

SUMMARY
Neuronal activity is highly dependent on energy metabolism; yet, the two processes have traditionally been regarded as independently regulated at the transcriptional level. Recently, we found that the same transcription factor, nuclear respiratory factor 1 (NRF-1) co-regulates an important energy-generating enzyme, cytochrome c oxidase, as well as critical subunits of glutamatergic receptors. The present study tests our hypothesis that the co-regulation extends to the next level of glutamatergic synapses, namely, neuronal nitric oxide synthase, which generates nitric oxide as a downstream signaling molecule. Using in silico analysis, electrophoretic mobility shift assay, chromatin immunoprecipitation, promoter mutations, and NRF-1 silencing, we documented that NRF-1 functionally bound to Nos1, but not Nos2 (inducible) and Nos3 (endothelial) gene promoters. Both COX and Nos1 transcripts were up-regulated by depolarizing KCl treatment and down-regulated by TTX-mediated impulse blockade in neurons. However, NRF-1 silencing blocked the up-regulation of both Nos1 and COX induced by KCl depolarization, and over-expression of NRF-1 rescued both Nos1 and COX transcripts downregulated by TTX. These findings are consistent with our hypothesis that synaptic neuronal transmission and energy metabolism are tightly coupled at the molecular level.
Keywords: gene regulation, EMSA, NRF-1 over-expression, rat primary neurons, transcription factor, TTX
INTRODUCTION
Nitric oxide (NO) is a short-lived radical that acts via a second messenger (cGMP) with many diverse actions in the nervous, vascular, and immune systems [1–2]. Nitric oxide synthase (NOS) is responsible for NO production by catalyzing the conversion of L-arginine to L-citrulline in an NADPH and calcium/calmodulin-dependent reaction [3]. NOS constitutes a family of three distinct isoforms: neuronal (NOS1 or nNOS), inducible (NOS2 or iNOS), and endothelial (NOS3 or eNOS) [3–5]. In many brain areas, NOS1 is activated by the influx of Ca2+ via glutamatergic NMDA receptors, inducing the binding of calmodulin and the generation of NO, which functions as a signaling molecule through the cGMP pathway [6–7]. Excitatory glutamatergic transmission is highly energy-dependent, as membrane repolarization after depolarization requires ATP-dependent Na+/K+ ATPase to actively pump cations against their concentration and electrical gradients, whereas repolarization after hyperpolarization is mainly passive [8]. Thus, neuronal activity, especially of the excitatory type, and energy metabolism are tightly coupled processes [8]. Regions with a higher concentration of glutamatergic synapses than GABAergic synapses are also rich in cytochrome c oxidase (COX) [9], a critical energy-generating enzyme that catalyzes the final step of oxidative metabolism [8,10]. When excitatory transmission is suppressed, such as with TTX-induced impulse blockade, the level of COX as well as of NMDA receptors and NOS1 are reduced [11–12].
Recently, we found that the coupling between glutamatergic synapses and oxidative metabolism exists at the transcriptional level. Essential NMDA receptor subunit 1 as well as subunit 2b and all subunits of COX are regulated by the same transcription factor, nuclear respiratory factor 1 (NRF-1) [13–14], a transcription factor known to be critical for mitochondrial biogenesis [15–16]. As a putative NRF-1 binding site has been reported for human Nos1 gene but not functionally characterized [17], we sought to determine if NRF-1 indeed directly regulates the expression of Nos1 in murine neurons. Using multiple approaches, we document in the present study that NRF-1 functionally regulates the expression of Nos1 in rodent neurons, but via a site different from the one reported by Hall et al. in humans (1994).
MATERIALS AND METHODS
All experiments were carried out in accordance with the US National Institutes of Health Guide for the care and use of laboratory animals and the Medical College of Wisconsin regulations. All efforts were made to minimize the number of animals and their suffering.
Cell culture
Murine neuroblastoma (N2a) cells (ATCC, CCL-131) were grown in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum, 50 units/ml penicillin, and 100 µg/ml streptomycin (Invitrogen, Carlsbad, CA) at 37°C in a humidified atmosphere with 5% CO2.
Sprague Dawley rats (Harlan, Indianapolis, IN) at 1 day of age were used for primary cultures. Rat primary visual cortical neurons were cultured as described previously [18]. Briefly, 1-day-old neonatal rat pups were sacrificed by decapitation. Brains were removed from the skull and the meninges were removed. Visual cortical tissue was dissected, trypsinized, and triturated to release individual neurons. Neurons were plated in 35 mm poly-L-lysine-coated dishes at a density of 50,000 cells/dish. Cells were maintained in Neurobasal-A media supplemented with B27 (Invitrogen). Ara-C (Sigma, St. Louis, MO) was added to the media to suppress the proliferation of glial cells.
Murine NOS isoform gene promoters analysed by in silico analysis
DNA sequences surrounding the transcription start points (TSPs) of NOS isoform genes (Nos1, 2, and 3) were derived from the mouse genome database in GenBankTM using Genomatix Gene2promoter software as well as NCBI GenBank. These promoter sequences encompassed 1 kb upstream and up to 1kb downstream of the TSP of each gene analyzed. Computer-assisted search for putative NRF-1 core binding sequences “GCGCA(T/C)GC” or “GCGCA(G/C)GC” was conducted on each promoter sequence. Alignment of human, mouse, and rat promoter sequences were done as previously described, using the Genome VISTA genome alignment tool [13,18]. Mouse Nos1, 2, and 3 promoter sequences were compared with rat and human genomic sequences using a 5-bp calculation window. Regions of high homology and/or contain putative NRF-1 binding sites were compared for the conservation of NRF-1 binding.
In vitro interactions via electrophoretic mobility shift (EMSA) and supershift assays
EMSAs to assay NRF-1 interactions with putative binding elements on NOS isoform genes were carried out with methods as previously described [13]. Briefly, oligonucleotide probes with putative NRF-1 binding site on each promoter (Nos1–3) (Table 2A, based on in silico analysis) were synthesized. These oligonucleotides were annealed, and labeled by a Klenow fragment fill-in reaction with [α-32P] dATP (50 µCi/200 ng). Each labeled probe was incubated with 2 µg of calf thymus DNA and 5 µg of HeLa nuclear extract (Promega, Madison, WI) and processed for EMSA. Supershift assays were also performed and, in each reaction, 1–1.5 µg of NRF-1-specific antibodies (polyclonal goat antibodies, gift of Dr. Richard Scarpulla, Northwestern University, Chicago, IL) were added to the probe/nuclear extract mixture and incubated for 20 min at room temperature. For competition, 100-fold excess of unlabeled oligonucleotides were incubated with nuclear extract before adding labeled oligonucleotides. Shift reactions were loaded onto 4% polyacrylamide gel and ran at 200 V for 2.5 h in 0.25X TBE buffer. Results were visualized by autoradiography. Rat cytochrome c with NRF-1 binding site at position −172/−147 was designed as previously described [15] and used as a positive control. NRF-1 mutants with mutated sequences as shown in Table 2A were used as negative controls.
