to placebo group | (Black et al. 2004) |
| - | NAC (600 mg /d) | Effect of NAC on cytokine and exhaled breath condensate (ECD) | COPD | Decrease IL-8 and ECP level in NAC group | (Van Overveld et al. 2005) |
| - | NAC (600 mg × 2/d × 2 months) | Effect of NAC on H2O2 | COPD | Decrease H2O2 level in NAC group | (De Benedetto et al. 2005) |
| - | NAC (600 mg once a day for 12 months) | Effect of NAC on H2O2 and TBARS in EBC | COPD | No change in TBARS reduce H2O2 levels | (Kasielski and Nowak 2001) |
| - | β-carotene (20 mg/day) and vitamin E (50 mg/day) | Effect on symptoms (chronic cough, phlegm, or dyspnea) | COPD | No benefit on symptoms | (Habib et al. 1999) |
| - | β-carotene (20 mg daily for 4 weeks) | Effect on lipid peroxides levels in exhaled breath | Smokers | Reduce lipid peroxidation (pentane levels) in exhaled breath | (Rautalahti et al. 1997; Allard et al. 1994; Steinberg and Chait 1998) |
| The MORGEN study | Diet rich in polyphenols/biofla vanoids (catechin, flavonol, and flavone) (58mg/d) | Effect on FEV1, chronic cough, breathlessness and chronic phlegm | COPD | Positively associated with decline in FEV1 and inversely associated with chronic cough and breathlessness, but not chronic phlegm | (Tabak et al. 2001) |
| European countries (Finnish, Italian and Dutch cohorts) | Diet rich in fruits, vegetables, and fish intake | Effect on 20-year COPD mortality | COPD | A 24% lower COPD mortality risk | (Walda et al. 2002) |
| EQUALIFE studies | Vectrine-thiol compound, 300mg bid for months | Blect exacerbation rate, hospitalization, lung function and quality of life | COPD | Decreased exacerbations and fewer days in hospital. No loss of lung function and improvement in hyalth-related quality of life | (Moretti et al. 2004) |
| - | Vitamin C (500 mg daily for 4 weeks) | Effect on lipid peroxides Levels in breath and plasma | Smokers | No change in lung function No change in lipid peroxidation (breath pentane and plasma MDA levels) | (Allard et al. 1994) |
| - | Vitamin C (600 mg) Vitamin E (400 IU) β-carotene (30 mg) | Effect on lipid peroxidation | Smokers | Reduce lipid peroxidation | (Aghdassi et al. 1999, Lykkesfeldt et al. 2000) |
| - | Vitamin E (400 IU twice daily for 3 weeks) | Effect on breath ethane levels | Smokers | No effect on breath ethane levels | (Habib et al. 1999) |
| PEACE study | Carbocysteine (carbocisteine) | Effect on rate of exacerbations | COPD | Long-term (one year) use of carbocysteine (1500 mg/day) produced reduction in numbers of exacerbations in patients with COPD | (Zheng et al. 2008) |
BONCUS- bronchitis randomized on NAC cost utility study
While there is some evidence that the administration of NAC provides benefit for some COPD patients, it is not clear whether this could be used as maintenance therapy [Decramer et al. 2001]. A phase III multicentre Bronchitis Randomized on NAC Cost-Utility Study (BRONCUS) has been completed, with the aim of addressing this question and determining whether the effectiveness of NAC as an “antioxidant” results in an alteration in the rate of decline in FEV1, exacerbation rate and quality of life (QoL) in patients with moderate-to-severe COPD [Gerrits et al. 2003] (Table 3). Although the results of this trial showed no effect on decline in FEV1, the trial did show a reduction in lung over-inflation in patients with severe COPD and in exacerbation rate in patients who were not treated with inhaled glucocorticoids [Decramer et al. 2005]. The variability in all the current studies using NAC (600 mg p.o. q.d.) may simply reflect the fact that the dose is not appropriate enough to achieve the said goal. Since NAC becomes hydrolyzed in biological systems, the measured bioavailability of the drug is also thought to be low. Also NAC has been reported to enhance nitric oxide (NO)-mediated systemic vasodilation in a variety of experimental models, including the spontaneously hypertensive rat [Girouard et al. 2003]. It causes vasodilation by increasing cyclic guanosine monophosphate levels, inhibits platelet aggregation, acts as a sulphydryl donor to regenerate endothelial-derived relaxing factor and reduces IL-8 and TNF-alpha production [Atkinson 2002]. NAC may also decrease adrenergic systemic vasoconstriction. Thus, further studies are required at higher doses (1,200 or 1,800 mg/day) or using other thiol agents with greater bioavailability in order to observe any clinical benefit on lung function, reduced exacerbation rate and improved health status. Overall, NAC can be perceived to play a therapeutic role in the management of chronic bronchitis due to its ability increase intracellular GSH along with its mucolytic properties. Therefore, NAC may be more likely to be of benefit in subjects with mucus hypersecretion. However, the results were not as encouraging when NAC was used against the gamut of COPD. This may be due to the possibility that oxidative stress might be less important in development of other COPD phenotypes. This warrants further incisive clinical studies regarding NAC as a therapeutic alternative against COPD.
N-acystelyn (NAL)
Another alternative to NAC is the lysine salt of NAC, N-acystelyn (NAL). It has been used as a mucolytic agent for cystic fibrosis. However, it also possess antioxidant properties and can reduce both ROS levels and ROS-mediated inflammatory events both in vitro [Antonicelli et al. 2002] and in vivo [Antonicelli et al. 2004]. NAL has several advantages over NAC, firstly it can enhance GSH levels twice as effectively as NAC and secondly it forms a neutral pH when in solution, unlike NAC, which is acidic [Gillissen et al. 1997]. These properties were tested in astudy wherein, when delivered directly into the lung by aerosol in healthy volunteers, NAL did not cause any irritation or other side effects [App et al. 2002]. Therefore NAL may be more promising than NAC in reducing the oxidant burden in the lung in chronic airways disease.
N-isobutyrylcysteine (NIC)
In view of the lower bioavalability of NAC, it was speculated that a drug might be synthesised that possessed greater bioavailability than NAC, and could be used as a more effective treatment for chronic bronchitis. N-isobutyrylcysteine (NIC) was such a compound synthesized, which is a NAC-like thiol compound that does not undergo effective first-pass hydrolysis and hence has a higher oral bioavailability than NAC [Ekberg-Jansson et al. 1999]. The oral bioavailability can be as high as 80%, depending on the food intake. However, when evaluated as a therapy for exacerbations of chronic bronchitis, NIC performed no better than placebo drugs, and not as well as NAC [Gillissen et al. 1997]. Furthermore, a study of NIC also failed to reduce exacerbation rates in patients with COPD [Ekberg-Jansson et al. 1999], thus obliterating its potential therapeutic role in amelioration of COPD.
Erdosteine, fudosteine, and carbocysteine
In view of the apparent problems that were observed for NAC, NAL and NIC, there was an attempt to identify/synthesise novel thiols that may have better and improved abililty over the previously described thiols. Such novel thiols include erdosteine, fudosteine, and carbocysteine, which have mucoactive properties (mucolytic) and the ability to reduce bacterial adhesiveness. Erdosteine is a new thiol compound that also acts as an antioxidant, but in addition has mucoactive properties and reduces bacterial adhesiveness [Moretti and Marchioni 2007]. In the “Equalife” randomized placebo controlled trial, erdosteine was orally dosed at 300 mg b.i.d. for a period of 8 months [Moretti et al. 2004]. Patients receiving erdosteine had significantly fewer exacerbations and showed lesser hospitalization time compared to the placebo group. In addition, patients receiving erdosteine showed no reduction in lung function over this period and showed a significant improvement in health related quality of life. Dal Negro et al. showed that erdosteine at 900 mg/day proved effective in significantly reducing reactive oxygen species (ROS) level in peripheral blood of stable COPD patients who smoke, together with the level of some chemotactic cytokines (IL-6 and IL-8) in their bronchial secretions [Dal Negro et al. 2005]. A clinical trial on the combination of steroids and erdosteine in patients with COPD is needed.
Fudosteine, [(-)-(R)-2-amino-3-(3-hydroxypropylthio)] propionic acid, has been used as mucoactive agent with indications for chronic respiratory diseases such as bronchial asthma, chronic bronchitis, pulmonary emphysema, COPD, and bronchiectasis, [Komatsu et al. 2005]. Fudosteine is a novel mucoactive agent [Komatsu et al. 2005; Rhee et al. 2008]. Fudosteine are a cysteine donating compounds that increase the cysteine levels of the cells and have greater bioavailability than NAC. Fudosteine inhibits MUC5AC triggered mucin hypersecretion by reducing the expression of the MUC5AC gene [Rhee et al. 2008]. Although, little is known about its mode of action, fudosteine has shown to possess promising potential in treatment of patients with COPD [Nagaoka et al. 2002]. These findings suggest that fudosteine may be useful in controlling stress-related mucus secretion states in patients with asthma, bronchiectasis or COPD.
S-carboxymethylcysteine (carbocysteine or S-CMC) is another muco-active drug with in vitro free radical scavenging and anti-inflammatory properties. Carbocysteine is well absorbed when taken orally and peak serum concentrations are achieved at 1–1.7 hours with a plasma half life of 1.33 hours (a well-telerated drug). It achieves good translocation into lung tissue and bronchial secretions [Braga et al. 1982]. Animal studies have demonstrated the anti-inflammatory action of carbocysteine in models of pulmonary inflammation exhibiting different cytokine profiles. Oral pre-treatment with S-CMC-lys appears to attenuate neutrophil recruitment in acute IL-1β-induced airway inflammation and neutrophil, macrophage and eosinophil migration to the pleural space in carrageenan-induced pleurisy [Asti et al. 1995]. Carbocysteine has been clinically used to treat patients with COPD. A recent study has shown that long-term use of carbocysteine reduced the rate of exacerbations in patients with COPD, but there was no difference in exacerbation rate between the carbocysteine group and placebo group during a short-term treatment (3 months) [Zheng et al. 2008]. However, 1-year treatment of carbocysteine (1,500 mg) was effective for COPD patients in terms of reductions in numbers of exacerbations and improvements in quality of life. Hence, it was suggested that longer use of carbocysteine may be more effective for preventing exacerbations in patients with COPD.
