

Abstract
Processes for the functionalization of carbon–hydrogen bonds are the focus of significant attention in organic synthesis[1] in response to the need to streamline molecular assembly. As a continuation of our efforts to generate carbocations through single-electron oxidation reactions,[2] we recently reported[3] DDQ-mediated cyclization reactions of benzylic and allylic ethers (Scheme 1; DDQ =2,3-dichloro-4,5-dicyanoquinone).
Keywords: carbocations, C–H activation, macrocycles, natural products, oxidation
These reactions proceed through oxidative cleavage of a carbon–hydrogen bond to form oxocarbenium ions that react with appended nucleophiles.[4] The use of relatively inert ethers as precursors to reactive electrophiles is strategically attractive, because facile substrate preparation makes etherification a useful fragment-coupling process for convergent syntheses. Oxidative carbocation generation enables the inclusion of acid-sensitive functional groups in starting materials and products, and provides an opportunity to incorporate multiple precursors to electrophiles in a synthetic intermediate. These electrophiles can later be revealed through the use of chemically orthogonal conditions.[5]
Macrocyclic oxocarbenium ions are interesting synthetic intermediates as a result of their capacity to engage in transannular nucleophilic addition reactions. The viability of these intermediates was demonstrated by the Wender research group in the synthesis of bryostatin analogues.[6] Scheidt and co-workers recently reported[7] carbon–carbon bond formation via a macrocyclic oxocarbenium ion formed in a Lewis acid mediated condensation, and related examples followed.[8] However, the generality of this remarkable protocol is not clear: In the absence of conformational preorganization, the process requires the formation of highly reactive intermediates through kinetically unfavorable pathways. A potentially more general approach to the formation of macrocyclic oxocarbenium ions for carbon–hydrogen-bond functionalization is the oxidation of preformed macrocyclic ethers with DDQ (Scheme 2). Herein, we report cyclization reactions that proceed through oxidatively generated macrocyclic oxocarbenium ions (3→4→5) and demonstrate the applicability of the protocol to the construction of complex molecules through a brief formal synthesis of neopeltolide.
Scheme 2.

Ring formation via an oxidatively generated macrocyclic oxocarbenium ion.
The concept was initially validated through the synthesis and cyclization of the macrocyclic ether 11, which was prepared from aldehyde 6[9] (Scheme 3). The exposure of 11 to DDQ under our previously reported conditions[3] resulted in a smooth reaction to form tetrahydropyrone 12 in 73% yield. The cis relationship between the substituents at the 2- and 6-positions of the tetrahydropyrone ring was confirmed by a strong cross-peak between the axial hydrogen atoms in the NOESY spectrum.
Scheme 3.

Substrate synthesis and cyclization: a) 9-BBN, THF, [Pd-(dppf)Cl2], K2CO3, DMF, 50 °C;[10] b) NaBH4, MeOH, 50 % (two steps); c) Cl3CCN, DBU, CH2Cl2, 90%; d) 9, La(OTf)3, toluene;[11] e) p-TsOH, MeOH, 60% (two steps); f) LiOH, THF, MeOH, H2O; g) trichlorobenzoyl chloride, Et3N, DMAP, toluene, 65 °C, 75% (two steps);[12] h) HOAc, Na2CO3, [{Ru(p-cymene)Cl2}2], 1-decyne, toluene, 80 °C, 75%;[13] i) DDQ, 2,6-dichloropyridine, DCE, 2 h, 73 %. 9-BBN =9-borabicyclo[3.3.1]nonane, DBU =1,8-diazabicyclo[5.4.0]undec-7-ene, DCE =1,2-dichloroethane, DMAP =4-dimethylaminopyridine, DMF =N,N-dimethylformamide, dppf =1,1′-bis(diphenylphosphanyl)-ferrocene, TBS =tert-butyldimethylsilyl, Tf =trifluoromethanesulfonyl, Ts =p-toluenesulfonyl.
The scope of this protocol is indicated by the examples in Table 1. Stereogenic centers can be incorporated into the macrocycle without significantly impacting the efficiency of the transformation (Table 1, entries 1 and 2). Changes in the size of the macrocycle alter the rate of the reaction but not the yield (Table 1, entries 3 and 4). The para-methoxy group promotes faster reactions, though benzolactone substrates that do not contain an electron-donating group can be coerced to cyclize in acceptable yields with gentle heating (Table 1, entry 5). A decrease in the distance between the oxygen atom of the oxocarbenium ion and the lactone carbonyl group results in a rate reduction (compare entries 4 and 6 in Table 1) due to inductive destabilization of the cationic intermediate.