Table 2.
| Table 2A EMSA Probes | ||
|---|---|---|
| Positions of probes are given relative to TSP. Putative NRF-1 binding sites are in boldface. Mutated nucleotide sequences are underlined. | ||
| Gene Promoter | Position | Sequence |
| Nos1 | +150/+164 | F 5′TTTTGTGCCGCGGAGCAGAGCGGCCTT 3′ R 3′ CACGGCGCCTCGTCTCGCCGGAATTTT 5′ |
| Nos2 | −98/−73 | F 5′TTTTAGTTATGCAAAATAGCTCTGCAGAG 3′ R 3′ TCAATACGTTTTATCGAGACGTCTCTTTT 5′ |
| Nos3 | −114/−89 | F 5′TTTTCCACATTAAATACGCAACAAATAGA 3′ R 3′ GGTGTAATTTATGCGTTGTTTATCTTTTT 5′ |
| Nos1 with mutated NRF-1 site | −49/−24 | F 5′TTTTGTAAAGCGGAAAAGAGCGGCCTT 3′ R 3′ CATTTCGCCTTTTCTCGCCGGAATTTT 5′ |
| Rat Cyt C | −172/−147 | F 5′TTTTCTGCTAGCCCGCATGCGCGCGCACCTTA3′ R 3′ GACGATCGGGCGTACGCGCGCGTGGAATTTTT5′ |
| Table 2B Chip Primers | |||
|---|---|---|---|
| Positions of amplicons are given relative to TSP. | |||
| Gene Promoter | Position | Sequence | Amplicon length |
| Nos1 | −134 to +68 | F 5′AAACGCAAAGTGGGAGGTCT 3′ R 5′GCATGGCTGGTTTACGTTTT 3′ |
202 bps |
| Nos2 | −140 to +5 | F 5′AGCTAACTTGCACACCCAACT 3′ R 5′GGGCCAGAGTCTCAGTCTTC 3′ |
145 bps |
| Nos3 | −171 to +17 | F 5′GGTATTTGATGCTCGGGACT 3′ R 5′CACTGTGATGGCTGAACTGA 3′ |
188 bps |
| TFB2M promoter | −64 to +115 | F 5'GAAGCGAGTGAGCAAAGGAC 3' R 5'GGTCCCCTCATCCTCCTCTA 3' |
179 bps |
| β-Actin exon 5 | −134 to +53 | F 5'GCTCTTTTCCAGCCTTCCTT 3' R 5'CGGATGTCAACGTCACACTT 3' |
187 bps |
| Table 2C PCR cloning primers | |
|---|---|
| NRF-1 mutated nucleotide sequences are in boldface. | |
| Cloning Primers | Primer sequence |
| Nos1 | F 5′AAGGTACCGCCAGGTGACCACCACTAAC 3′ R 5′AAAAGCTTCTGACGCATGGCTGGTTTAC 3′ |
| MCOX6b1 | F 5'AAGGTACCGCCAGCCCTTAATTGTTTTC3' R 5'AAAAGCTTTCGCAACTAAAAGCTCCACA 3' |
| Mutagenesis Primers |
|
| Nos1Mut | F 5′ GAGGTGCCGCGGATTTGAGCGTTCTTATCCAAGCC 3′ R 5′ GGCTTGGATAAGAACGCTCAAATCCGCGGCACCTC 3′ |
| MCOX6b1Mut | F 5'CAGCACTAGTTAGGCAGAGTTTGGCGGATTTCTGAGTCTAC3' R 5'GTAGACTCAGAAATCCGCCAAACTCTGCCTAACTAGTGCTGG 3' |
In vivo interactions using chromatin immunoprecipitation (ChIP) assays
ChIP assays were performed similar to those described previously [13]. Briefly, ∼750,000 N2a cells were used for each immunoprecipitation and were fixed with 1% formaldehyde for 10 min at room temperature. ChIP assay kit (Upstate, Charlottesville, VA) was used with minor modifications. Following formaldehyde fixation, cells were resuspended in a swelling buffer (5 mM PIPES, pH 8.0, 85 mM KCl, and 1% Nonidet P-40, and protease inhibitors added right before use) and homogenized 10 times in small pestle Dounce tissue homogenizer (7 ml). Nuclei were then isolated by centrifugation before being subjected to sonication. The sonicated lysate was immunoprecipitated with either 0.2 µg of NRF-1 polyclonal rabbit antibodies (gift of Dr. Scarpulla) or 2 µg of anti-nerve growth factor receptor (NGFR) p75 polyclonal goat antibodies (C20, sc-6188, from Santa Cruz Biotechnology, Santa Cruz, CA). Semi-quantitative PCR was performed using 1/20th of precipitated chromatin. Primers targeting promoter sequences near TSP of Nos1–3 genes were designed (Table 2B) as previously described [18]. The promoter of transcription factor B2 of mitochondria (TFB2M), previously found to be activated by NRF1 [19–20], was used as a positive control, whereas exon 5 of β-actin gene was used as a negative control (Table 2B). PCR reactions were carried out with the EX Taq hot-start polymerase (Takara Mirus Bio, Madison, WI) with the following cycling parameters: 30-s denaturation at 94°C, 30-s annealing at 59.5°C, and 20-s extension at 72°C (32–36 cycles per reaction). All reactions were hot-started by heating to 94°C for 120 s. Use of hot-start polymerase and PCR additives significantly improved the quality and reproducibility of ChIP. PCR products were visualized on 2% agarose gels stained with ethidium bromide.
Promoter mutagenesis study
Luciferase reporter constructs of Nos1 promoters were made by PCR cloning the proximal promoter sequences using genomic DNA prepared from mouse N2a cells as template, digested with HindIII and KpnI restriction enzymes, and ligated directionally into pGL3 basic vector (Promega).
Sequences of primers used for PCR cloning and mutagenesis are provided in Table 2C. Subunit COX6b1 clone was used as previously described [13]. Site-directed mutagenesis of putative NRF-1 binding site on each promoter was generated using QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA). Both constructs were verified by sequencing.
Each promoter construct (Nos1 and COX6b1) was transfected into N2a cells in a 24-well plate using Lipofectamine 2000. Each well received 0.6 µg of promoter construct and 0.03 µg of pCMVβgal, which constitutively expressed β-galactosidase. After 48 h of transfection, cell lysates were harvested and measured for luciferase activity as described previously [13].
To further investigate the effect of KCl stimulation after mutating the NRF-1 binding site, transfected neurons were stimulated with KCl at a final concentration of 20 mM in the culture media for 5 h as previously described [21]. Cell lysates were then harvested and measured for luciferase activity as described previously [13].
NRF-1 shRNA plasmid construction
NRF-1 silencing was carried out using small hairpin RNAs (shRNA) against murine NRF-1 (GenBankTM accession no. for NRF-1: NM_010938) cloned into pLVTHM vector with H1 promoter and green fluorescent protein reporter (gift of Dr. P. Aebischer, Swiss Federal Institute of Technology). Four shRNA sequences were selected: 5’-GAAAGCTGCAAGCCTATCT-3’; 5’-GCCACAGGAGGTTAATTCA-3’; 5’-GCATTACGGACCATAGTTA-3’; and 5’-AGAGCATGATCCTGGAAGA-3’, with a linker sequence (5’-TTCAAGAGA- 3’) and complementary sequence for each to form the NRF-1-shRNA cassette. Multiple shRNA sequences enhance the efficiency of gene silencing [22–23]. The pLL3.7/U6 promoter vector with puromycin resistance (Addgene, Cambridge, MA) vector was used concurrently for puromycin selection. The empty vector pLVTHM or scrambled shRNA in pLVTHM served as negative controls. The basic gene cloning method was followed as described previously [13,20]. N2a cells or primary neurons were plated in 35-mm dishes at a density of 5 to 8 × 106 cells/dish. They were co-transfected 3 days post-plating with NRF-1 shRNA expression vectors (four sequences at equal amounts; 4 µg total for N2a cells and 1 µg total for primary neurons) and the pLL3.7/U6 vector for puromycin resistance (1.5 µg for N2a cells and 1 µg for primary neurons) via Lipofectamine 2000. Empty vectors or scrambled shRNA vectors alone were used at the same concentrations as vectors with shRNA against NRF-1. Puromycin at a final concentration of 0.5 µg/ml was added to the culture medium on the second day after transfection to select for purely transfected cells. Green fluorescence was observed to monitor transfection efficiency. Transfection efficiency for N2a cells ranged from 40% to 75%, while that for primary cortical neurons was from 40% to 60%. However, puromycin selection effectively yielded 100% of transfected cells. Cells were harvested after 48 h of silencing and lysed for either protein or total RNA preparation.
To determine the effect of KCl stimulation, N2a cells transfected with shRNA against NRF-1 were exposed to KCl at a final concentration of 20 mM in the culture media for 5 h as previously described [21]. Cells were then harvested for RNA isolation.