Nrf2 activators
Nrf2 is a redox-sensitive basic leucin zipper transcription factor, which is essential for the antioxidant responsive element (ARE)-mediated induction of phase II detoxifying genes. It plays a pivotal role in cellular defense against electrophiles and ROS present in cigarette smoke. Kelch-like ECH-associated protein 1 (Keap1) negatively regulates Nrf2 activity by targeting it to proteasomal degradation. Studies have shown that acute cigarette smoke exposure increased but chronic exposure decreased the levels of phase II drug metabolizing enzymes in both rat lung and nasal epithelium, despite Nrf2 protein level remaining stable [Gebel et al. 2004]. The authors believe that Nrf2 becomes less sensitive to the oxidants from smoke with repeated exposures, although no mechanism for this decreased sensitivity has been demonstrated. Nrf2 deficient (Nrf2−/−) mice have been shown to be more susceptible to airway inflammation caused by bleomycin [Cho et al. 2004], ovalbumin challenge [Rangasamy et al. 2005] and hyperoxia [Hayes and McLellan 1999]. A significant reduction in the mRNA expression of ARE-responsive lung antioxidant and phase 2 enzymes, some of which included heme-oxygenase-1, GPx2, and NAD(P)H: quinone oxidoreductase, was also observed in Nrf2−/− mice compared to normal mice. Thus Nrf2 appears to protect against pulmonary hyperoxia/bleomycin injury and ovalbumin challenge in mice, presumably by upregulating the transcription of lung antioxidant defense enzymes.
Studies have also shown increased susceptibility of Nrf2−/− mice to CS-induced emphysema compared with wild type mice [Rangasamy et al. 2004; Iizuka et al. 2005], suggesting the protective role of Nrf2. Studies from our laboratory revealed that treatment of human alveolar/airway epithelial cells and macrophages with CS extract (CSE) had no effect on nuclear translocation of Nrf2 due to the post-translational modifications of Nrf2 and Keap1 caused by aldehydes present in CSE. Immunohistochemistry data on human peripheral lung tissue also showed sequestration of Nrf2 predominantly in the cytoplasm of airway epithelium, alveolar type II cells and macrophages in smokers and patients with COPD compared with non-smokers, suggesting that CS causes sequestration of Nrf2 in the cytoplasm by its post-translational modifications. Recent studies have clearly highlighted the downregulation of Nrf2 (GSH levels) in pulmonary macrophages and in lungs of patients with COPD via loss of GCL and Nrf2 positive regulator DJ-1 and post-translational modifications of Keap1-Bach1 equilibrium [Malhotra et al. 2008; Goven et al. 2008; Suzuki et al. 2008]. Moreover, decreased GSH-levels may also have direct functional consequences leading to inflammation. Therapeutically, it has been shown that activating Nrf2 with the compound CDDO-imidazolide can reduce LPS induced inflammation and mortality, providing a novel therapeutic strategy for diseases where oxidative stress is implicated [Thimmulappa et al. 2006]. Hence, novel compounds can be developed which are potent activators of Nrf2 leading to buffering the antioxidant potential in the lung against cigarette smoke, particularly in patients with COPD.
Spin traps
Spin traps are based around a nitrone or nitroxide containing molecule such as isoindole based nitrones [Bottle and Micallef 2003], and azulenyl-based nitrones [Becker et al. 2002]. One such azulenyl nitrone, STANZ, may prove promising for in vivo use as it exhibits very potent antioxidant activity. Suprisingly, no studies have been performed in COPD, looking at the impact of spin traps on clinical endpoints, such as FEV1. A phenyl-based nitrone spin trap developed by AstraZeneca, NXY-059, is due to enter Phase III clinical trials for use in acute ischemic stroke. The utility of this compound in SAINT II trial was unsuccessful though the drug was safe. Nevertheless, it remains to be seen whether such compounds could be developed for more long-term use in COPD. Very recently Rohr et al investigated the possible toxicity of 5-ethoxycarbonyl-3,5-dimethyl-pyrroline N-oxide (3,5-EDPO) and its derivatives earmarked for their possible biological use [Rohr-Udilova et al. 2007]. Out of the nine derivatives studied, only three were found suitable for biological application. Thus it would be prudent to consider that a chosen spin trap should be first screened for its toxicity in the biological system. Recently, the new spin trap 2-diphenylphosphinoyl-2-methyl-3,4-dihydro-2H-pyrrole N-oxide (DPhPMPO) was developed by Karakawa et al [Karakawa et al. 2008]. DPhPMPO was found to be more sensitive towards detection and binding of O2.− and formed a very stable product, a property of a good spin trap. There are reports of good spin traps which effectively trap a free radical but are quite unstable, yielding another form of a free radical [Shi et al. 2005]. It is therefore noteworthy that a chosen spin trap should be effective and specific for a given free radical and should be stable for a long time in a given biological environment.
Redox sensor inhibitors
The intracellular redox state is not only sensitive to the external environment, but may also be altered by the contribution that internal redox couples can make. The relative contribution of each of these reduced versus oxidised isoforms can in turn affect the overall redox state of the cell. Common redox sensors include the NADP/NADPH and GSH/GSSG systems. GSH being the most abundant redox sensor also plays an important protective role, as discussed earlier. The NADP/NADPH system can also play a protective role, particularly in the mitochondrial compartment, buffering against excessive redox shifts. Recently the oxidoreductase family of redox sensors, such as thioredoxin and redox effector factor-1 (ref-1) has attracted particular interest. Thioredoxin, has been shown to play an important role in regulating redox sensitive signaling pathways, such as NF-κB and AP-1, p38MAPK and JNK [Filomeni et al. 2002]. Thioredoxin has been reported to associate with other proteins such as hepatopoietin [Li et al. 2005] and the apoptosis signal regulating kinase, ASK-1 [Saitoh et al. 1998] as heterodimeric complexes. Under conditions of oxidative stress thioredoxin is released from these complexes liberating ASK-1 [Filomeni et al. 2002] and hepatopoietin [Li et al. 2005], causing multimerization of ASK-1 and dimerization of heptopoietin. This allows thioredoxin to reduce a key thiol group within cys62 of the RelA/p65 NF-κB subunit, leading to its full transcriptional activition [Qin et al. 1995]. At the same time, the free ASK-1 is also activated by oxidative stress and then able to activate the pro-inflammatory p38 MAPK and JNK pathways [Filomeni et al. 2002], two pathways known to be redox sensitive. It has been demonstrated that inhibiting the oxidoreductase activity of thioredoxin with small molecular weight inhibitor MOL-294, leads to suppression of both NF-κB and AP-1 dependent transcriptional activation. This is achieved through preventing reduction of the important cys62 thiol in the RelA/p65 subunit of NF-κB allowing it to remain s-nitrosylated thereby inhibiting transcriptional activity [Henderson Jr. et al. 2002a]. Recent studies have shown that recombinant thioredoxin-1 amerliorates cigarette smoke- and elastase-induced emphysema in mice [Kinoshita et al 2007; Sato et al 2007; Sato et al 2008). The clinical trials for this important endogenous thiol modifier and anti-inflammatory agent are required. Another redox sensor inhibitor, PNRI-299, has also been demonstrated to prevent AP-1 activation. This is achieved through inhibition of another oxidoreductase family member similar to thioredoxin, namely ref-1 [Nguyen et al. 2003]. Both inhibitors have been shown in vivo to reduce airway eosinophilia, mucus hypersecretion, airway hyper-resposiveness and cytokine release in a murine ovalbumin challenge model of asthma [Henderson Jr. et al. 2002b; Nguyen et al. 2003]. However, it remains to be seen whether these compounds will also prevent NF-κB/AP-1 activation and associated pro-inflammatory responses after cigarette smoke induced COPD in vivo.
Enzyme mimetics
Enzyme mimetics are generally small compounds, which exhibit catalytic properties similar to those of larger enzyme based molecules. In the case of anti-oxidants this includes the SOD, catalase and glutathione peroxidase like activities. A number of SOD mimetics based around organo-manganese complexes have been developed which retain their antioxidant properties in vivo. These include a series of manganese-based macrocyclic ligands, such as M40401, M40403 and M40419 [Salvemini et al. 1999; Tuder et al. 2003; Muscoli et al. 2004]. The second class of SOD mimetics are the manganese-metalloporphyrin based compounds as exemplified by AEOL-10113 and AEOL-10150 [Chang and Crapo 2002; Smith et al. 2002]. The third class of manganese-based SOD mimetic are the “Salens”. These are generally aromatic substituted ethylene-diamine metal complexes [Doctrow et al. 1997], such as EUK134 [Izumi et al. 2002; Chatterjee et al. 2004]. The “Salens” also posses some catalytic activity and can also scavenge hydrogen peroxide, a product of SOD activity. Moreover, they are also able to decompose peroxynitrite [Sharpe et al. 2002]. Of the various classes of SOD mimetic described here, only the metalloporphyrin-based compounds AEOL-10150 and AEOL-10113 (iron-containing porphyrin complexes that decomposes peroxynitrite into innocuous nitrate) have been studied in models of airway inflammation [Cuzzocrea et al. 2004]. In one study, AEOL-10113 was shown to inhibit both airway inflammation and bronchial hyperreactivity in an ovalbumin challenge model of airway inflammation [Chang and Crapo 2002]. This highlighted an important therapeutic benefit in asthma. In another study, AEOL-10150 was demonstrated to inhibit cigarette smoke-induced lung inflammation [Smith et al. 2002] suggesting a potential therapeutic benefit in COPD.
Another type of catalytic antioxidant is the glutathione peroxidase mimetic ebselen. This is a selenium-based organic complex and has been shown to be a very powerful antioxidant against the highly reactive and destructive peroxynitrite radical [Jozsef and Filep 2003]. It is able to prevent both NF-κB/AP-1 activation and pro-inflammatory gene expression in human leukocytes exposed to peroxynitrite. Other studies have shown that ebselen is also active in vivo in preventing LPS-induced airway inflammation [Haddad et al. 2002; Zhang et al. 2002b]. However, no studies have been reported so far on the protective effects of ebselen in cigarette smoke-induced lung inflammation. In contrast, the glutathione peroxidase mimetic BXT-51072 (Oxis, USA) and the lipid peroxidation inhibitor BO-653 (Chugai Pharma, Japan) are either currently in Phase I or at a stage of pre-clinical trials in patients with COPD. A more detailed review of the development and properties of these catalytic antioxidants is described elsewhere [Day 2004].