Table 1.
Reaction scope.a
| Entry | Substrate | Product | t [h] | Yield [%]b |
|---|---|---|---|---|
| 1 |
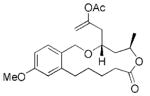 13 |
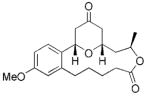 14 |
4 | 74 |
| 2 |
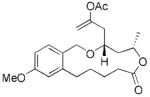 15 |
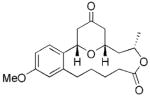 16 |
4 | 69 |
| 3 |
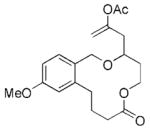 17 |
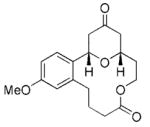 18 |
3.25 | 76 |
| 4 |
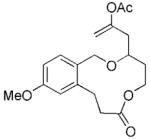 19 |
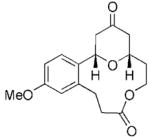 20 |
6 | 73 |
| 5c |
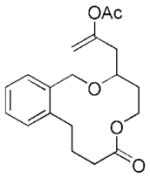 21 |
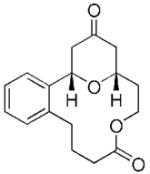 22 |
24 | 71 |
| 6 |
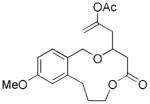 23 |
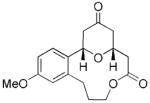 24 |
28 | 65 |
General procedure: DDQ (1.5 equiv), 2,6-Cl2Py, and 4 Å molecular sieves were added to a solution of the substrate in 1,2-dichloroethane at room temperature. The reaction mixture was stirred at room temperature for the indicated time. For specific details, see the Supporting Information.
Yield of the isolated, purified product.
The reaction was conducted at 50°C.
These cyclization reactions were uniformly slower than reactions that we had conducted previously with p-methoxybenzyl ethers.[3] To determine whether the rate retardation arises from the lactone structure or the presence of an ortho-alkyl group, we prepared substrate 25, an analogue of 17 with respect to inductive effects, and subjected it to the cyclization conditions (Scheme 4). Tetrahydropyrone 26 was formed in 76% yield within 0.5 h, which indicates that constraining the ether in a macrocycle slows oxidative carbocation formation. This effect can most likely be attributed to conformational restrictions associated with cyclization, for which overlap is required between the cleaving bond and the π orbitals of the radical cation.[14]
Scheme 4.
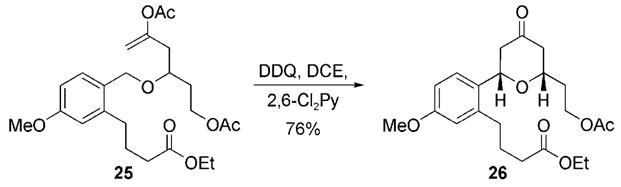
Cyclization of an acyclic analogue of 17. 2,6-Cl2Py = 2,6-dichloropyridine.
Allylic ethers undergo oxidative carbocation formation with an efficiency that rivals or exceeds that of benzylic ethers.[3] We chose to establish the viability of forming macrocyclic α,β-unsaturated oxocarbenium ions and to validate the applicability of the method in the construction of natural products through a formal synthesis of neopeltolide (27; Scheme 5). Neopeltolide was isolated from sponges of the genus Deadolapelta off the Jamaican coast and was shown to exhibit potent cytotoxic activity (IC50 values <10 nM) against a number of cancer cell lines.[15] The Panek[16] and Scheidt[7] research groups independently established the correct stereostructure of neopeltolide through total synthesis, and several total and formal syntheses followed these initial reports.[8a,17] The objective of our synthesis was to prepare tetrahydropyrone 28, which was converted into 27 in two steps by Paterson and Miller.[17d] This structure can be accessed from 29 through an oxidative cyclization followed by a stereoselective alkene reduction. The lactone derives from diester 30, which can be formed from enyne 31 through a regioselective alkyne-hydration reaction. Building blocks 32, 33, and 34 serve as the starting structures for the synthesis.
Scheme 5.

Retrosynthesis of neopeltolide (27).