RNA Isolation and cDNA Synthesis
Total RNA was isolated by RNeasy kits (Qiagen, Valencia, CA) according to the manufacturer’s instructions. Three micrograms of total RNA was treated with DNase I and purified by phenolchloroform. cDNA was synthesized using random hexamer primers and SuperScriptTM II RNase H-Reverse Transcriptase (Invitrogen) according to the manufacturer’s instructions.
Real-time Quantitative PCR
Real-time quantitative PCRs were carried out in a Cepheid Smart Cycler Detection system (Cepheid, Sunnyvale, CA). SyBr Green (BioWhittaker Molecular Application) and EX Taq realtime quantitative PCR hotstart polymerase were used following the manufacturer’s protocols and as described previously [13]. Primer sequences are shown in Table 3. PCR runs: hot start 2 min at 95°C, denaturation 10 s at 95°C, annealing 15 s according to the Tm of each primer, and extension 10 s at 72°C for 15–30 cycles. Melt curve analyses verified the formation of single desired PCR product. Mouse β-actin for N2a cells and rat 18s for primary neurons was used as the internal control and the 2-ΔΔCT method [24] was used for the relative amount of transcripts. Subunits COX2 (mitochondrial-encoded) and COX6c (nuclear-encoded) were used as previously described [13].
Table 3.
Primers for real-time PCR
| Gene | Sequence | Amplicon length |
Tm |
|---|---|---|---|
| Nos1 | F 5′CTGGAGGAAGTAGCCAAGAAG 3′ R 5′TTCTCCATGGTTTGATGAAGG 3′ |
154 | 60° |
| Nos2 | F 5′TGATCTTGTGCTGGAGGTGACCAT 3′ R 5′TGTAGCGCTGTGTGTCACAGAAGT 3′ |
200 | 60° |
| Nos3 | F 5′TATTTGATGCTCGGGACTGCAGGA 3′ R 5′ACGAAGATTGCCTCGGTTTGTTGC 3′ |
92 | 60° |
| NRF-1 | F 5′GGCACAGGCTGAGCTGATG 3′ R 5′CTAGTTCCAGGTCAGCCACCTTT 3′ |
90 | 59.5° |
| NRF-2α | F 5'CTCCCGCTACACCGACTAC 3' R 5'TCTGACCATTGTTTCCTGTTCTG 3' |
145 | 59.5° |
| COX2 | F 5′TGGCTTACAAGACGCTACATC 3′ R 5′GGAGGGAAGGGCAATTAGAA 3′ |
201 | 59.5° |
| COX6c | F 5'AGCGTCTGCGGGTTCATA 3' R 5'GCCTGCCTCATCTCTTCAAA 3' |
154 | 60° |
| Gucy1a2 | F 5′GTGCATATCCAGCAATGTTGAACGG 3′ R 5′CGTTGCAAACGGTGAATTATCCTGCC 3′ |
89 | 60° |
| β-Actin | F 5'GGCTGTATTCCCTCCATCG 3' R 5'CCAGTTGGTAACAATGCCATGT 3' |
154 | 59.5 |
| 18S | F 5′CGCGGTTCTATTTTGTTGGT 3′ R 5′AGTCGGCATCGTTTATGGTC 3′ |
219 | 59.5° |
Western Blot Assays
Control and NRF-1 shRNA samples were lysed with lysis buffer (0.5% Triton-X-100 and 5 mM ethylenediamine-tetraacetic acid (EDTA) in PBS with 1× complete protease inhibitor) and centrifuged. The concentration of the supernatant was measured with the Bio-Rad protein Assay Kit II (Bio-Rad, Hercules, CA, USA). A total of 50 µg proteins were loaded onto each lane of 10% SDS-PAGE gels and electrophoretically transferred onto polyvinylidene difluoride membranes (Bio-Rad, Hercules, CA). Subsequent to blocking, blots were incubated in primary antibodies (polyclonal antibodies against NRF-1 (1:500; gift of Dr. Scarpulla,), NOS1 (monoclonal antibodies, 1:200; N2280, Sigma), or NOS2 (1:200; sc-7271, C11 from Santa Cruz Biotechnology). Monoclonal antibodies against β-actin (A5316, Sigma) at 1:3,000 dilutions were used as loading controls. Blots were then incubated in secondary antibodies (goat-anti-rabbit, goat-anti-mouse, or rabbit-anti-goat; Chemicon), reacted with ECL, and exposed to autoradiographic film (Santa Cruz Biotechnology). Quantitative analyses of relative changes were done with Personal Molecular Imager, with Quantity one 1-D analysis software (Bio-Rad).
NRF-1 overexpression and TTX treatment
pSG5NRF-1 expression plasmid, a generous gift from Dr. Richard Scarpulla [25], was used for NRF-1 over-expression. N2a cells and primary neurons were each plated in 35 mm dish at a density of 2 to 5 × 105 cells/dish. N2a cells were co-transfected 3 days post-plating with either 2.5 µg of the pSG5NRF-1 plasmid or an empty vector plus 0.5 µg of pLL3.7 Puro vector, using Lipofectamine 2000 (Invitrogen) at a 1:3 ratio. Procedures were identical for the transfection of primary neurons, except that 2 µg pSG5NRF-1 plasmids were used. Puromycin at a final concentration of 0.5 µg/ml was added on the second day after transfection to select for purely transfected cells. After one day of over-expression, TTX at a final concentration of 0.4 µM was added to the culture media for 3 days. N2a cells and primary neurons were harvested on the 4th day for RNA isolation.
Statistical Analysis
Significance among group means was determined by analysis of variance (ANOVA). Significance between two groups was analyzed by Student’s t test. P-values of 0.05 or less were considered significant.
RESULTS
Promoter analysis of NOS genes by means of in silico analysis
The proximal promoters of murine Nos1, 2, and 3 genes were analyzed using in silico analysis with DNA sequence 1 kb 5’ upstream and 1 kb beyond 3’ of transcription start point (TSP). The sequence (GCGCGTGTGCGC) reported but not functionally characterized by Hall et al. (1994) to be the putative NRF-1 binding site in the human Nos1 gene has no homology in either the mouse or rat Nos1 gene; thus we were not able to use it for the present study. Moreover, it lacks a GCA core found by us to be invariant for NRF-1 binding in the genes that we have analyzed thus far in neurons [13,14,26] and is present in the traditional NRF-1 sequence [15]. In searching further for putative NRF-1 binding site even 2 kb upstream and downstream of TSP, we could find only a single site with GC-rich sequences flanking a GCA core (GCGAGCAGAGCGGCGC) in exon 1 coding region of murine Nos1 gene designated by Genomatix as a promoter region (Gene ID 18125, Genomatix GXP_435815, NCBI NT_078458, and ENSMUST00000102557). It bore a high (85–95%) homology with both the rat and human Nos1 genes (Table 1). This region was also rich in putative binding sites for other transcription factors, such as NRF-2, Sp1, and CREB (unpublished observations).
Table 1. Aligned partial sequences of Nos1 promoters from rat (R), mouse (M), and human (H) genomes show conservation of NRF-1 binding sites (in boldface).
Invariant GCA core sequences are underlined. Solid boxes highlight NRF-1 sites that are highly conserved in all three or at least two species.