Polyphenols
Several epidemiological studies have established a beneficial link between polyphenol intake and reduced risk of disease, which were attributed to both the antioxidant and anti-inflammatory properties of polyphenols [Arts and Hollman 2005]. In one Finnish study with over 10,000 participants, a significant inverse correlation was observed between polyphenol intake and the incidence of asthma [Knekt et al. 2002]. Similar beneficial associations were also observed for COPD in a study encompassing over 13,000 adults. This study reported that increased polyphenols, such as catechin (e.g. green tea polyphenols, epigallocatechin gallate), flavonol (e.g. quercetin and kaempferol), and flavone (such as apigenin and luteolin) intake correlated with improved symptoms, as assessed by cough, phlegm production, and breathlessness and improved lung function as measured by FEV1 [Tabak et al. 2001]. While, single component, such as catechin intake was independently associated with FEV1 and all three COPD symptoms, flavonol and flavone intake was independently associated with chronic cough only. Two further studies corroborated these findings. The first study showed a beneficial protective effect against COPD symptoms after increased intake of fruits high in polyphenol and vitamin E content [Walda et al. 2002]. In the second more recent study, a standardized polyphenol extract administered orally was shown to be effective in reducing oxidant stress and increasing PaO2, as well as improvements in FEV1 between enrolment and the end of the study [Santus et al. 2005]. These important studies certainly encourage further multi-national studies to validate the beneficial effects of a high intake of nutraceuticals (polyphenols/bioflavanoids) against COPD symptoms.
While the above studies would appear to demonstrate an epidemiological link between polyphenol intake and clinical benefit in asthma and COPD, other studies have demonstrated a direct impact of specific polyphenolic compounds on inflammation in vitro and in vivo. For example, the flavanoid resveratrol, a constituent of red wine, inhibits inflammatory cytokine release from macrophages isolated from COPD patients [Culpitt et al. 2003]. Birrel and collegues have confirmed this effect of resveratrol in rat lungs challenged with LPS [Birrell et al. 2005]. Furthermore, in both monocytic U937 cells and alveolar epithelial A549 cells, resveratrol inhibits NF-κB and AP-1 activation [Manna et al. 2000; Donnelly et al. 2004]. It has been recently shown that resveratrol, a red wine polyphenol, induces GSH synthesis via activation of Nrf2 in human lung epithelial cells [Biswas et al. 2005; Kode et al. 2008]. However, the clinical utility of resveratrol in terms of inhibiting inflammation and/or inducing GSH biosynthesis in smokers and in patients with COPD is not known.
Another well-studied polyphenol is curcumin. It is the active constituent of Curcuma longa, commonly known as tumeric. Like resveratrol, it has also been reported to inhibit NF-κB activation, along with IL-8 release, cyclooxygenase-2 expression, and neutrophil recruitment in the lungs [Biswas et al. 2005]. Interestingly, one study postulates that curcumin prevents cigarette smoke-induced NF-κB activation through inhibition of IκBα kinase in human lung epithelial cells [Shishodia et al. 2003]. Recently, we have observed that curcumin can inhibit inflammation and restore glucocorticoid efficacy in response to oxidative stress, through upregulation/restoration of HDAC2 activity in macrophages (U937 and MonoMac6 cells) [Meja et al. 2008]. This would facilitate steroid-mediated HDAC2 recruitment in attenuating NF-κB-mediated chromatin acetylation and subsequent pro-inflammatory gene expression. As glucocorticoids are the main thrust of anti-inflammatory treatment, any therapeutic agent that can be used as an add-on to improve steroid responsiveness in COPD would be of significant clinical benefit. Clearly clinical trials by using a combination approach of a steroid with polyphenols are warranted. Despite many successful reports of beneficial effects of polyphenols in vitro, it is important to note that many polyphenols are either very less bioavailable in vivo and/or are biotransformed in the gastrointestinal tract into compounds that may lose the bioactivity or may also be toxic. Future studies employing polyphenols should be designed keeping in view of the preceding observations.
Antioxidant modulation of REDOX sensitive transcription factors and inflammatory pathways
A number of transcription factors involved in the regulation of a variety of inflammatory genes, such as NF-κB and AP-1 are activated by oxidative stress. Recently, Di Stefano and colleagues have demonstrated increased expression of the RelA/p65 subunit of NF-κB in bronchial epithelium of smokers and patients with COPD [Di Stefano et al. 2002], which correlated with the degree of airflow limitation in such patients. Similarly, Caramori and co-workers have shown that p65 subunit of NF-κB was increased in sputum macrophages but not in sputum neutrophils during exacerbations of COPD, suggesting cell specific activation of this factor [Caramori et al. 2003]. The activation of NF-κB in monocytes/macrophages can then trigger the release of pro-inflammatory mediators in lung epithelial fluid, leading to amplification of the inflammatory cascade by activation of epithelial cells as well as recruitment of neutrophils to the airways. That NAC could inhibit activation of NF-κB by oxidative stress in vitro, provided the evidence that activation of this transcription factor is due to oxidative stress [Schreck et al. 1992]. Small molecule inhibition of this pathway is currently of great interest as a potential means to down regulate the inflammation profile of COPD. I-κB kinase-2 (IKK), which mediates phosphorylation of I-κB (an inhibitory subunit of NF-κB) is required for subsequent ubiquitination and degradation of I-κB and consequent nuclear translocation of NF-κB. Therefore small molecule inhibitors of IKK would be expected to block the nuclear translocation of NF-κB and hence inflammation. A variety of compounds are in preclinical development for this target from different pharmaceutical companies, but as yet no compound suitable for clinical studies has been reported in the literature. Alternative means to inhibit NF-κB activation, such as inhibitors of ubiquitin ligase [Wertz et al. 2004], peptide inhibitors of NEMO (a protein that forms a complex with IKK-2) [Choi et al. 2003] and direct inhibition of NF-κB transport through nuclear pores, are being investigated in preclinical studies.
Another study reported on the potential therapeutic effect of the small molecule antagonist siRNA against NF-κB subunit RelA/p65 in airway epithelial cell lines [McIntyre et al. 2003]. In this in vitro study, cells treated with TNF-α showed a reduced NF-κB RelA/p65 expression and concomitant IL-6 and CXCL8 expression. An antisense antagonist for NF-κB RelA/p65 may thus be beneficial in inhibiting the NF-κB-mediated inflammatory genes in patients with COPD.
Oxidative stress and steroid resistance in COPD: reversing by antioxidants
It has been suggested that oxidative stress may have a role in the poor efficacy of corticosteroids and corticosteroid resistance in COPD. Ito and co-workers have shown a role for histone acetylation and deacetylation in IL-1β-induced TNF-α release in alveolar macrophages derived from cigarette smokers [Ito et al. 2001]. They have also suggested that oxidants play an important role in the modulation of HDAC and inflammatory cytokine gene transcription. Furthermore, we have shown that both cigarette smoke/H2O2 and TNF-α caused an increase in histone acetylation (HAT activity) leading to IL-8 expression in monocytes and alveolar epithelial cells both in vitro and in vivo in rat lungs [Rahman et al. 2002b; Moodie et al. 2004; Marwick et al. 2004].
Glucocorticoid suppression of inflammatory genes requires recruitment of histone deacetylase-2 (HDAC2) to the transcription activation complex by the glucocorticoid receptor [Rahman et al. 2004; Ito et al. 2001]. This results in deacetylation of histones and a decrease in inflammatory gene transcription. A reduced level of HDAC2 was associated with increased proinflammatory response and reduced responsiveness to glucocorticoids in alveolar macrophages obtained from smokers [Ito et al. 2001; Rahman et al. 2002a; Moodie et al. 2004; Marwick et al. 2004; Rahman et al. 2004]. Culpitt and co-workers have shown that CS solution stimulated release of IL-8 and GM-CSF, which was not inhibited by dexamethasone, in alveolar macrophages obtained from patients with COPD compared to that of smokers [Culpit et al. 2003]. They suggested that the lack of efficacy of corticosteroids in COPD might be due to steroid insensitivity of macrophages in the respiratory tract. Thus, the cigarette smoke/oxidant-mediated reductions in HDAC2 levels in alveolar epithelial cells and macrophages will not only increase inflammatory gene expression but will also cause a decrease in glucocorticoid function in patients with COPD. HDAC2 activity has also been measured in bronchial biopsies and alveolar macrophages from COPD patients and smoking controls, demonstrating a significant decrease in HDAC2 activity, the magnitude of which increased with severity of disease [Ito et al. 2005]. Moreover, protein expression of HDAC2 was decreased in a similar manner in COPD patients. Consequently, a potential means by which to treat COPD would be to increase HDAC2 expression and activity such that steroids regain their anti-inflammatory activity. We have shown that co-incubation of cells with NAC and H2O2 protects HDAC2 from down regulation and reduction of specific activity [Moodie et al. 2004]. In addition, it has been reported that theophylline has a similar effect in lung macrophage cells, increasing HDAC2 expression and re-sensitizing the cells to steroids [Borja et al. 2004]. Alternative means of upregulating HDAC2 activity by polyphenols would be of great interest for potential combination therapies of the future [Meja et al. 2008].
Conclusions
There is now considerable evidence of increased oxidative stress in COPD, which is pivotal in the pathogenesis and progression of the disease. Several small molecule compounds under clinical trials have either targeted component(s) of oxidant signaling or quench oxidants derived from cigarette smoke. Antioxidant and/or anti-inflammatory agents such as thiol molecules/donors, spin traps, dietary polyphenols, antioxidant mimetics and inhibitors of oxidiative stress induced signaling pathways present potential means by which to treat this element of COPD. However, the clinical trials so far have failed to elaborately define a) the type of antioxidant, b) regimen and c) time period of treatment for successfully combating exacerbations and pathogenesis of COPD. This may be largely due to incomplete understanding of the pathophysiology of COPD and subtle differences in COPD phenotypes. Antioxidant compounds may also enhance the efficacy of glucocorticoids by quenching oxidants and aldehydes, increasing HDAC2 activity in COPD patients. An effective wide spectrum antioxidant therapy that has good bioavailability and potency is urgently needed to control the localized oxidative and inflammatory processes that occur in the pathogenesis of COPD. Although thiol antioxidant treatments have shown promising effects in targetting ROS and oxidant-mediated cellular alterations, development of novel small molecular antioxidants with dual activities (antioxidant/anti-inflammatory) are urgently needed to validate these compounds as clinical therapies. In the case of antioxidants, most oxidative tissue damage will already have taken place by the time therapy is administered. Moreover, some of the antioxidants will not reach the correct cellular/tissue compartment where the oxidative damage is occurring. Therefore, late-stage therapeutic intervention may be insufficient to reverse all the existing pathology and will at best prevent further damage. Hence early management of the the disease is warranted based on clinical phenotypes and stages of COPD diagnosis. At the same time, endeavours in identifying new and more efficacious anti-oxidants as a therapeutic strategy should continue. In this way, at least the therapeutic benefits would be to dampen down and curailing the severity and chronicity of the oxidative stress/inflammatory response in pathogenesis of COPD. Effect of combination of antioxidants along with thiols, spin traps or polyphenols may be an interesting proposition worth investigating. Indeed, elucidating the mechanism of action for some of the naturally occurring anti-oxidants, such as the potent enzyme mimetics and polyphenols and their derivates, may lead to new therapeutic targets that may be validated through more conventional pharmacological approaches. Furthermore, it may be pertinent to suggest that therapeutic research involving GSH or its analogues or thiol compounds must be carefully designed keeping in mind the species barrier, bioavailability, half life of the analogs, and generation of toxic byproducts by the analogues and interference with metabolic and signaling pathways in a cell or tissue. Since patients may have different level of antioxidants in them and since the oxidative states may differ considerably depending on nutritional, behavioral and occupational states, proper understanding of the above issues may lead to development of better thiol/GSH-dependent therapeutic strategies (e.g. Nrf2 activators) for pulmonary diseases. Finally, one has to be careful for excessive use of antioxidant therapy particularly therapies based on redox sensitive molecules which would be deleterious and impose pro-oxidative and inflammatory response in the lung.