The synthetic sequence commenced with the conversion of 32[18] into its trichloroacetimidate, followed by etherification with 33[19] in the presence of TfOH to give 35 as an inseparable 7.3:1 mixture of alkene stereoisomers (Scheme 6). Exploratory studies indicated that the Sonogashira coupling of 35 with derivatives of 34[20] proceeded most effectively when the homopropargylic alcohol was protected as a silyl ether. In consideration of downstream transformations, 34 was converted into a diisopropylsilyl ether and coupled to 35[21] to provide 36 in 69% overall yield as an inseparable 5.7:1 mixture of alkene stereoisomers. The diisopropylsilyl group was selected because it is stable during coupling and purification steps but is also sufficiently reactive to be useful in the impending alkyne-hydration step. Regioselective alkyne hydration through a sequence of [Pt(DVDS)]-mediated hydrosilylation[22] followed by oxidative cleavage of the intermediate vinylsilane and concomitant alkyne desilylation under conditions similar to those described by Tamao et al.[23] provided 37.[24] A single alkene stereoisomer was isolated after the reaction, and, remarkably, minimal migration of the alkene into conjugation with the ketone was observed. A stereoselective reduction of 37 with EtCHO and SmI2,[25] followed by methylation of the resulting hydroxy group, provided diester 38. Cleavage of both esters and lactonization of the seco acid under Yamaguchi conditions[12] provided macrolactone 39 in 72% yield. The synthesis of cyclization substrate 40 was completed by a ruthenium-mediated addition of HOAc across the alkyne.[13] This reaction provided an inseparable 5:1 mixture of enol acetate regioisomers in 82% yield.
Scheme 6.

Reagents and conditions: a) NaH, Cl3CCN, Et2O, 0 °C, 100%; b) 33, TfOH, cyclohexane, 77%; c) iPr2Si(H)Cl, imidazole, THF, 77%; d) 35, [(Ph3P)2PdCl2], CuI, iPr2NH, 89%; e) [Pt(DVDS)], THF, then H2O2, KF, Bu4NF, KHCO3, DMF, 40 °C, 57 % (67% based on starting-material purity); f) EtCHO, SmI2, THF, −10 °C, 77%; g) Me3OBF4, proton sponge, CH2Cl2, 0 °C, 93%; h) LiOH, H2O, MeOH, 45°C; i) Et3N, 2,4,6-Cl3BzCl, THF, then DMAP, toluene, 65 °C, 72% (two steps); j) HOAc, Na2CO3, [{Ru(p-cymene)Cl2}2], (2-furyl)3P, 1-decyne, toluene, 80°C, 82 %, 5:1 regioisomer mixture. Bz =benzoyl, DVDS =1,3-divinyl-1,1,3,3-tetramethyldisiloxane.
The exposure of 40 to DDQ in the presence of LiClO4 (which has been shown to minimize side reactions of allylic ether substrates)[3] and 2,6-dichloropyridine resulted in the formation of tetrahydropyrone 41 as a single stereoisomer in 58% yield (Scheme 7). The yield of 41 from pure 40 can be extrapolated to 65%, since the cyclization substrate contained an unproductive regioisomer. The structure of 41 was confirmed by crystallographic analysis.[26] The conformation of 41 suggested that alkene hydrogenation should occur preferentially from the top face of the molecule to provide the correct stereochemical orientation at C9. Indeed, when 41 was treated with H2 and Pd/C, 28 was obtained in 74% yield to complete the formal synthesis. A negligible amount (<10%) of the separable undesired C9 stereoisomer of 28 was formed. All spectral data matched those reported[17d] by Paterson and Miller. The capacity to functionalize the alkene in 41 with predictable stereocontrol presents numerous options for the preparation of neopeltolide analogues.
Scheme 7.

Reagents and conditions: a) DDQ, 2,6-Cl2Py, LiClO4, DCE, 58% (65% based on starting-material purity); b) H2, Pd/C, EtOH, 74%.
We have demonstrated that oxocarbenium ions can be formed in macrocycles through oxidative carbon–hydrogen-bond functionalization, and that these intermediates react with appended nucleophiles to form bridged bicyclic structures. The reactions are somewhat slower than related cyclization reactions of noncyclic precursors as a result of conformational constraints, but the overall efficiency and stereocontrol are comparable. A formal synthesis of neopeltolide on the basis of this process enables access to the natural product in 13 steps from 2-butyn-1-ol. In addition to the oxidative cyclization reaction, key steps in the synthesis include the use of etherification and Sonogashira reactions as fragment-coupling processes, the application of an intramolecular hydrosilylation reaction to effect a regioselective alkyne hydration, and the use of a stereoselective alkene hydrogenation to establish the C9 stereocenter. Early formation of the ether group as an oxocarbenium ion precursor enables minimal reliance upon protecting groups in the sequence. This feature is a significant benefit of selective carbon–hydrogen-bond activation in complex-molecule synthesis.