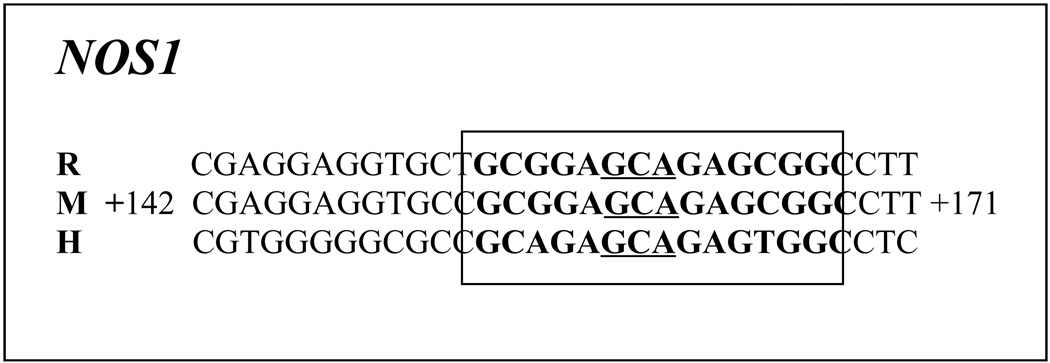 |
At least twelve alternative exon 1 have been identified in the human Nos1 gene, and transcription is partially controlled by exon 1- specific promoter sequences [27,28]. In mice, five alternatively spliced variants and one unspliced form encoding five different isoforms of protein have been reported, and our NRF-1 core binding sequence matched two of the splice variants, aSep07Nos1 and bSep07Nos1, found also in vivo [29]. This diversity of alternative promoters and Nos1 transcripts probably helps to control the diversity of Nos1 expression in many different organs [27]. Transcriptional regulatory element has been found in the coding sequence in a number of genes [30–32].
Nos2 and Nos3 promoters, on the other hand, lack both typical and atypical NRF-1 binding sites. Thus, we selected from a region close to the TSP with some GC and a GCA sequence for each of the genes as probes for electrophoretic mobility shift assays.
In vitro NRF-1 interactions with NOS promoters
Electrophoretic mobility shift assays (EMSAs) for NRF-1 interactions were carried out in vitro using 32P-labeled oligonucleotide probes to determine the specificity of NRF-1 binding to murine NOS promoters (Nos1, Nos2 and Nos3) (Fig. 1). Rat cytochrome c promoter with known NRF-1 site at positions −172/−147 [15] served as a positive control and it formed specific DNA/NRF-1 shift and supershift complexes (Fig. 1, lanes 1 and 3, respectively). When an excess of unlabelled probe was added as a competitor, no shift band was formed (Fig. 1, lane 2). Nos1 promoter formed a specific DNA-protein shift complex when incubated with HeLa nuclear extract (Fig. 1, lane 5) and a DNA-protein-antibody supershift complex with the addition of NRF-1 antibodies (Fig. 1, lane 8). Competition with excess unlabelled probe eliminated the shift complex (Fig. 1, lanes 6 and l0, respectively), whereas an excess of unlabeled mutant NRF-1 probes was not able to compete (Fig. 1, lane 7). To rule out any non-specific antibody and oligonucleotide interaction, labeled probes were incubated with NRF-1 antibody without HeLa nuclear extract, and no shift bands were observed (Fig. 1, lane 4). Nos1 probe with mutated NRF-1 site showed neither shift nor supershift complex (Fig. 1, lanes 9 and 11, respectively). Nos2 and Nos3 lacked both typical and atypical NRF-1 binding sites and did not yield any shift bands (Fig. 1, lanes 12–13), verifying our previous report that the presence of a GCA core flanked by GC-rich sequences are essential for NRF-1 binding in neurons [13–14,26].
Fig. 1. NRF-1 interactions in-vitro and in-vivo with NOS isoform genes and promoter mutational analysis.

EMSAs for NRF-1. 32P- labeled oligonucleotides, excess unlabeled oligos as competitors, excess unlabeled mutant NRF-1 as competitors, HeLa extract, and NRF-1 antibodies are indicated by a + or a − sign. Arrowheads indicate NRF-1 shift and supershift complexes. The positive control, cytochrome c, shows a shift (A, lane 1) and a supershift (A, lane 3) band. When excess unlabeled competitor was added, it did not yield any band (A, lane 2). Nos1 subunit showed specific shift and supershift bands that were eliminated by excess unlabeled competitors (A, lanes 5, 8 & 6, respectively), while Nos2 and Nos3 showed no shift bands (A, lanes 12–13). Labeled mutated NRF-1 site on Nos1 was used as a negative control, and it did not yield any band (A, lanes 9–11). Excess unlabeled but mutated NRF-1 site could not compete (A, lane 7). Labeled oligos with NRF-1 antibodies alone with no HeLa extract did not yield any band (A, lane 4).
In vivo interaction of NRF-1 with NOS isoform genes
To verify NRF-1 interactions with NOS isoform genes in vivo, chromatin immunoprecipitation assays (ChIP) were performed. PCRs that targeted NOS isoform promoters surrounding putative NRF-1 binding sites were carried out in parallel on chromatin immunoprecipitated from N2a cells. A 0.5 and 0.1 % dilution of input chromatin (i.e. prior to immunoprecipitation) was used as a standard to indicate the efficiency of the PCRs. The proximal promoters of Nos1–3 isoforms in the presence of nuclear extract were subjected to chromatin immunoprecipitation with NRF-1 antibodies. Nos1 and TFB2M each produced a band from DNA immunoprecipitated with anti-NRF-1 antibodies at a position identical to that of the genomic DNA control (input) (Fig. 2). On the other hand, Nos2, Nos3 and β-actin yielded no bands (Fig. 2). In all cases, immunoprecipitation with NGFR antibodies, an additional negative control, did not yield any PCR product, confirming the specificity of the ChIP reaction.
Fig. 2. ChIP assays.

Input lanes represent 0.5% and 0.1% of chromatin. TFB2M promoter was the positive control and β-actin was the negative control. Nos1 promoter immunoprecipitated with NRF-1, while Nos2 and Nos3 did not. Anti-nerve growth factor receptor p75 antibodies (NGFR) represent a negative control.
Mutational analysis for Nos1 and COX6b1 promoters
Based on EMSA probes (Table 2A) that formed NRF-1 specific complexes (Fig. 1), site-directed mutagenesis of these same putative NRF-1 binding sites on Nos1 and known murine COX6b1 [13] promoters were constructed (Table 2C), generated in luciferase reporter plasmids, and analyzed by gene transfection. As shown in Figure 3, mutation of NRF-1 binding sites led to ∼ 61 – 66% reduction in promoter activity of Nos1 and COX6b1 genes, respectively (P < 0.05 – 0.001). COX6b1 subunit promoter served as a positive control and confirmed our previous report [13].
Fig. 3. Site-directed mutations of NRF-1 binding sites on Nos1 and COX6b1 promoters.

Mutated NRF-1 binding sites on Nos1 and COX6b1 subunit resulted in significant reductions in luciferase activity as compared to wild type (wt). (N = 6 for each construct). *, P < 0.05, ***, P < 0.001.
Knockdown of NRF-1 by RNA interference
To determine the effect of NRF-1 knockdown on the expression of NOS, plasmid vectors expressing small hairpin RNA (shRNA) against target sequences of NRF-1 mRNA were used. These vectors were previously found to suppress NRF-1 expression in N2a cells [13]. Transfection of neurons with shRNA vectors resulted in ∼ 70 – 85% decrease in levels of NRF-1 and NOS1, but not of NOS2, proteins as measured by western blots (P < 0.05 – 0.01) (Fig. 4A). cDNAs from N2a cells (Fig. 4B) and primary neurons (Fig. 4C) transfected with NRF-1 shRNA vectors, scrambled shRNA vectors, or empty vectors were analyzed with quantitative real-time PCRs. As shown in Figures 4B and 4C, relative mRNA levels of NRF-1, Nos1, and two positive controls (COX2 and COX6c) were significantly reduced in both N2a cells and primary neurons transfected with shRNA as compared to those transfected with empty vectors. The extent of reduction ranged between 70 to 85% (P < 0.01 – 0.001). On the other hand, the expression of Nos2, Nos3, and nuclear respiratory factor 2α (another transcription factor) remained unchanged. The normal levels of Nos2 and Nos3 mRNA in both N2a cells and primary neurons were ∼ 14 – 30% and 25 – 50%, respectively, as compared to that of Nos1 (data not shown). The scrambled shRNA also did not have any effect on any mRNA level tested (Fig. 4B–C).