Acknowledgements
This work was supported by the National Institutes of Health (NIH)-NHLBI R01-HL085613 and National Institute of Environmental Health Sciences Center (NIEHS) Grant ES-01247.
Footnotes
Conflict of interest statement
None. The author has no conflict of interest.
References
- Aaron SD, et al. Granulocyte inflammatory markers and airway infection during acute exacerbation of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2001;163(2):349–355. doi: 10.1164/ajrccm.163.2.2003122. [DOI] [PubMed] [Google Scholar]
- Adam Odhiambo DHP. Identification of oxidative post-translational modification of serum albumin in patients with idiopathic pulmonary arterial hypertension and pulmonary hypertension of sickle cell anemia. Rapid Communications in Mass Spectrometry. 2007;21(14):2195–2203. doi: 10.1002/rcm.3074. [DOI] [PubMed] [Google Scholar]
- Aghdassi E, et al. Oxidative stress in smokers supplemented with vitamin C. Int J Vitam Nutr Res. 1999;69:45–51. doi: 10.1024/0300-9831.69.1.45. [DOI] [PubMed] [Google Scholar]
- Allard JP, et al. Critical assessment of body-composition measurements in malnourished subjects with Crohn's disease: the role of bioelectric impedance analysis. Am J Clin Nutr. 1994;59:884–890. doi: 10.1093/ajcn/59.2.325. [DOI] [PubMed] [Google Scholar]
- American Thoracic Society Standards for the diagnosis and care of patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 1995;152:S77–S120. [PubMed] [Google Scholar]
- Antonicelli F, et al. Nacystelyn inhibits oxidant-mediated interleukin-8 expression and NF-kappaB nuclear binding in alveolar epithelial cells. Free Radic Biol Med. 2002;32(6):492–502. doi: 10.1016/s0891-5849(01)00820-6. [DOI] [PubMed] [Google Scholar]
- Antonicelli F, et al. Regulation of LPS-mediated inflammation in vivo and in vitro by the thiol antioxidant Nacystelyn. Am J Physiol Lung Cell Mol Physiol. 2004;286(6):L1319–L1327. doi: 10.1152/ajplung.00329.2003. [DOI] [PubMed] [Google Scholar]
- Antwerpen LV, et al. Cigarette smoke-mediated oxidant stress, phagocytes, vitamin C, vitamin E, and tissue injury. Ann NY Acad Sci. 1993;686:53–65. doi: 10.1111/j.1749-6632.1993.tb39153.x. [DOI] [PubMed] [Google Scholar]
- Aoshiba K, et al. Immunohistochemical evaluation of oxidative stress in murine lungs after cigarette smoke exposure. Inhal Toxicol. 2003;15:1029–1038. doi: 10.1080/08958370390226431. [DOI] [PubMed] [Google Scholar]
- App EM, et al. Dose-finding and 24-h monitoring for efficacy and safety of aerosolized Nacystelyn in cystic fibrosis. Eur Respir J. 2002;19(2):294–302. doi: 10.1183/09031936.02.00025802. [DOI] [PubMed] [Google Scholar]
- Arts IC, Hollman PC. Polyphenols and disease risk in epidemiologic studies. Am J Clin Nutr. 2005;81(1 Suppl):317S–325S. doi: 10.1093/ajcn/81.1.317S. [DOI] [PubMed] [Google Scholar]
- Asti C, et al. Effectiveness of carbocysteine lysine salt monohydrate on models of airway inflammation and hyperresponsiveness. Pharmacol Res. 1995;31:387–392. doi: 10.1016/1043-6618(95)80094-8. [DOI] [PubMed] [Google Scholar]
- Atkinson The use of N-acetylcysteine in intensive care. Crit Care Resusc. 2002;4(1):21–27. [PubMed] [Google Scholar]
- Aylward M, et al. Clinical evaluation of acetylcysteine in the treatment of patients with chronic obstructive bronchitis: a balanced double-blind trial with placebo control. Eur J Respir Dis. 1980;61:81–89. [PubMed] [Google Scholar]
- Balint B, et al. Increased nitrotyrosine in exhaled breath condensate in cystic fibrosis. European Respiratory Journal. 2001;17(6):1201–1207. doi: 10.1183/09031936.01.00072501. [DOI] [PubMed] [Google Scholar]
- Barreiro E, et al. Oxidative Stress and Respiratory Muscle Dysfunction in Severe Chronic Obstructive Pulmonary Disease. Am J Respir Crit Care Med. 2005;171(10):1116–1124. doi: 10.1164/rccm.200407-887OC. [DOI] [PubMed] [Google Scholar]
- Barton GM, Roath OS. Leucocyte ascorbic acid in abnormal leucocyte states. Int J Vitam Nutr Res. 1976;46:271–274. [PubMed] [Google Scholar]
- Becker DA, et al. Stilbazulenyl nitrone (STAZN): a nitronyl-substituted hydrocarbon with the potency of classical phenolic chain-breaking antioxidants. J Am Chem Soc. 2002;124(17):4678–4684. doi: 10.1021/ja011507s. [DOI] [PubMed] [Google Scholar]
- Bindoli A, et al. Thiol Chemistry in Peroxidase Catalysis and Redox Signaling. Antioxidants & Redox Signaling. 2008;10(9):1549–1564. doi: 10.1089/ars.2008.2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birrell MA, et al. Resveratrol, an extract of red wine, inhibits lipopolysaccharide induced airway neutrophilia and inflammatory mediators through an NF-kappaB-independent mechanism. FASEB J. 2005;19(7):840–841. doi: 10.1096/fj.04-2691fje. [DOI] [PubMed] [Google Scholar]
- Biswas SK, et al. Curcumin induces glutathione biosynthesis and inhibits NF-kappaB activation and interleukin-8 release in alveolar epithelial cells: mechanism of free radical scavenging activity. Antioxid Redox Signal. 2005;7(1–2):32–41. doi: 10.1089/ars.2005.7.32. [DOI] [PubMed] [Google Scholar]
- Black PN, et al. Randomised, controlled trial of N-acetylcysteine for treatment of acute exacerbations of chronic obstructive pulmonary disease [ISRCTN21676344] BMC Pulm Med. 2004;4:13. doi: 10.1186/1471-2466-4-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borgstrom L, Kagedal B. Dose dependent pharmacokinetics of N-acetylcysteine after oral dosing to man. Biopharm Drug Dispos. 1990;11:131–136. doi: 10.1002/bdd.2510110205. [DOI] [PubMed] [Google Scholar]
- Borgstrom L, et al. Pharmacokinetics of N-acetylcysteine in man. Eur J Clin Pharmacol. 1984;31(2):217–222. doi: 10.1007/BF00606662. [DOI] [PubMed] [Google Scholar]
- Borja GC, et al. Theophylline Restores Histone Deacetylase Activity and Steroid Responses in COPD Macrophages. J Exp Med. 2004;200:689–695. doi: 10.1084/jem.20040416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bottle SE, Micallef AS. Synthesis and EPR spin trapping properties of a new isoindole-based nitrone: 1,1,3-trimethylisoindole N-oxide (TMINO) Org Biomol Chem. 2003;1(14):2581–2584. doi: 10.1039/b300642e. [DOI] [PubMed] [Google Scholar]
- Braga PC, et al. Pharmacokinetic behaviour of S-carboxymethyl-cysteine-lys in patients with chronic bronchitis. Clin. Ther. 1982;4:480–488. [PubMed] [Google Scholar]
- Bridgemen MME, et al. Cysteine and glutathione concentrations in plasma and bronchoalveolar lavage fluid after treatment with N-acetylcysteine. Thorax. 1991;46(1):39–42. doi: 10.1136/thx.46.1.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridgemen MME, et al. Effect of N-acetyl cysteine on the concentrations of thiols in plasma, bronchoalveolar lavage fluid, and lung tissue. Thorax. 1994;49:670–675. doi: 10.1136/thx.49.7.670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridges AB, et al. Age, sex, cigarette smoking and indices of free radical activity in healthy humans. Eur J Med. 1993;2:205–208. [PubMed] [Google Scholar]
- Bridges RB, et al. Micronutrient status and immune function in smokers. Ann N Y Acad Sci. 1990;587:218–231. doi: 10.1111/j.1749-6632.1990.tb00149.x. [DOI] [PubMed] [Google Scholar]
- Britton JR, et al. Dietary antioxidant vitamin intake and lung function in the general population. Am J Respir Crit Care Med. 1995;151:1383–1387. doi: 10.1164/ajrccm.151.5.7735589. [DOI] [PubMed] [Google Scholar]
- Burgunder JM, et al. Effect of N-acetylcysteine on plasma cysteine and glutathione following paracetamol administration. Eur J Clin Pharmacol. 1989;36(2):127–131. doi: 10.1007/BF00609183. [DOI] [PubMed] [Google Scholar]
- Cantin AM, et al. Normal alveolar epithelial lung fluid contains high levels of glutathione. J Appl Physiol. 1987;63:152–157. doi: 10.1152/jappl.1987.63.1.152. [DOI] [PubMed] [Google Scholar]
- Caramori G, et al. Nuclear localization of p65 in sputum macrophages but not in sputum neutrophils during COPD exacerbations. Thorax. 2003;58:348–351. doi: 10.1136/thorax.58.4.348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang LY, Crapo JD. Inhibition of airway inflammation and hyperreactivity by an antioxidant mimetic. Free Radic Biol Med. 2002;33(3):379–386. doi: 10.1016/s0891-5849(02)00919-x. [DOI] [PubMed] [Google Scholar]
- Chatterjee PK. EUK-134 reduces renal dysfunction and injury caused by oxidative and nitrosative stress of the kidney. Am J Nephrol. 2004;24(2):165–177. doi: 10.1159/000076547. [DOI] [PubMed] [Google Scholar]
- Cho HY, et al. The transcription factor NRF2 protects against pulmonary fibrosis. FASEB J. 2004;18(11):1258–1260. doi: 10.1096/fj.03-1127fje. [DOI] [PubMed] [Google Scholar]
- Choi M, et al. Inhibition of NF-kappaB by a TAT-NEMO-binding domain peptide accelerates constitutive apoptosis and abrogates LPS-delayed neutrophil apoptosis. Blood. 2003;102:2259–2267. doi: 10.1182/blood-2002-09-2960. [DOI] [PubMed] [Google Scholar]