Supplementary Material
Scheme 1.
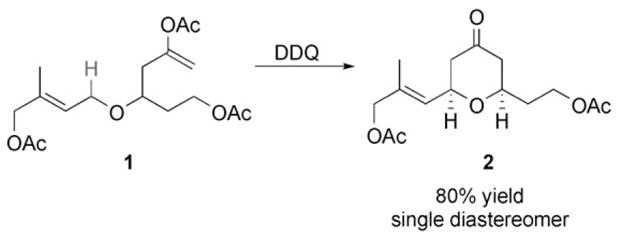
Cyclization through oxidative C–H bond functionalization.
Footnotes
We thank the National Institutes of Health (GM062924) and the National Science Foundation (CHE-139851) for generous support of this research, and the NIH for the 700 MHz NMR instrument at the University of Pittsburgh (S10RR023404). We thank Dr. Steve Geib for crystallographic analysis.
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/anie.200901489.
References
- 1.For reviews and commentary, see: Kakiuchi F, Kochi T. Synthesis. 2008:3013.Godula K, Sames D. Science. 2006;312:67. doi: 10.1126/science.1114731.Davies HML. Angew Chem. 2006;118:6574.Angew Chem Int Ed. 2006;45:6422.Murai S. Adv Synth Catal. 2003;345:1033.
- 2.Floreancig PE. Synlett. 2007:191. [Google Scholar]
- 3.Tu W, Liu L, Floreancig PE. Angew Chem. 2008;120:4252. doi: 10.1002/anie.200706002. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2008;47:4184. [Google Scholar]
- 4.For other examples of carbon–carbon bond formation through DDQ-mediated ether oxidation, see: Hayashi Y, Mukaiyama T. Chem Lett. 1987:1811.Xu YC, Kohlman DT, Liang SX, Erikkson C. Org Lett. 1999;1:1599.Ying BP, Trogden BG, Kohlman DT, Liang SX, Xu YC. Org Lett. 2004;6:1523. doi: 10.1021/ol036314j.Zhang Y, Li CJ. Angew Chem. 2006;118:1983.Angew Chem Int Ed. 2006;45:1949.Zhang Y, Li CJ. J Am Chem Soc. 2006;128:4242. doi: 10.1021/ja060050p.
- 5.For examples of this strategy in complex-molecule synthesis, see: Green ME, Rech JC, Floreancig PE. Angew Chem. 2008;120:7427. doi: 10.1002/anie.200802548.Angew Chem Int Ed. 2008;47:7317.Jung HH, Seiders JR, II, Floreancig PE. Angew Chem. 2007;119:8616. doi: 10.1002/anie.200702999.Angew Chem Int Ed. 2007;46:8464.Wan S, Gunaydin H, Houk KN, Floreancig PE. J Am Chem Soc. 2007;129:7915. doi: 10.1021/ja0709674.
- 6.Wender PA, De Brabander J, Harran PG, Jimenez JM, Koehler MFT, Lippa B, Park CM, Shiozaki M. J Am Chem Soc. 1998;120:4534. [Google Scholar]
- 7.Custar DW, Zabawa TP, Scheidt KA. J Am Chem Soc. 2008;130:804. doi: 10.1021/ja710080q. [DOI] [PubMed] [Google Scholar]
- 8.a) Woo SK, Kwon MS, Lee E. Angew Chem. 2008;120:3286. doi: 10.1002/anie.200800386. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2008;47:3242. [Google Scholar]; b) Wender PA, DeChristopher BA, Schrier AJ. J Am Chem Soc. 2008;130:6658. doi: 10.1021/ja8015632. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Bahnck KB, Rychnovsky SD. J Am Chem Soc. 2008;130:13177. doi: 10.1021/ja805187p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lear Y, Durst T. Can J Chem. 1997;75:817. [Google Scholar]
- 10.Miyaura N, Ishiyama T, Sasaki H, Ishikawa M, Sato M, Suzuki A. J Am Chem Soc. 1989;111:314. [Google Scholar]
- 11.Rai AN, Basu A. Tetrahedron Lett. 2003;44:2267. [Google Scholar]
- 12.Inanaga J, Hirata K, Saeki H, Katzuki T, Yamaguchi M. Bull Chem Soc Jpn. 1979;52:1989. [Google Scholar]
- 13.a) Goossen LJ, Paetzold J, Koley D. Chem Commun. 2003:706. [PubMed] [Google Scholar]; b) Neveux M, Bruneau C, Dixneuf PH. J Chem Soc Perkin Trans. 1;1991:1197. [Google Scholar]
- 14.For discussions of the stereoelectronic requirements for efficient bond-cleavage reactions of radical cations, see: Tolbert LM, Khanna RK, Popp AE, Gelbaum L, Bottomley LA. J Am Chem Soc. 1990;112:2373.Perrott AL, de Lijser HJP, Arnold DR. Can J Chem. 1997;75:384.Freccero M, Pratt A, Albini A, Long C. J Am Chem Soc. 1998;120:284.