Fig. 4. NRF-1 silencing suppresses Nos1 and COX subunit mRNAs.

(A) Western blot reveals a down-regulation of NRF-1 and NOS1 protein levels in NRF-1 shRNA-transfected neuron, whereas NOS2 protein levels were not affected. β-Actin served as a loading control. (B–C) N2a cells and primary neurons were transfected with shRNA against NRF-1 (light gray bars), or with empty vectors (black bars), or with scrambled shRNA (dark gray bars). NRF-2α served as a negative control. NRF-1, Nos1, COX2, and COX6c subunit mRNAs show significant decreases in shRNA-treated samples as compared to those with empty vectors, whereas Nos2, Nos3, and NRF-2α mRNA remained unchanged. N = 6 for each data point. *, P < 0.05; **, P < 0.01 as compared to empty vectors.
Response of Nos1 gene promoter to KCl depolarizing stimulation
To determine if depolarizing stimulation altered the expression of Nos1 gene in N2a cells, 20 mM of potassium chloride was added to the culture media for 5 hours, a mild regimen previously found to activate NRF-1 and COX gene expression in primary neurons [21,33–34]. As shown in Figure 5A for N2a cells, depolarizing stimulation resulted in a significant increase (∼120%) in the activity of Nos1 promoter as monitored by luciferase assays (P < 0.05 – 0.01). This increase was abolished by mutating the NRF-1 site, confirming a link between KCl–induced depolarization and the activation of Nos1 via NRF-1 binding.
Fig. 5. Depolarization-induced up-regulation of promoter gene expression and mRNA levels of NOS and COX subunits in neurons.

(A) Site-directed mutations of NRF-1 binding sites on Nos1 promoter resulted in a significant reduction in luciferase activity as compared to the wild type (wt). KCl depolarization increased promoter activity in the wild type but not in the mutated Nos1. (N = 6 for each construct). *, P < 0.05, **, P < 0.01 as compared to Nos1 wild type. X = P < 0.01 as compared to Nos1 wt with KCl depolarization. (B) Data from real time quantitative PCR indicate that Nos1, COX2, and COX6c gene expression in N2a cells were increased by KCl depolarization as compared to controls, whereas those of Nos2 and Nos3 were not affected by KCl. NRF-1 silencing with shRNA prevented the up-regulation of Nos1, COX2, and COX6c mRNAs by KCl. Again, Nos2 and Nos3 were not affected. Values represent mean ± S.E.M of combined data from 3 independent experiments. * P < 0.05, ** P < 0.01 versus controls. All # P values were compared to 20 mM KCl-treated samples (# P < 0.05). Nos2 and Nos3 levels were not significantly different from controls.
Effect of NRF-1 silencing on KCl stimulation of NOS1 and COX subunit gene expression
To determine if NRF-1 silencing affected the expression of NOS and COX subunit genes, N2a cells were transfected with NRF-1 shRNA for 48 hours and then subjected to 20 mM of KCl for 5 hours. As shown in Figure 5B, depolarizing stimulation resulted in a 185% to 198% increase in the expression of Nos1, COX2, and COX6c genes as monitored by real time quantitative PCR (P < 0.05 – 0.01) without affecting the expression of Nos2 and Nos3. In the presence of NRF-1 silencing, however, KCl depolarization could no longer up-regulate the message levels of Nos1, COX2, and COX6c. These results confirmed the essential role of NRF-1 in the regulation of Nos1 and COX subunits in response to changing neuronal activity.
NRF-1 over-expression increased Nos and COX subunit mRNA levels and rescued neurons from tetrodotoxin-induced transcript reduction
A low concentration of TTX (0.4 µM) has been shown to decrease levels of COX subunit mRNAs as well as COX enzyme activity in vivo and in primary neurons [11,34].To determine if over-expression of NRF-1 could rescue not only COX but also Nos1 transcript, a pSG5NRF-1 construct (gift of Dr. Richard Scarpulla) for NRF-1 over-expression was transfected into primary neurons that were then exposed to TTX (0.4 µM) for 3 days. When neurons were transfected with empty vectors, exposure to TTX led to a 52% reduction in NRF-1 and a 71 – 75% reduction in Nos1, Nos2, Nos3, COX2, and COX6C mRNA levels (Figs. 6A–F), indicating an overall suppressive effect of TTX on gene expression in neurons. Neurons transfected with the pSG5NRF-1 construct had a 600% increase in mRNA levels of NRF-1 (P < 0.001) (Fig. 6A) and a 64 – 78% increase in those of Nos1, COX2, and COX6C (P < 0.05 – 0.01) as compared to empty vector controls (Figs. 6B–D). No change in Nos2 and Nos3 levels was evident (Fig. 6E – F). When exposed to TTX, neurons transfected with pSG5NRF-1 expressed 45 – 57% more Nos1, COX2, and COX6C transcripts as compared to those with empty vectors (P < 0.05– 0.01) (Fig. 6A–D). No increase was found for Nos2 and Nos3 (Fig. 6E–F). The expression of NRF-1 itself was 286% higher in pSG5NRF-1-transfected neurons than those with empty vectors in the presence of TTX (P < 0.001) and 234% higher in the absence of TTX (P < 0.001). These results confirmed that NRF-1 could rescue Nos1 and COX subunit mRNAs but not those of Nos2 and Nos3 in the presence of TTX.
Fig. 6. NRF-1 over-expression in primary neurons significantly increased mRNA levels for Nos1, COX2, and COX6c genes and rescued them from TTX-induced suppression.

NRF-1, Nos1, COX2, COX6c, Nos2, and Nos3 mRNA levels (A–F) were all reduced by TTX as compared to controls. Over-expression of NRF-1 significantly increased transcript levels of NRF-1, Nos1, COX2, and COX6c, but not of Nos2 and Nos3. Over-expression of NRF-1 was able to rescue NRF-1, Nos1, COX2, and COX6c, but not Nos2 and Nos3. Group means were analyzed for overall statistical significance using the Student's t-test (N = 6 for each group). All * P values were compared to empty vectors (* P < 0.05, ** P < 0.01, *** P < 0.001). All # P values were compared to empty vector + TTX (# P < 0.05, ## P < 0.01, ### P < 0.001), and all X P values were compared to NRF-1 over-expression (X P < 0.05, XXX P < 0.001).
Effect of NRF-1 silencing on guanylyl cyclase activity
To determine if NRF-1 also played an important role in regulating guanylyl cyclase, a downstream target of NO pathway, the promoter region of soluble guanylyl cyclase gene (Gucy1a2) was analyzed in silico, and a potential NRF-1 binding was found. shRNA knockdown of NRF-1 resulted in an 80% decrease in the expression of Gucy1a2 as compared to empty vector controls (P < 0.01) (Fig. 7). Scrambled shRNA had no effect. Future studies will determine if NRF-1 regulates Gucy1a2 directly or indirectly.
Fig. 7. NRF-1 silencing suppresses mRNA level in guanylyl cyclase gene.

N2a cells were transfected with shRNA against NRF-1, or with empty vector, or with scrambled shRNA. Guanylyl cyclase (Gucy1a2) mRNA shows significant decrease in shRNA-treated samples as compared to those with empty vectors. N = 6 for each data point. *, P < 0.01. Scrambled shRNA did not cause any change in Gucy1a2 levels.
DISCUSSION
The present study documents with multiple approaches that NRF-1 co-regulates COX and Nos1, but not Nos2 and Nos3 genes, thereby linking glutamatergic NOS-mediated synaptic transmission and energy metabolism at the transcriptional level of regulation in neurons. There is high homology in NRF-1 binding sites for Nos1 (Table 1) and COX subunit promoters [13] among mice, rats, and humans, emphasizing the conservation of such co-regulation through evolution. With depolarizing neuronal activity, NRF-1 itself is activated at both protein and mRNA levels [21, the present study], and it, in turn, synchronizes the transcriptional activation of both Nos1 and COX subunit genes, thereby ensuring that neuronal activity and energy metabolism remain tightly coupled.