- Church DF, Pryor WA. Free radical chemistry of cigarette smoke and its toxicological implications. Environ Health Perspect. 1985;64:111–126. doi: 10.1289/ehp.8564111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corradi M, et al. Aldehydes in exhaled breath condensate of patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2003;167:1380–1386. doi: 10.1164/rccm.200210-1253OC. [DOI] [PubMed] [Google Scholar]
- Cotgreave IA. N-acetylcysteine: pharmacological considerations and experimental and clinical applications. Adv Pharmacol. 1997;38:205–227. [PubMed] [Google Scholar]
- Cotgreave IA, et al. Gastrointestinal metabolism of N-acetylcysteine in the rat, including an assay for sulfite in biological systems. Biopharm Drug Dispos. 1987;8:377–386. doi: 10.1002/bdd.2510080408. [DOI] [PubMed] [Google Scholar]
- Couillard A, et al. Evidence of local exercise-induced systemic oxidative stress in chronic obstructive pulmonary disease patients. Eur Respir J. 2002;20(5):1123–1129. doi: 10.1183/09031936.02.00014302. [DOI] [PubMed] [Google Scholar]
- Cross CE, et al. Oxidants, antioxidants, and respiratory tract lining fluids. Environ Health Perspec. 1994;102:185–191. doi: 10.1289/ehp.94102s10185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crouch BI, Rusho WJ. Intravenous Administration of N-Acetylcysteine. Ann Emerg Med. 2005;46:207–208. doi: 10.1016/j.annemergmed.2005.02.027. [DOI] [PubMed] [Google Scholar]
- Culpitt SV, et al. Inhibition by red wine extract, resveratrol, of cytokine release by alveolar macrophages in COPD. Thorax. 2003;58(11):942–946. doi: 10.1136/thorax.58.11.942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuzzocrea S, et al. Potential therapeutic effect of antioxidant therapy in shock and inflammation. Curr Med Chem. 2004;11(9):1147–1162. doi: 10.2174/0929867043365396. [DOI] [PubMed] [Google Scholar]
- Dal Negro RW, et al. Erdosteine 900mg/day leads to substantial changes in blood ROS, e-NO and some chemotactic cytokines in human secretions of current smokers. Am J Respir Crit Care Med. 2005 Suppl 2:A89. [Google Scholar]
- Day BJ. Catalytic antioxidants: a radical approach to new therapeutics. tDrug Discov Today. 2004;9(13):557–566. doi: 10.1016/S1359-6446(04)03139-3. [DOI] [PubMed] [Google Scholar]
- De Benedetto F, et al. Long-term oral n-acetylcysteine reduces exhaled hydrogen peroxide in stable COPD. Pulm Pharmacol Ther. 2005;18:41–47. doi: 10.1016/j.pupt.2004.09.030. [DOI] [PubMed] [Google Scholar]
- Decramer M, et al. The Bronchitis Randomized On NAC Cost-Utility Study (BRONCUS): hypothesis and design. BRONCUS-trial Committee. Eur Respir J. 2001;17:329–336. doi: 10.1183/09031936.01.17303290. [DOI] [PubMed] [Google Scholar]
- Decramer M, et al. Effects of N-acetylcysteine on outcomes in chronic obstructive pulmonary disease (Bronchitis Randomized on NAC Cost-Utility Study, BRONCUS): a randomized placebo-controlled trial. Lancet. 2005;365:1552–1560. doi: 10.1016/S0140-6736(05)66456-2. [DOI] [PubMed] [Google Scholar]
- Dekhuijzen PN. Antioxidant properties of N-acetylcysteine: their relevance in relation to chronic obstructive pulmonary disease. Eur Respir J. 2004;23:629–636. doi: 10.1183/09031936.04.00016804. [DOI] [PubMed] [Google Scholar]
- Dekhuijzen PN, Van Beurden WJ. The role for N-acetylcysteine in the management of COPD. Int J Chron Obstruct Pulmon Dis. 2006;1(2):99–106. doi: 10.2147/copd.2006.1.2.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Stefano A, et al. Increased expression of nuclear factor-kB in bronchial biopsies from smokers and patients with COPD. Eur Respir J. 2002;20:556–563. doi: 10.1183/09031936.02.00272002. [DOI] [PubMed] [Google Scholar]
- Doctrow SR, et al. Salen-manganese complexes: combined superoxide dismutase/catalase mimics with broad pharmacological efficacy. Adv Pharmacol. 1997;38:247–269. doi: 10.1016/s1054-3589(08)60987-4. [DOI] [PubMed] [Google Scholar]
- Donnelly LE, et al. Anti-inflammatory effects of resveratrol in lung epithelial cells: molecular mechanisms. Am J Physiol Lung Cell Mol Physiol. 2004;287(4):L774–L783. doi: 10.1152/ajplung.00110.2004. [DOI] [PubMed] [Google Scholar]
- Drost EM, et al. Oxidative stress and airway inflammation in severe exacerbations of COPD. Thorax. 2005;60(4):293–300. doi: 10.1136/thx.2004.027946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dueholm M, et al. N-acetylcysteine by metered dose inhaler in the treatment of chronic bronchitis: a multi-centre study. Respir Med. 1992;86:89–92. doi: 10.1016/s0954-6111(06)80220-9. [DOI] [PubMed] [Google Scholar]
- Duthie GG, et al. Effects of smoking and vitamin E on blood antioxidant status. Am J Clin Nutr. 1991;53:1061S–1063S. doi: 10.1093/ajcn/53.4.1061S. [DOI] [PubMed] [Google Scholar]
- Dworski R. Oxidant stress in asthma. Thorax. 2000;55 Suppl 2:S51–S53. doi: 10.1136/thorax.55.suppl_2.S51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekberg-Jansson A, et al. N-isobutyrylcysteine, a donor of systemic thiols, does not reduce the exacerbation rate in chronic bronchitis. Eur Respir J. 1999;13:829–834. doi: 10.1034/j.1399-3003.1999.13d22.x. [DOI] [PubMed] [Google Scholar]
- Filomeni, et al. Cell signalling and the glutathione redox system. Biochem Pharmacol. 2002;64(5–6):1057–1064. doi: 10.1016/s0006-2952(02)01176-0. [DOI] [PubMed] [Google Scholar]
- Gebel S, et al. Gene expression profiling in respiratory tissues from rats exposed to mainstream cigarette smoke. Carcinogenesis. 2004;25(2):169–178. doi: 10.1093/carcin/bgg193. [DOI] [PubMed] [Google Scholar]
- Gerrits CMGM, et al. N-acetylcysteine reduces risk of rehospitalisation among patients with COPD. Eur Respir J. 2003;21:795–798. doi: 10.1183/09031936.03.00063402. [DOI] [PubMed] [Google Scholar]
- Gillissen A, et al. Nacystelyn, a novel lysine salt of N-acetylcysteine, to augment cellular antioxidant defence in vitro. tRespir Med. 1997;91(3):159–168. doi: 10.1016/s0954-6111(97)90052-4. [DOI] [PubMed] [Google Scholar]
- Girouard H, et al. N-acetylcysteine improves nitric oxide and alpha-adrenergic pathways in mesenteric beds of spontaneously hypertensive rats. Am J Hypertens. 2003;16:577–584. doi: 10.1016/s0895-7061(03)00863-x. [DOI] [PubMed] [Google Scholar]
- Gompertz S, et al. Relationship between airway inflammation and the frequency of exacerbations in patients with smoking related COPD. Thorax. 2001;56(1):36–41. doi: 10.1136/thorax.56.1.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goven D, et al. Altered Nrf2/Keap1-Bach1 equilibrium in pulmonary emphysema. Thorax. 2008 doi: 10.1136/thx.2007.091181. (In press) [DOI] [PubMed] [Google Scholar]
- Grandjean EM, et al. Efficacy and oral long-term N-acetylcysteine in chronic bronchopulmonary disease: a meta-analysis of published double-blind, placebo-controlled clinical trials. Clin Ther. 2000;22:209–221. doi: 10.1016/S0149-2918(00)88479-9. [DOI] [PubMed] [Google Scholar]
- Grievink L, et al. Dietary intake of antioxidant (pro)-vitamins, respiratory symptoms and pulmonary function: the MORGEN study. Thorax. 1998;53:166–171. doi: 10.1136/thx.53.3.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutteridge JM, Halliwell B. Free radicals and antioxidants in the year 2000. A historical look to the future. Ann N Y Acad Sci. 2000;899:136–147. doi: 10.1111/j.1749-6632.2000.tb06182.x. [DOI] [PubMed] [Google Scholar]
- Guyton KZ, et al. Activation of mitogen-activated protein kinase by H2O2. Role in cell survival following oxidant injury. J Biol Chem. 1996;271(8):4138–4142. doi: 10.1074/jbc.271.8.4138. [DOI] [PubMed] [Google Scholar]
- Habib MP, et al. Effect of vitamin E on exhaled ethane in cigarette smokers. Chest. 1999;115:684–690. doi: 10.1378/chest.115.3.684. [DOI] [PubMed] [Google Scholar]
- Haddad E, et al. Differential effects of ebselen on neutrophil recruitment, chemokine, and inflammatory mediator expression in a rat model of lipopolysaccharide-induced pulmonary inflammation. J Immunol. 2002;169(2):974–982. doi: 10.4049/jimmunol.169.2.974. [DOI] [PubMed] [Google Scholar]
- Harju T, et al. Diminished immunoreactivity of gamma-glutamylcysteine synthetase in the airways of smokers' lung. Am J Respir Crit Care Med. 2002;166:754–759. doi: 10.1164/rccm.2112014. [DOI] [PubMed] [Google Scholar]
- Hatch GE. Asthma, inhaled oxidants, and dietary antioxidants. Am J Clin Nutr. 1995;61(3 Suppl):625S–630S. doi: 10.1093/ajcn/61.3.625S. [DOI] [PubMed] [Google Scholar]
- Hayes JD, McLellan LI. Glutathione and glutathione-dependent enzymes represent a co-ordinately regulated defence against oxidative stress. Free Radic Res. 1999;31(4):273–300. doi: 10.1080/10715769900300851. [DOI] [PubMed] [Google Scholar]