- 15.Wright AE, Botelho JC, Guzman E, Harmody D, Linley P, McCarthy PJ, Pitts TP, Pomponi SA, Reed JK. J Nat Prod. 2007;70:412. doi: 10.1021/np060597h. [DOI] [PubMed] [Google Scholar]
- 16.Youngsaye W, Lowe JT, Pohlki F, Ralifo P, Panek JS. Angew Chem. 2007;119:9371. doi: 10.1002/anie.200704122. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2007;46:9211. [Google Scholar]
- 17.a) Vintonyak VV, Maier ME. Org Lett. 2008;10:1239. doi: 10.1021/ol8001255. [DOI] [PubMed] [Google Scholar]; b) Fuwa H, Naito S, Goto T, Sasaki M. Angew Chem. 2008;120:4815. doi: 10.1002/anie.200801399. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2008;47:4737. [Google Scholar]; c) Ulanovskaya OA, Janjic J, Suzuki M, Sabharwal SS, Schumacker PT, Kron SJ, Kozmin SA. Nat Chem Biol. 2008;4:418. doi: 10.1038/nchembio.94. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Paterson I, Miller NA. Chem Commun. 2008:4708. doi: 10.1039/b812914b. [DOI] [PubMed] [Google Scholar]; e) Kartika R, Gruffi TR, Taylor RE. Org Lett. 2008;10:5047. doi: 10.1021/ol802254z. [DOI] [PubMed] [Google Scholar]
- 18.Compound 32 was prepared by treating 2-butyn-1-ol with sodium bis(2-methoxyethoxy)aluminum hydride (Red-Al), followed by quenching with I2: Denmark SE, Jones TK. J Org Chem. 1982;47:4595.
- 19.Compound 33 was prepared in one step from an enantiomerically pure commercially available epoxide according to a previously reported procedure: Shen R, Lin CT, Porco JA., Jr J Am Chem Soc. 2002;124:5650. doi: 10.1021/ja026025a.The epoxide can be prepared more economically through a hydrolytic kinetic resolution: Schaus SE, Brandes BD, Larrow JF, Tokunaga M, Hansen KB, Gould AE, Furrow ME, Jacobsen EN. J Am Chem Soc. 2002;124:1307. doi: 10.1021/ja016737l.
- 20.Compound 34 was prepared from an enantiomerically pure commercially available epoxide in one step according to a previously reported procedure: Schmidt DR, Park PK, Leighton JL. Org Lett. 2003;5:3535. doi: 10.1021/ol035431b.This epoxide can also be prepared economically through hydrolytic kinetic resolution.
- 21.a) Sonogashira K, Tohda Y, Hagihara N. Tetrahedron Lett. 1975;16:4467. [Google Scholar]; b) Chinchilla R, NQjera C. Chem Rev. 2007;107:874. doi: 10.1021/cr050992x. [DOI] [PubMed] [Google Scholar]
- 22.Denmark SE, Wang Z. Org Lett. 2001;3:1073. doi: 10.1021/ol0156751. [DOI] [PubMed] [Google Scholar]
- 23.Tamao K, Maeda K, Tanaka T, Ito Y. Tetrahedron Lett. 1988;29:6955. [Google Scholar]
- 24.For other examples of regioselective alkyne hydration through intramolecular hydrosilylation, see: Marshall JA, Yanik MM. Org Lett. 2000;2:2173. doi: 10.1021/ol0061182.Burova SA, McDonald FE. J Am Chem Soc. 2004;126:2495. doi: 10.1021/ja039618+.Robles O, McDonald FE. Org Lett. 2008;10:1811. doi: 10.1021/ol800659z.
- 25.Evans DA, Hoveyda AH. J Am Chem Soc. 1990;112:6447. [Google Scholar]
- 26.CCDC 721650 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.