NOS enzymes constitute a family of proteins with varying functions [35]. NOS1 is neuronal and is present at low concentrations in many cells but at high concentrations in a select group of neurons, primarily GABAergic ones [36–37]. NOS2 is typically not present in unstimulated cells, but is expressed in response to cytokines, lipopolysaccharides, and immunological challenges in macrophages [38–40]. NOS3 is expressed mainly in vascular endothelial cells and plays an important role in the regulation of vascular tone and tissue perfusion [39–41]. NRF-1 functionally binds to Nos1 but not Nos2 and Nos3 promoters in neurons, and NRF-1 silencing down-regulates both protein and mRNA expression of COX and NOS1 without affecting NOS2 and NOS3 [the present study]. Thus, NRF-1’s regulation of Nos1 is linked to neuronal functioning and not to the production of NO per se. It also suggests that the transcriptional regulation of Nos1 is very different from those of Nos2 and Nos3. Each isoform, in turn, mediates different cellular functions, including the regulation of various gene expression [35]. A fourth type of NOS is reportedly present in the mitochondria [42]. However, NO-dependent mitochondrial biogenesis in the murine subcortex is mediated by the NOS1 isoform [43]. Moreover, cardiomyocytes from neuronal NOS-knockout mice do not produce NO in the mitochondria, unlike those of the wild-type, confirming that the mitochondrial NOS (mtNOS) is actually a neuronal NOS [44]. MtNOS is reportedly involved in the regulation of oxidative phosphorylation, as NO can react with COX [42]. However, recent reports indicate that NO produced in the mitochondria by mtNOS (NOS1) in fact protects COX from external inhibitors [6,45]. The current study confirms the relation between COX and NOS1, and not with NOS2 and NOS3.
The level of COX activity in the normal monkey retina, monkey visual cortex, and cultured rat primary neurons are positively correlated with the density of distribution of excitatory neurotransmitters and their receptors, such as glutamate, NMDA receptor subunit NR1, and NOS1 [11–12,46–49]. This is consistent with the fact that repolarization after excitation-induced depolarization is highly energy-dependent and consumes the bulk of ATP produced in the CNS [8]. However, when neuronal activity is altered, such as afferent impulse blockade by TTX or depolarizing treatment with KCl, neuronal COX activity is adjusted to match the new energy demand [8,11]. NOS1 expression is also regulated by changes in neuronal activity [37,50]. At the transcriptional level, this regulation involves NRF-1 (present study). Thus, KCl-medited depolarization increases neuronal activity and up-regulates both Nos1 and COX transcripts, but NRF-1 silencing with RNA interference prevented the up-regulation of Nos1 and COX in the presence of KCl stimulation. The up-regulation of Nos1 could be limited, as excess production of NO may induce glutamate toxicity [7]. On the other hand, TTX-mediated impulse blockade down-regulates both Nos1 and COX transcripts, and over-expression of NRF-1 is able to rescue both Nos1 and COX mRNAs from the suppressive effect of TTX. These results confirm the significant role of NRF-1 in regulating both Nos1 and COX gene expression in neurons [13,the present study].
NRF-1, a phosphoprotein, is a transcriptional activator of nuclear genes encoding a number of mitochondrial respiratory enzymes, including subunits of the five respiratory chain complexes [51–52]. In fact, all ten nuclear-encoded subunit genes of complex IV (COX) are regulated by NRF-1 in neurons [13]. NRF-1 also activates the promoters of mitochondrial transcription factor A (Tfam), transcription specificity factors (TFB1M and TFB2M), and RNA-processing proteins required for mtDNA transcription and replication [16,19]. Thus, NRF-1 indirectly regulates the expression of the largest 3 COX subunit genes encoded in the mtDNA, and effectively regulates all 13 COX subunits. NRF-1 is vital for normal cell growth, cell function, and mitochondrial biogenesis [51–52], and its knockout is embryonically lethal [53]. Based on the present and our previous studies [13–14,26], the traditional consensus sequence for NRF-1 binding (YGCGCAYGCGCR) needs to be modified to include an invariant GCA core flanked by GC-rich sequences on either side, with the exact sequence of GC being variable, especially in rodents.
In response to glutamate, NOS1 is activated by the influx of calcium via NMDA receptors, and NO is produced to stimulate guanylyl cyclase to convert GTP to cGMP, an important second messenger that mediates neurotransmission [3–4,7]. The fact that NRF-1 regulates critical subunit genes of NMDA receptors [14], NOS1 (present study), and possibly guanylyl cyclase via either direct or indirect pathway (present study) strongly indicates that NRF-1 is intimately associated with the coordinated regulation of key neurochemicals associated with a major glutamatergic synaptic pathway in neurons (Fig. 8).
Fig. 8. Schematic representation of Nos1 regulation via NRF-1.

A working model indicating that energy metabolism and neuronal activity are tightly coupled. NMDAR (NR1 and NR2B) (Dhar and Wong-Riley, 2009), COX subunits (Dhar et al., 2008) and Nos1 (present study) are all co-regulated by the same transcription factor, NRF-1. A downstream mediator of NOS1 pathway, guanylyl cyclase, also has a potential NRF-1 binding site. Thus, NRF-1 is intimately associated with the coordinated regulation of energy metabolism as well as key neurochemicals associated with a major glutamatergic synaptic pathway in neurons.
In conclusion, our findings confirm the tight coupling between neuronal activity and energy metabolism beyond the cellular level [8] to the molecular level. By having the same transcription factor participate in the regulation of both processes, this tight coupling can be initiated and/or maintained at the transcriptional level. NRF-1 can effectively coordinate the expression of both Nos1 and COX subunit genes, thereby coordinating a controlled and stable interplay between energy utilization of synaptic transmission and energy production. These findings beg many other questions. For example, do other transcription factors participate in the co-regulation of Nos1 and COX? AP1, Sp1, CREB or NF-kB binding sites have been reported for Nos1 promoters in humans and rats [17,54], but they have not been functionally characterized in neurons. Their roles in regulating energy metabolism, if present, have not been defined. Other likely candidates for co-regulation are nuclear respiratory factor 2 (NRF-2) and the coactivator peroxisome proliferator-activated receptor-γ coactivator 1α (PGC-1α). Previously, we have shown that NRF-2 also regulates all 13 COX subunits from the two genomes [18,20]. NRF-2 binding sites appear to be present on the murine Nos1 promoter (our unpublished observations). PGC-1α is known to stimulate a powerful induction of both NRF-1 and NRF-2 [55] as well as binds to and co-activates NRF-1 in stimulating Tfam expression [20]. In primary neurons, PGC-1α responds to changing neuronal activity earlier than those of NRF-1 and NRF-2 [33,56]. Thus, for co-regulating neuronal activity and energy metabolism, the transcriptional mechanism is likely to engage many, if not all, of these key factors. Research is underway to explore these possibilities.