- Henderson WR, Jr, et al. A role for cysteinyl leukotrienes in airway remodeling in a mouse asthma model. Am J Respir Crit Care Med. 2002a;165(1):108–116. doi: 10.1164/ajrccm.165.1.2105051. [DOI] [PubMed] [Google Scholar]
- Henderson WR, Jr, et al. A small molecule inhibitor of redox-regulated NF-kappa B and activator protein-1 transcription blocks allergic airway inflammation in a mouse asthma model. J Immunol. 2002b;169(9):5294–5299. doi: 10.4049/jimmunol.169.9.5294. [DOI] [PubMed] [Google Scholar]
- Iizuka T, et al. Nrf2-deficient mice are highly susceptible to cigarette smoke-induced emphysema. Genes Cells. 2005;10(12):1113–1125. doi: 10.1111/j.1365-2443.2005.00905.x. [DOI] [PubMed] [Google Scholar]
- Inci I, et al. N-acetylcysteine attenuates lung ischemia-reperfusion injury after lung transplantation. Ann Thorac Surg. 2007;84:240–246. doi: 10.1016/j.athoracsur.2007.03.082. [DOI] [PubMed] [Google Scholar]
- Ito K, et al. Cigarette smoking reduces histone deacetylase 2 expression, enhances cytokine expression, and inhibits glucocorticoid actions in alveolar macrophages. FASEB J. 2001;15:1110–1112. [PubMed] [Google Scholar]
- Izumi M, et al. Superoxide dismutase mimetics with catalase activity reduce the organ injury in hemorrhagic shock. Shock. 2002;18(3):230–235. doi: 10.1097/00024382-200209000-00005. [DOI] [PubMed] [Google Scholar]
- Janssen-Heininger YMW, et al. Reactive Nitrogen Species and Cell Signaling: Implications for Death or Survival of Lung Epithelium. Am J Respir Crit Care Med. 2002;166(12):9S–16S. doi: 10.1164/rccm.2206008. [DOI] [PubMed] [Google Scholar]
- Jozef L, Filep JG. Selenium-containing compounds attenuate peroxynitrite-mediated NF-kappaB and AP-1 activation and interleukin-8 gene and protein expression in human leukocytes. Free Radic Biol Med. 2003;35(9):1018–1027. doi: 10.1016/s0891-5849(03)00439-8. [DOI] [PubMed] [Google Scholar]
- Kasielski M, Nowak D. Long-term administration of N-acetylcysteine decreases hydrogen peroxide exhalation in subjects with chronic obstructive pulmonary disease. Respir Med. 2001;95:448–456. doi: 10.1053/rmed.2001.1066. [DOI] [PubMed] [Google Scholar]
- Karakawa T, et al. Applicability of new spin trap agent, 2-diphenylphosphinoyl-2-methyl-3,4-dihydro-2H-pyrrole N-oxide, in biological system. Biochem Biophys Res Commun. 2008;370(1):93–97. doi: 10.1016/j.bbrc.2008.03.048. [DOI] [PubMed] [Google Scholar]
- Keatings VM, et al. Differences in interleukin-8 and tumor necrosis factor-alpha in induced sputum from patients with chronic obstructive pulmonary disease or asthma. Am J Respir Crit Care Med. 1996;153(2):530–534. doi: 10.1164/ajrccm.153.2.8564092. [DOI] [PubMed] [Google Scholar]
- Kharitonov SA, Barnes PJ. Exhaled markers of pulmonary disease. Am J Respir Crit Care Med. 2001;163:1693–1722. doi: 10.1164/ajrccm.163.7.2009041. [DOI] [PubMed] [Google Scholar]
- Kinoshita T, et al. Thioredoxin prevents the development and progression of elastase-induced emphysema. Biochem Biophys Res Commun. 2007;354:712–719. doi: 10.1016/j.bbrc.2007.01.053. [DOI] [PubMed] [Google Scholar]
- Kirkham PA, et al. Cigarette smoke triggers macrophage adhesion and activation: role of lipid peroxidation products and scavenger receptor. Free Radic Biol Med. 2003;35(7):697–710. doi: 10.1016/s0891-5849(03)00390-3. [DOI] [PubMed] [Google Scholar]
- Kirkham PA, et al. Macrophage phagocytosis of apoptotic neutrophils is compromised by matrix proteins modified by cigarette smoke and lipid peroxidation products. Biochem Biophys Res Commun. 2004;318(1):32–37. doi: 10.1016/j.bbrc.2004.04.003. [DOI] [PubMed] [Google Scholar]
- Knekt P, et al. Flavonoid intake and risk of chronic diseases. Am J Clin Nutr. 2002;76(3):560–568. doi: 10.1093/ajcn/76.3.560. [DOI] [PubMed] [Google Scholar]
- Kode A, et al. Resveratrol induces glutathione synthesis by activation of Nrf2 and protects against cigarette smoke-mediated oxidative stress in human lung epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2008;294:L478–L488. doi: 10.1152/ajplung.00361.2007. [DOI] [PubMed] [Google Scholar]
- Koechlin C, et al. Does systemic inflammation trigger local exercise-induced oxidative stress in COPD? Eur Respir J. 2004;23:538–544. doi: 10.1183/09031936.04.00069004. [DOI] [PubMed] [Google Scholar]
- Komatsu H, et al. Inhibition of endotoxin- and antigen-induced airway inflammation by fudosteine, a mucoactive agent. Pulm Pharmacol Ther. 2005;18(2):121–127. doi: 10.1016/j.pupt.2004.11.002. [DOI] [PubMed] [Google Scholar]
- Lander HM, et al. A molecular redox switch on p21(ras). Structural basis for the nitric oxide-p21(ras) interaction. J Biol Chem. 1997;272(7):4323–4326. doi: 10.1074/jbc.272.7.4323. [DOI] [PubMed] [Google Scholar]
- Lee KS, et al. A novel thiol compound, N-acetylcysteine amide, attenuates allergic airway disease by regulating activation of NF-kappaB and hypoxia-inducible factor-1alpha. Exp Mol Med. 2007;39:756–768. doi: 10.1038/emm.2007.82. [DOI] [PubMed] [Google Scholar]
- Lenz AG, et al. Oxidatively modified proteins in bronchoalveolar lavage fluid of patients with ARDS and patients at-risk for ARDS. European Respiratory Journal. 1999;13(1):169–174. doi: 10.1034/j.1399-3003.1999.13a31.x. [DOI] [PubMed] [Google Scholar]
- Li XY, et al. Mechanisms of cigarette smoke induced increased airspace permeability. Thorax. 1996;51:465–471. doi: 10.1136/thx.51.5.465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, et al. Direct association of hepatopoietin with thioredoxin constitutes a redox signal transduction in activation of AP-1/NF-kappaB. Cell Signal. 2005;17(8):985–996. doi: 10.1016/j.cellsig.2004.11.016. [DOI] [PubMed] [Google Scholar]
- Linden M, et al. Airway inflammation in smokers with nonobstructive and obstructive chronic bronchitis. Am Rev Respir Dis. 1993;148:1226–1232. doi: 10.1164/ajrccm/148.5.1226. [DOI] [PubMed] [Google Scholar]
- Lykkesfeldt J, Moos T. Age-dependent change in Vitamin C status: a phenomenon of maturation rather than of ageing. Mech Ageing Dev. 2005;126(8):892–898. doi: 10.1016/j.mad.2005.03.010. [DOI] [PubMed] [Google Scholar]
- Lykkesfeldt J, et al. Ascorbate is depleted by smoking and repleted by moderate supplementation: a study in male smokers and nonsmokers with matched dietary antioxidant intakes. Am J Clin Nutr. 2000;71:530–536. doi: 10.1093/ajcn/71.2.530. [DOI] [PubMed] [Google Scholar]
- Makita H, et al. Characterisation of phenotypes based on severity of emphysema in chronic obstructive pulmonary disease. Thorax. 2007;62(11):932–937. doi: 10.1136/thx.2006.072777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malhotra D, et al. Decline in Nrf2 regulated antioxidants in COPD lungs due toloss of its positive regulator DJ-1. Am J Respir Crit Care Med. 2008 doi: 10.1164/rccm.200803-380OC. (In press) [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Manna SK, et al. Resveratrol suppresses TNF-induced activation of nuclear transcription factors NF-kappa B, activator protein-1, and apoptosis: potential role of reactive oxygen intermediates and lipid peroxidation. J Immunol. 2000;164(12):6509–6519. doi: 10.4049/jimmunol.164.12.6509. [DOI] [PubMed] [Google Scholar]
- Marwick JA, et al. Cigarette smoke alters chromatin remodeling and induces proinflammatory genes in rat lungs. Am J Respir Cell Mol Biol. 2004;31(6):633–642. doi: 10.1165/rcmb.2004-0006OC. [DOI] [PubMed] [Google Scholar]
- McIntyre KW, et al. A highly selective inhibitor of I kappa B kinase, BMS-345541, blocks both joint inflammation and destruction in collagen-induced arthritis in mice. A highly selective inhibitor of I kappa B kinase, BMS-345541, blocks both joint inflammation and destruction in collagen-induced arthritis in mice. Arthritis Rheum. 2003;48:2652–2659. doi: 10.1002/art.11131. [DOI] [PubMed] [Google Scholar]
- Meja KK, et al. Curcumin Restores Corticosteroid Function in Monocytes Exposed to Oxidants by Maintaining HDAC2. Am J Respir Cell Mol Biol. 2008 doi: 10.1165/rcmb.2008-0012OC. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mezzetti A, et al. Vitamins E, C and lipid peroxidation in plasma and arterial tissue of smokers and non-smokers. Atherosclerosis. 1995;112:91–99. doi: 10.1016/0021-9150(94)05403-6. [DOI] [PubMed] [Google Scholar]
- Montuschi P, Barnes PJ. Analysis of exhaled breath condensate for monitoring airway inflammation. Trends in Pharmacological Sciences. 2002;23:232–237. doi: 10.1016/s0165-6147(02)02020-5. [DOI] [PubMed] [Google Scholar]
- Montuschi P, et al. Exhaled 8-isoprostane as an in vivo biomarker of lung oxidative stress in patients with COPD and healthy smokers. Am J Respir Crit Care Med. 2000;162:1175–1177. doi: 10.1164/ajrccm.162.3.2001063. [DOI] [PubMed] [Google Scholar]
- Moodie, et al. Oxidative stress and cigarette smoke alter chromatin remodeling but differentially regulate NF-kappaB activation and proinflammatory cytokine release in alveolar epithelial cells. FASEB J. 2004;18:1897–1899. doi: 10.1096/fj.04-1506fje. [DOI] [PubMed] [Google Scholar]