ACKNOWLEDGMENTS
It gives us great pleasure to thank Dr. Richard Scarpulla for his generous gift of NRF-1 antibodies and pSG5NRF-1 plasmid and for his critical reading of the paper. We thank Dr. P. Aebischer for his gift of pLVTHM and Drs. S. Ongwijitwat and H. Meng for assisting in the construction of shRNA vectors. Supported by NIH Grant EY018441.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Ignarro LJ. A novel signal transduction mechanism for transcellular communication. Hypertension. 1990;5:477–483. doi: 10.1161/01.hyp.16.5.477. [DOI] [PubMed] [Google Scholar]
- 2.Moncada S, Higgs A. The L-arginine-nitric oxide pathway. N. Engl. J. Med. 1993;329:20002–20012. doi: 10.1056/NEJM199312303292706. [DOI] [PubMed] [Google Scholar]
- 3.Garthwaite J. Glutamate, nitric oxide and cell-cell signalling in the nervous system. Trends Neurosci. 1991;14:60–67. doi: 10.1016/0166-2236(91)90022-m. [DOI] [PubMed] [Google Scholar]
- 4.Garthwaite J. Concepts of neural nitric oxide-mediated transmission. Eur.J.Neurosci. 2008;11:2783–2802. doi: 10.1111/j.1460-9568.2008.06285.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nathan C, Xie QW. Nitric oxide synthases: roles, tolls, and controls. Cell. 1994;78:915–918. doi: 10.1016/0092-8674(94)90266-6. [DOI] [PubMed] [Google Scholar]
- 6.Garthwaite J, Boulton CL. Concepts of neural nitric oxide-mediated transmission. Annu. Rev. Physiol. 1995;57:683–706. doi: 10.1146/annurev.ph.57.030195.003343. [DOI] [PubMed] [Google Scholar]
- 7.Dawson TM, Dawson VL. Nitric oxide synthase: Role as a transmitter/mediator in the brain and endocrine system. Ann. Rev. Med. 1996;47:219–227. doi: 10.1146/annurev.med.47.1.219. [DOI] [PubMed] [Google Scholar]
- 8.Wong-Riley MT. Cytochrome oxidase: an endogenous metabolic marker for neuronal activity. Trends Neurosci. 1989;12:94–101. doi: 10.1016/0166-2236(89)90165-3. [DOI] [PubMed] [Google Scholar]
- 9.Wong-Riley MTT. Primate visual cortex: dynamic metabolic organization and plasticity revealed by cytochrome oxidase. In: Peters A, Rockland K, editors. Cerebral Cortex, Primary Visual Cortex in Primates. Vol. 10. New York: Plenum Press; 1994. pp. 141–200. [Google Scholar]
- 10.Wikström M, Krab K, Saraste M. A Synthesis. New York: Academic Press; 1981. Cytochrome Oxidase. [Google Scholar]
- 11.Wong-Riley MT, Huang Z, Liebl W, Nie F, Xu H, Zhang C. Neurochemical organization of the macaque retina: effect of TTX on levels and gene expression of cytochrome oxidase and nitric oxide synthase and on the immunoreactivity of Na+ K+ ATPase and NMDA receptor subunit I. Vision Res. 1998;38:1455–1477. doi: 10.1016/s0042-6989(98)00001-7. [DOI] [PubMed] [Google Scholar]
- 12.Zhang C, Wong-Riley MT. Do nitric oxide synthase, NMDA receptor subunit R1 and cytochrome oxidase co-localize in the rat central nervous system? Brain Res. 1996;729:205–215. [PubMed] [Google Scholar]
- 13.Dhar SS, Ongwijitwat S, Wong-Riley MT. Nuclear respiratory factor 1 regulates all ten nuclear-encoded subunits of cytochrome c oxidase in neurons. J. Biol. Chem. 2008;283:3120–3129. doi: 10.1074/jbc.M707587200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dhar SS, Wong-Riley MT. Coupling of energy metabolism and synaptic transmission at the transcriptional level: role of nuclear respiratory factor 1 in regulating both cytochrome c oxidase and NMDA glutamate receptor subunit genes. J. Neurosci. 2009;29:483–492. doi: 10.1523/JNEUROSCI.3704-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Evans MJ, Scarpulla RC. NRF-1, a trans-activator of nuclear-encoded respiratory genes in animal cells. Genes Dev. 1990;4:1023–1034. doi: 10.1101/gad.4.6.1023. [DOI] [PubMed] [Google Scholar]
- 16.Scarpulla RC. Nuclear control of respiratory chain expression by nuclear respiratory factors and PGC-1-related coactivator. Ann. N. Y. Acad. Sci. 2008;1147:321–334. doi: 10.1196/annals.1427.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hall AV, Antoniou H, Wang Y, Cheung AH, Arbus AM, Olson SL, Lu WC, Kau CL, Marsden PA. Structural organization of the human neuronal nitric oxide synthase gene (NOS1) J. Biol. Chem. 1994;52:33082–33090. [PubMed] [Google Scholar]
- 18.Ongwijitwat S, Wong-Riley MT. Is nuclear respiratory factor 2 a master transcriptional coordinator for all ten nuclear-encoded cytochrome c oxidase subunits in neurons? Gene. 2005;360:65–77. doi: 10.1016/j.gene.2005.06.015. [DOI] [PubMed] [Google Scholar]
- 19.Gleyzer N, Vercauteren K, Scarpulla RC. Control of mitochondrial transcription specificity factors (TFB1M and TFB2M) by nuclear respiratory factors (NRF-1 and NRF-2) and PGC-1 family coactivators. Mol Cell Biol. 2005;25:1354–1366. doi: 10.1128/MCB.25.4.1354-1366.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ongwijitwat S, Liang HL, Graboyes EM, Wong-Riley MT. Nuclear respiratory factor 2 senses changing cellular energy demands and its silencing down-regulates cytochrome oxidase and other target gene mRNAs. Gene. 2006;374:39–49. doi: 10.1016/j.gene.2006.01.009. [DOI] [PubMed] [Google Scholar]
- 21.Yang SJ, Liang HL, Wong-Riley MT. Activity-dependent transcriptional regulation of nuclear respiratory factor-1 in cultured rat visual cortical neurons. Neuroscience. 2006;141:1181–1192. doi: 10.1016/j.neuroscience.2006.04.063. [DOI] [PubMed] [Google Scholar]
- 22.Tomlinson DC, Hurst CD, Knowles MA. Knockdown by shRNA identifies S249C mutant FGFR3 as a potential therapeutic target in bladder cancer. Oncogene. 2007;26:5889–5899. doi: 10.1038/sj.onc.1210399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sun D, Melegari M, Sridhar S, Rogler CE, Zhu L. Multi-miRNA hairpin method that improves gene knockdown efficiency and provides linked multi-gene knockdown. Biotechniques. 2006;41:59–63. doi: 10.2144/000112203. [DOI] [PubMed] [Google Scholar]
- 24.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using realtime quantitative PCR and the 2(-delta delta C(T)) Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 25.Virbasius CA, Virbasius JV, Scarpulla RC. NRF-1, an activator involved in nuclear-mitochondrial interactions, utilizes a new DNA-binding domain conserved in a family of developmental regulators. Genes Dev. 1993;7:2431–2445. doi: 10.1101/gad.7.12a.2431. [DOI] [PubMed] [Google Scholar]
- 26.Dhar SS, Liang HL, Wong-Riley MT. Nuclear respiratory factor 1 co-regulates AMPA glutamate receptor subunit 2 and cytochrome c oxidase: Tight coupling of glutamatergic transmission and energy metabolism in neurons. J Neurochem. 2009;108:1595–1606. doi: 10.1111/j.1471-4159.2009.05929.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Boissel JP, Schwarz PM, Förstermann U. Neuronal-type NO synthase: transcript diversity and expressional regulation. Nitric Oxide. 1998;5:337–349. doi: 10.1006/niox.1998.0189. [DOI] [PubMed] [Google Scholar]
- 28.Bros M, Boissel JP, Gödtel-Armbrust U, Förstermann U. Transcription of human neuronal nitric oxide synthase mRNAs derived from different first exons is partly controlled by exon 1-specific promoter sequences. Genomics. 2006;4:463–473. doi: 10.1016/j.ygeno.2005.11.013. [DOI] [PubMed] [Google Scholar]