- Moretti M, et al. The effect of long-term treatment with erdosteine on chronic obstructive pulmonary disease: the EQUALIFE Study. Drugs Exp Clin Res. 2004;30(4):143–152. [PubMed] [Google Scholar]
- Moretti M, Marchioni CF. An overview of drdosterine antioxidant activity in experimental research. Pharmacol Res. 2007;55:249–254. doi: 10.1016/j.phrs.2006.12.006. [DOI] [PubMed] [Google Scholar]
- Morrison D, et al. Epithelial permeability, inflammation, and oxidant stress in the air spaces of smokers. Am J Respir Crit Care Med. 1999;159:473–479. doi: 10.1164/ajrccm.159.2.9804080. [DOI] [PubMed] [Google Scholar]
- Muscoli C, et al. The protective effect of superoxide dismutase mimetic M40401 on balloon injury-related neointima formation: role of the lectin-like oxidized low-density lipoprotein receptor-1. J Pharmacol Exp Ther. 2004;311(1):44–50. doi: 10.1124/jpet.104.068205. [DOI] [PubMed] [Google Scholar]
- Nagaoka S, et al. Phase III clinical study of SS320A-double blind trial in comparison with placebo. J Clin Ther. 2002:109–140. [Google Scholar]
- Neurohr C, et al. Glutamate-cysteine ligase modulatory subunit in BAL alveolar macrophages of healthy smokers. Eur Respir J. 2003;22:82–87. doi: 10.1183/09031936.03.00080403. [DOI] [PubMed] [Google Scholar]
- Nguyen C, et al. Chemogenomic identification of Ref-1/AP-1 as a therapeutic target for asthma. Proc Natl Acad Sci U S A. 2003;100(3):1169–1173. doi: 10.1073/pnas.0437889100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishida M, et al. G[alpha]i and G[alpha]o are target proteins of reactive oxygen species. Nature. 2000;408(6811):492–495. doi: 10.1038/35044120. [DOI] [PubMed] [Google Scholar]
- Nowak D, et al. Increased content of thiobarbituric acid-reactive substances and hydrogen peroxide in the expired breath condensate of patients with stable chronic obstructive pulmonary disease: no significant effect of cigarette smoking. Resp Med. 1999;93:389–396. doi: 10.1053/rmed.1999.0574. [DOI] [PubMed] [Google Scholar]
- Pacht ER, et al. Deficiency of vitamin E in the alveolar fluid of cigarette smokers. Influence on alveolar macrophage cytotoxicity. J Clin Invest. 1988;77:789–796. doi: 10.1172/JCI112376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel B, et al. Airway and parenchymal disease in chronic obstructive pulmonary disease are distinct phenotypes. tProc Am Thorac Soc. 2006;3(6):533. doi: 10.1513/pats.200603-087MS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauwels RA, et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease. NHLBI/WHO Global Initiative for Chronic Obstructive Lung Disease (GOLD) Workshop summary. Am J Respir Crit Care Med. 2001;163:1256–1276. doi: 10.1164/ajrccm.163.5.2101039. [DOI] [PubMed] [Google Scholar]
- Petruzzelli S, et al. Pulmonary lipid peroxidation in cigarette smokers and lung cancer patients. Chest. 1990;98:930–935. doi: 10.1378/chest.98.4.930. [DOI] [PubMed] [Google Scholar]
- Pinamonti S, et al. Detection of xanthine oxidase activity products by EPR and HPLC in bronchoalveolar lavage fluid from patients with chronic obstructive pulmonary disease. Free Radic Biol Med. 1998;25(7):771–779. doi: 10.1016/s0891-5849(98)00128-2. [DOI] [PubMed] [Google Scholar]
- Poli G, et al. Oxidative Stress and Cell Signalling. Current Medicinal Chemistry. 2004;11:1163–1182. doi: 10.2174/0929867043365323. [DOI] [PubMed] [Google Scholar]
- Poole PJ, Black PN. Oral mucolytic drugs for exacerbations of chronic obstructive pulmonary disease: systematic review. BMJ. 2001;322(7297):1271–1274. doi: 10.1136/bmj.322.7297.1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poole PJ, Black PN. Preventing exacerbations of chronic bronchitis and COPD: therapeutic potential of mucolytic agents. Am J Respir Med. 2003;2:367–370. doi: 10.1007/BF03256664. [DOI] [PubMed] [Google Scholar]
- Pratico D, et al. Chronic obstructive pulmonary disease is associated with an increase in urinary levels of isoprostane F2alpha-III, an index of oxidant stress. Am J Respir Crit Care Med. 1998;158:1709–1714. doi: 10.1164/ajrccm.158.6.9709066. [DOI] [PubMed] [Google Scholar]
- Qin J, et al. Solution structure of human thioredoxin in a mixed disulfide intermediate complex with its target peptide from the transcription factor NF kappa B. Structure. 1995;3(3):289–297. doi: 10.1016/s0969-2126(01)00159-9. [DOI] [PubMed] [Google Scholar]
- Rahman I. Oxidative stress and gene transcription in asthma and chronic obstructive pulmonary disease: antioxidant therapeutic targets. Curr Drug Targets Inflammation and Allergy. 2002;1:291–315. doi: 10.2174/1568010023344607. [DOI] [PubMed] [Google Scholar]
- Rahman I. The role of oxidative stress in the pathogenesis of COPD: implications for therapy. Treat Respir Med. 2005;4:175–200. doi: 10.2165/00151829-200504030-00003. [DOI] [PubMed] [Google Scholar]
- Rahman I, Adcock IM. Oxidative stress and redox regulation of lung inflammation in COPD. Eur Respir J. 2006;28:219–242. doi: 10.1183/09031936.06.00053805. [DOI] [PubMed] [Google Scholar]
- Rahman I, Biswas SK. Non-invasive biomarkers of oxidative stress: reproducibility and methodological issues. Redox Report. 2004;9:125–143. doi: 10.1179/135100004225005219. [DOI] [PubMed] [Google Scholar]
- Rahman I, Kelly F. Biomarkers in breath condensate: a promising new non-invasive technique in free radical research. Free Rad Res. 2003;37:1253–1266. doi: 10.1080/10715760310001623331. [DOI] [PubMed] [Google Scholar]
- Rahman I, MacNee W. Role of oxidants/antioxidants in smoking-induced lung diseases. Free Radic Biol Med. 1996;21(5):669–681. doi: 10.1016/0891-5849(96)00155-4. [DOI] [PubMed] [Google Scholar]
- Rahman I, MacNee W. Role of transcription factors in inflammatory lung diseases. Thorax. 1998;53(7):601–612. doi: 10.1136/thx.53.7.601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman I, MacNee W. Lung glutathione and oxidative stress: implications in cigarette smoke-induced airway disease. Am J Physiol. 1999;277(6 Pt 1):L1067–L1088. doi: 10.1152/ajplung.1999.277.6.L1067. [DOI] [PubMed] [Google Scholar]
- Rahman I, MacNee W. Regulation of redox glutathione levels and gene transcription in lung inflammation: therapeutic approaches. Free Rad Biol Med. 2000a;28:1405–1420. doi: 10.1016/s0891-5849(00)00215-x. [DOI] [PubMed] [Google Scholar]
- Rahman I, MacNee W. Oxidative stress and regulation of glutathione in lung inflammation. Eur Respir J. 2000b;16(3):534–554. doi: 10.1034/j.1399-3003.2000.016003534.x. [DOI] [PubMed] [Google Scholar]
- Rahman I, et al. Systemic oxidative stress in asthma, COPD, and smokers. Am J Respir Crit Care Med. 1996;154(4 Pt 1):1055–1060. doi: 10.1164/ajrccm.154.4.8887607. [DOI] [PubMed] [Google Scholar]
- Rahman I, et al. Is there any relationship between plasma antioxidant capacity and lung function in smokers and in patients with chronic obstructive pulmonary disease? Thorax. 2000;55:189–193. doi: 10.1136/thorax.55.3.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman I, et al. 4-Hydroxy-2-nonenal, a specific lipid peroxidation product, is elevated in lungs of patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2002a;166(4):490–495. doi: 10.1164/rccm.2110101. [DOI] [PubMed] [Google Scholar]
- Rahman I, et al. Oxidative stress and TNF-alpha induce histone acetylation and NF-kappaB/AP-1 activation in alveolar epithelial cells: potential mechanism in gene transcription in lung inflammation. Mol Cell Biochem. 2002b;234:239–248. [PubMed] [Google Scholar]
- Rahman I, et al. Redox modulation of chromatin remodeling: impact on histone acetylation and deacetylation, NF-kappaB and pro-inflammatory gene expression. Biochem Pharmacol. 2004;68:1255–1267. doi: 10.1016/j.bcp.2004.05.042. [DOI] [PubMed] [Google Scholar]
- Rahman I, et al. Oxidant and antioxidant balance in the airways and airway diseases. European Journal of Pharmacology. 2006;533(1–3):222–239. doi: 10.1016/j.ejphar.2005.12.087. [DOI] [PubMed] [Google Scholar]
- Rangasamy T, et al. Genetic ablation of Nrf2 enhances susceptibility to cigarette smoke-induced emphysema in mice. J Clin Invest. 2004;114(9):1248–1259. doi: 10.1172/JCI21146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rangasamy T, et al. Disruption of Nrf2 enhances susceptibility to severe airway inflammation and asthma in mice. J Exp Med. 2005;202(1):47–59. doi: 10.1084/jem.20050538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmusse JB, Glennow C. Reduction in days of illness after long-term treatment with N-acetylcysteine controlled-release tablets in patients with chronic bronchitis. Eur J Respir Dis. 1988;1:351–355. [PubMed] [Google Scholar]
- Rautalahti M, et al. The effect of alpha-tocopherol and beta-carotene supplementation on COPD symptoms. Am J Respir Crit Care Med. 1997;156:1447–1452. doi: 10.1164/ajrccm.156.5.96-11048. [DOI] [PubMed] [Google Scholar]
- Reddy S, et al. Identification glutathione modifications by cigarette smoke. Free Radic Biol Med. 2002;33:1490–1498. doi: 10.1016/s0891-5849(02)01079-1. [DOI] [PubMed] [Google Scholar]
- Rhee CK, et al. Effect of fudosteine on mucin production. Eur Respir J. 2008 doi: 10.1183/09031936.00018508. In press. [DOI] [PubMed] [Google Scholar]
- Richter C, et al. Oxidants in mitochondria: from physiology to diseases. Biochim Biophys Acta. 1995;1271(1):67–74. doi: 10.1016/0925-4439(95)00012-s. [DOI] [PubMed] [Google Scholar]
- Rochelle LG, et al. Concurrent production of reactive oxygen and nitrogen species by airway epithelial cells in vitro. Free Radic Biol Med. 1998;24(5):863–868. doi: 10.1016/s0891-5849(97)00375-4. [DOI] [PubMed] [Google Scholar]
- Rohr-Udilova N, et al. Cytotoxicity of the novel spin trapping compound 5-ethoxycarbonyl-3,5-dimethyl-pyrroline N-oxide (3,5-EDPO) and its derivatives. tBioorg Med Chem Lett. 2007;17(20):5698–5703. doi: 10.1016/j.bmcl.2007.07.054. [DOI] [PubMed] [Google Scholar]
- Romieu I, Trenga C. Diet and obstructive lung diseases. Epidemiol Rev. 2001;23(2):268–287. doi: 10.1093/oxfordjournals.epirev.a000806. [DOI] [PubMed] [Google Scholar]
- Saitoh, et al. Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK)1. EMBO J. 1998;17(9):2596–2606. doi: 10.1093/emboj/17.9.2596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvemini D, et al. A nonpeptidyl mimic of superoxide dismutase with therapeutic activity in rats. Science. 1999;286(5438):304–306. doi: 10.1126/science.286.5438.304. [DOI] [PubMed] [Google Scholar]
- Santus, et al. Lipid peroxidation and 5-lipoxygenase activity in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2005;171(8):838–843. doi: 10.1164/rccm.200404-558OC. [DOI] [PubMed] [Google Scholar]
- Sargeant LA, et al. Interaction of vitamin C with the relation between smoking and obstructive airways disease in EPIC Norfolk. European Prospective Investigation into Cancer and Nutrition. Eur Respir J. 2000;16:397–403. doi: 10.1034/j.1399-3003.2000.016003397.x. [DOI] [PubMed] [Google Scholar]
- Sato A, et al. Thioredoxin-1 suppresses systemic inflammatory responses against cigarette smoking. Antioxid Redox Signal. 2006;8:1891–1896. doi: 10.1089/ars.2006.8.1891. [DOI] [PubMed] [Google Scholar]
- Sato A, et al. Thioredoxin-1 ameliorates cigarette smoke-induced lung inflammation and emphysema in mice. J Pharmacol Exp Ther. 2008;325:380–388. doi: 10.1124/jpet.107.134007. [DOI] [PubMed] [Google Scholar]
- Schreck R, et al. Nuclear factor kappa B: an oxidative stress-responsive transcription factor of eukaryotic cells. Free Radic Res Commun. 1992;17:221–237. doi: 10.3109/10715769209079515. [DOI] [PubMed] [Google Scholar]
- Shahar E, et al. Docosahexaenoic acid and smoking-related chronic obstructive pulmonary disease. The Atherosclerosis Risk in Communities Study Investigators. Am J Respir Crit Care Med. 1999;159:1780–1785. doi: 10.1164/ajrccm.159.6.9810068. [DOI] [PubMed] [Google Scholar]
- Sharpe MA. Oxidation of nitric oxide by oxomanganese-salen complexes: a new mechanism for cellular protection by superoxide dismutase/catalase mimetics. Biochem J. 2002;366(Pt 1):97–107. doi: 10.1042/BJ20020154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi H, et al. Evaluation of spin trapping agents and trapping conditions for detection of cell-generated reactive oxygen species. Arch Biochem Biophys. 2005;437(1):59–68. doi: 10.1016/j.abb.2005.02.028. [DOI] [PubMed] [Google Scholar]
- Shishodia S, et al. Curcumin (diferuloylmethane) down-regulates cigarette smoke-induced NF-kappaB activation through inhibition of IkappaBalpha kinase in human lung epithelial cells: correlation with suppression of COX-2, MMP-9 and cyclin D1. Carcinogenesis. 2003;24(7):1269–1279. doi: 10.1093/carcin/bgg078. [DOI] [PubMed] [Google Scholar]
- Smith KR, et al. Inhibition of tobacco smoke-induced lung inflammation by a catalytic antioxidant. Free Radic Biol Med. 2002;33(8):1106–1114. doi: 10.1016/s0891-5849(02)01003-1. [DOI] [PubMed] [Google Scholar]
- Snider GL. Chronic obstructive pulmonary disease: risk factors, pathophysiology and pathogenesis. Annu Rev Med. 1989;40:411–429. doi: 10.1146/annurev.me.40.020189.002211. [DOI] [PubMed] [Google Scholar]
- Steinberg FM, Chait A. Antioxidant vitamin supplementation and lipid peroxidation in smokers. Am J Clin Nutr. 1998;68:319–327. doi: 10.1093/ajcn/68.2.319. [DOI] [PubMed] [Google Scholar]
- Stey C, et al. The effect of oral N-acetylcysteine in chronic bronchitis: a quantitative systematic review. Eur Respir J. 2000;16:253–262. doi: 10.1034/j.1399-3003.2000.16b12.x. [DOI] [PubMed] [Google Scholar]
- Suzuki M, et al. Downregulated NF-E2-related Factor 2 in pulmonary macrophages of aged smokers and COPD patients. Am J Respir Cell Mol Biol. 2008 doi: 10.1165/rcmb.2007-0424OC. (In press) [DOI] [PubMed] [Google Scholar]
- Tabak C, et al. Chronic obstructive pulmonary disease and intake of catechins, flavonols, and flavones: the MORGEN Study. Am J Respir Crit Care Med. 2001;164:61–64. doi: 10.1164/ajrccm.164.1.2010025. [DOI] [PubMed] [Google Scholar]
- Theron AJ, et al. Investigation of the role of phagocytes and anti-oxidant nutrients in oxidant stress mediated by cigarette smoke. Int J Vitam Nutr Res. 1990;60:261–266. [PubMed] [Google Scholar]
- Thimmulappa RK, et al. Nrf2-dependent protection from LPS induced inflammatory response and mortality by CDDO-Imidazolide. Biochem Biophys Res Commun. 2006;351(4):883–889. doi: 10.1016/j.bbrc.2006.10.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuder RM, et al. Oxidative stress and apoptosis interact and cause emphysema due to vascular endothelial growth factor receptor blockade. Am J Respir Cell Mol Biol. 2003;29(1):88–97. doi: 10.1165/rcmb.2002-0228OC. [DOI] [PubMed] [Google Scholar]
- Tug T, et al. Antioxidant vitamins (A, C and E) and malondialdehyde levels in acute exacerbation and stable periods of patients with chronic obstructive pulmonary disease. Clin Invest Med. 2004;27:123–128. [PubMed] [Google Scholar]
- Van der Toorn, et al. Cigarette smoke irreversibly modifies glutathione in airway epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2007;293:L1156–L1162. doi: 10.1152/ajplung.00081.2007. [DOI] [PubMed] [Google Scholar]
- Van Overveld FJ, et al. Induced sputum of patients with chronic obstructive pulmonary disease (COPD) contains adhesion-promoting, therapy-sensitive factors. Inflamm Res. 2000;49(1):8–13. doi: 10.1007/PL00000203. [DOI] [PubMed] [Google Scholar]
- Van Overveld FJ, et al. New developments in the treatment of COPD: comparing the effects of inhaled corticosteroids and N-acetylcysteine. J Physiol Pharmacol. 2005;56 Suppl 4:135–142. [PubMed] [Google Scholar]
- Van Schooten FJ, et al. Effects of oral administration of N-acetyl-L-cysteine: a multi-biomarker study in smokers. Cancer Epidemiol Biomarkers Prev. 2002;11:167–175. [PubMed] [Google Scholar]
- Vecchiarelli A, et al. Macrophage activation by N-acetyl-cysteine in COPD patients. Chest. 1994;105:806–811. doi: 10.1378/chest.105.3.806. [DOI] [PubMed] [Google Scholar]
- Walda IC, et al. Diet and 20-year chronic obstructive pulmonary disease mortality in middle-aged men from three European countries. Eur J Clin Nutr. 2002;56(7):638–643. doi: 10.1038/sj.ejcn.1601370. [DOI] [PubMed] [Google Scholar]
- Wertz IE, et al. De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-kappaB signalling. Nature. 2004;5:694–699. doi: 10.1038/nature02794. [DOI] [PubMed] [Google Scholar]
- Wilson JX. Regulation of vitamin C transport. Annu Rev Nutr. 2002;25:105–125. doi: 10.1146/annurev.nutr.25.050304.092647. [DOI] [PubMed] [Google Scholar]
- Zhang M, et al. Ebselen suppresses late airway responses and airway inflammation in guinea pigs. Free Radic Biol Med. 2002a;32(5):454–464. doi: 10.1016/s0891-5849(01)00825-5. [DOI] [PubMed] [Google Scholar]
- Zhang P, et al. Compartmentalization of macrophage inflammatory protein-2, but not cytokine-induced neutrophil chemoattractant, in rats challenged with intratracheal endotoxin. Shock. 2002b;17(2):104–108. doi: 10.1097/00024382-200202000-00004. [DOI] [PubMed] [Google Scholar]
- Zheng JP, et al. Effect of carbocisteine on acute exacerbation of chronic obstructive pulmonary disease (PEACE Study): a randomised placebo-controlled study. tLancet. 2008;371:2013–2018. doi: 10.1016/S0140-6736(08)60869-7. [DOI] [PubMed] [Google Scholar]
- Zuin R, et al. High-dose N-acetylcysteine in patients with exacerbations of chronic obstructive pulmonary disease. Clin Drug Investig. 2005;25(6):401–408. doi: 10.2165/00044011-200525060-00005. [DOI] [PubMed] [Google Scholar]