- 29.Thierry-Mieg D, Thierry-Mieg J. AceView: a comprehensive cDNA-supported gene and transcripts annotation. Genome Biol. 2006;7 Suppl 1:1–4. doi: 10.1186/gb-2006-7-s1-s12. S12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lang G, Gombert WM, Gould HJ. A transcriptional regulatory element in the coding sequence of the human Bcl-2 gene. Immunology. 2005;114:25–36. doi: 10.1111/j.1365-2567.2004.02073.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Barthel KK, Liu X. A transcriptional enhancer from the coding region of ADAMTS5. PLoS ONE. 2008;3:e2184. doi: 10.1371/journal.pone.0002184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tumpel S, Cambronero F, Sims C, Krumlauf R, Wiedemann LM. A regulatory module embedded in the coding region of Hoxa2 controls expression in rhombomere 2. Proc Natl Acad Sci U S A. 2008;105:20077–20082. doi: 10.1073/pnas.0806360105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liang HL, Wong-Riley MT. Activity-dependent regulation of nuclear respiratory factor-1, nuclear respiratory factor-2, and peroxisome proliferator-activated receptor gamma coactivator-1 in neurons. Neuroreport. 2006;17:401–405. doi: 10.1097/01.wnr.0000204980.98876.11. [DOI] [PubMed] [Google Scholar]
- 34.Liang HL, Ongwijitwat S, Wong-Riley MT. Bigenomic functional regulation of all 13 cytochrome c oxidase subunit transcripts in rat neurons in vitro and in vivo. Neuroscience. 2006;140:177–190. doi: 10.1016/j.neuroscience.2006.01.056. [DOI] [PubMed] [Google Scholar]
- 35.Bogdan C. Nitric oxide and the regulation of gene expression. Trends Cell Biol. 2001;11:66–75. doi: 10.1016/s0962-8924(00)01900-0. [DOI] [PubMed] [Google Scholar]
- 36.Seress L, Abrahám H, Hajnal A, Lin H, Totterdell S. NOS-positive local circuit neurons are exclusively axo-dendritic cells both in the neo- and archicortex of the rat brain. Brain Res. 2005;1056:183–190. doi: 10.1016/j.brainres.2005.07.034. [DOI] [PubMed] [Google Scholar]
- 37.Wong-Riley MTT, Nie F, Hevner RF, Liu S. Brain cytochrome oxidase: functional significance and bigenomic regulation in the CNS. In: Gonzalez-Lima F, editor. Cytochrome oxidase in Neuronal Metabolism and Alzheimer's Disease. New York: Plenum Press; 1998. pp. 1–53. [Google Scholar]
- 38.de Vera ME, Shapiro RA, Nussler AK, Mudgett JS, Simmons RL, Morris SM, Billiar TR, Geller DA. Transcriptional regulation of human inducible nitric oxide synthase (NOS2) gene by cytokines: Initial analysis of the human NOS2 promoter. Proc. Natl. Acad. Sci. USA. 1996;93:1054–1059. doi: 10.1073/pnas.93.3.1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kröncke KD, Fehsel K, Kolb-Bachofen V. Inducible nitric oxide synthase in human diseases. Clin. Exp. Immunol. 1998;113:147–156. doi: 10.1046/j.1365-2249.1998.00648.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mungrue IN, Bredt DS, Stewart DJ, Husain M. From molecules to mammals: what's NOS got to do with it? Acta Physiol. Scand. 2003;179:123–135. doi: 10.1046/j.1365-201X.2003.01182.x. [DOI] [PubMed] [Google Scholar]
- 41.Li H, Wallerath T, Forstermann U. Physiological mechanisms regulating the expression of endothelial-type NO synthase. Nitric Oxide. 2002;7:132–147. doi: 10.1016/s1089-8603(02)00127-1. [DOI] [PubMed] [Google Scholar]
- 42.Bates TE, Loesch A, Burnstock G, Clark JB. Mitochondrial nitric oxide synthase: a ubiquitous regulator of oxidative phosphorylation? Biochem Biophys Res Commun. 1996;218:40–44. doi: 10.1006/bbrc.1996.0008. [DOI] [PubMed] [Google Scholar]
- 43.Gutsaeva DR, Carraway MS, Suliman HB, Demchenko IT, Shitara H, Yonekawa H, Piantadosi CA. Transient hypoxia stimulates mitochondrial biogenesis in brain subcortex by a neuronal nitric oxide synthase-dependent mechanism. J. Neurosci. 2008;28:2015–2024. doi: 10.1523/JNEUROSCI.5654-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kanai AJ, Pearce LL, Clemens PR, Birder LA, VanBibber MM, Choi SY, de Groat WC, Peterson J. Identification of a neuronal nitric oxide synthase in isolated cardiac mitochondria using electrochemical detection. Proc. Natl. Acad. Sci. U S A. 2001;98:14126–14131. doi: 10.1073/pnas.241380298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Collman JP, Dey A, Decreau RA, Yang Y, Hosseini A, Solomon EI, Eberspacher TA. Interaction of nitric oxide with a functional model of cytochrome c oxidase. Proc. Natl. Acad. Sci. U S A. 2008;105:9892–9896. doi: 10.1073/pnas.0804257105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nie F, Wong-Riley MT. Mitochondrial-, nuclear-encoded subunits of cytochrome oxidase in neurons: differences in compartmental distribution, correlation with enzyme activity, and regulation by neuronal activity. J Comp. Neurol. 1996;373:139–155. doi: 10.1002/(SICI)1096-9861(19960909)373:1<139::AID-CNE12>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 47.Wong-Riley MTT, Jacobs P. AMPA glutamate receptor subunit 2 in normal and visually deprived macaque visual cortex. Vis. Neurosci. 2002;19:563–573. doi: 10.1017/s0952523802195022. [DOI] [PubMed] [Google Scholar]
- 48.Zhang C, Wong-Riley M. Expression and regulation of NMDA receptor subunit R1 and neuronal nitric oxide synthase in cortical neuronal cultures: correlation with cytochrome oxidase. J Neurocytol. 1999;28:525–539. doi: 10.1023/a:1007053204929. [DOI] [PubMed] [Google Scholar]
- 49.Zhang C, Wong-Riley MT. Depolarizing stimulation upregulates GA-binding protein in neurons: a transcription factor involved in the bigenomic expression of cytochrome oxidase subunits. Eur. J. Neurosci. 2000;12:1013–1023. doi: 10.1046/j.1460-9568.2000.00997.x. [DOI] [PubMed] [Google Scholar]
- 50.Tascedda F, Molteni R, Racagni G, Riva MA. Acute and chronic changes in K+-induced depolarization alter NMDA and nNOS gene expression in cultured cerebellar granule cells. Mol. Brain Res. 1996;40:171–174. doi: 10.1016/0169-328x(96)00111-8. [DOI] [PubMed] [Google Scholar]
- 51.Scarpulla RC. Nuclear control of respiratory gene expression in mammalian cells. J Cell Biochem. 2006;97:673–683. doi: 10.1002/jcb.20743. [DOI] [PubMed] [Google Scholar]
- 52.Scarpulla RC. Transcriptional paradigms in mammalian mitochondrial biogenesis and function. Physiol. Rev. 2008;88:611–638. doi: 10.1152/physrev.00025.2007. [DOI] [PubMed] [Google Scholar]
- 53.Huo L, Scarpulla RC. Mitochondrial DNA instability and peri-implantation lethality associated with targeted disruption of nuclear respiratory factor 1 in mice. Mol Cell Biol. 2001;21:644–654. doi: 10.1128/MCB.21.2.644-654.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sasaki M, Gonzalez-Zulueta M, Huang H, Herring WJ, Ahn S, Ginty DD, Dawson VL, Dawson TM. Dynamic regulation of neuronal NO synthase transcription by calcium influx through a CREB family transcription factor-dependent mechanism. Proc. Natl. Acad. Sci. U S A. 2000;97:8617–8622. doi: 10.1073/pnas.97.15.8617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wu Z, Puigserver P, Andersson U, Zhang C, Adelmant G, Mootha V, Troy A, Cinti S, Lowell B, Scarpulla RC, Spiegelman BM. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell. 1999;98:115–124. doi: 10.1016/S0092-8674(00)80611-X. [DOI] [PubMed] [Google Scholar]
- 56.Meng H, Liang HL, Wong-Riley M. Quantitative immuno-electron microscopic analysis of depolarization-induced expression of PGC-1alpha in cultured rat visual cortical neurons. Brain Res. 2007;1175:10–16. doi: 10.1016/j.brainres.2007.07.063. [DOI] [PubMed] [Google Scholar]