

Abstract
Considerable data supports the hypothesis that mitochondrial abnormalities link gene defects and/or environmental insults to the neurodegenerative process The interaction of oxidants with calcium and the mitochondrial enzymes of the tricarboxylic acid (TCA) cycle are central to that relationship. Abnormalities that were discovered in brains or fibroblasts from patients with Alzheimer's Disease (AD) have been modeled in vitro and in vivo to assess their pathophysiological importance and to determine how they might be reversed. The conclusions are consistent with the hypothesis that the AD-related abnormalities result from oxidative stress. The selection of compounds for reversal is complex because the actions of the relevant compounds vary under different conditions such as cell redox states and acute vs chronic changes. However, the models that have been developed are useful for testing the effectiveness of the potential medications. The results suggest that the reversal of the mitochondrial deficits and a reduction in oxidative stress will reduce the clinical and pathological changes and benefit patients.
Keywords: Ketoglutarate dehydrogenase, Calcium, Alzheimer's disease, Oxidative stress, Fibroblasts
Introduction
Alzheimer's disease (AD) is a common neurodegenerative disease that effects one in eight persons over the age of 65 and more than half of the people over the age of 85. Approximately 5.2 million Americans have AD and the cost is estimated to be 148 billion dollars a year. As the population ages, the problem will become exaggerated. Although a small percentage of AD cases (about 5%) are genetically linked, the cause is generally unknown (Alz.org). Even in the small percentage where the cause is known to be genetic defects, the linkage of gene defects to neuronal dysfunction is still unknown. The studies that are described here suggest that mitochondrial abnormalities are central to the disease process.
Impaired metabolism and oxidative stress in AD
Evidence for impaired mitochondrial function and a reduction in brain glucose metabolism in AD is overwhelming. AD does not occur without a reduction in metabolism. Reduced metabolism in AD has been known for decades. Reductions correlate with APOEε4 gene dose and are progressive in late-middle-aged persons.1 These reductions are also apparent in young adult APOEε4 heterozygotes for more than four decades before the anticipated median onset of dementia,2 years before the expected onset of the major histopathological features (i.e., the plaques and tangles), and prior to the initial regional appearance of fibrillar amyloid deposition.1, 3 Metabolic changes have been reported in brains of patients who are mildly cognitively impaired.4 Reductions in metabolism can be readily related to cognitive decline. Even mild hypoglycemia or hypoxia diminishes brain function; memory and judgment are the most sensitive.5 (For more discussion of the effects of reductions in cerebral metabolic rate and cognitions, see the chapter by Mosconi in this volume).
Evidence for oxidative stress in AD is overwhelming. Oxidative stress is closely coupled to metabolism since disruptions in metabolism lead to increased free radical production and probably impair free radical removal (“quenching”). There are many types and sources of oxidative stress in the brain. These have been summarized by Brookes,6 and Starkov (this volume) and Perry.7 Common radicals or reactive oxygen species (ROS) include superoxide, hydroxyl radical, nitric oxide, peroxynitrite and hydroperoxyl radicals. Mitochondrial ROS can originate from multiple reactions in the TCA cycle and/or the respiratory chain. Perturbations in metabolism generally exaggerate ROS production. However, ROS cannot be measured in human brain so investigations are limited to the consequences of oxidative stress. Autopsied brains reveal extensive evidence of oxidized proteins, lipids, DNA and RNA.7-9 In general, these modifications are more widespread in the brain than the plaques and tangles, the hallmark pathologies of AD. For example, acrolein, the most reactive among the α, β-unsaturated aldehyde products of lipid peroxidation, can be rapidly incorporated into proteins, thereby generating a carbonyl derivative which is a stable marker of oxidative stress in proteins. Antibodies to acrolein reveals oxidatively modified proteins around the areas of plaques and tangles but also in other areas.10 Lipoxygenase, another marker of oxidative stress, is increased in brains of AD patients.11 In humans, markers of oxidative stress are also increased in Alzheimer CSF.12 In transgenic “amyloid” mice, markers of oxidative stress precede plaque formation in the brain and even occur in the urine before they occur in brain. (For more discussion of oxidative stress, see the chapter by Pratico in this volume).
Oxidative stress and impaired metabolism in the production of AD-like pathology
Considerable data indicates that these changes in oxidative stress and mitochondrial dysfunction are important causative factors in the development and progression of AD.13, 14 Knockout of one allele of manganese superoxide dismutase (MnSOD) in mice crossed with Tg19959 mice elevates protein carbonyl levels, Aβ levels and increase plaque burden in cortex and hippocampus.15 Inhibition of energy metabolism by various pharmacological agents such as insulin, 2-deoxyglucose, 3-nitropropionic acid, or kainic acid elevates β-secretase levels and enhances plaque pathology in Tg2576 transgenic mice.16 Intraocular administration of sub-lethal doses of mitochondrial respiratory inhibitors such as rotenone, 3-nitropropionic acid, sodium azide and oxidant FeCl3 and Aβ42 fibrils, elevate β-secretase levels and β-carboxy terminal fragments in retinal lysates.17 Also, amyloid-β-peptide selectively complexes with divalent cations, copper and zinc; those metal-protein complexes can generate free radicals.9, 18 Thus, a variety of experimental approaches suggest that oxidative stress and impaired metabolism are involved in the pathophysiology of AD.
Changes in the tricarboxylic acid (TCA) cycle and the diminished metabolism in AD
The underlying basis for the decline in glucose metabolism in AD is unknown. The activities of the enzymes of the TCA cycle change in brains from patients that died with autopsy-confirmed AD. The primary source of reducing equivalents in brain is the TCA cycle. The changes in the TCA cycle with AD form an unexpected and intriguing pattern [Figure 1]. The activities of the pyruvate dehydrogenase complex (PDHC) (-41%), isocitrate dehydrogenase complex (ICDHC) (-27%), and the α-ketoglutarate dehydrogenase complex (KGDHC) (-57%) decline significantly (p < 0.01). These enzymes are the entry steps of substrates into the TCA cycle and form the first half of the TCA cycle. They are dehydrogenases and decarboxylases. On the other hand, activities of succinate dehydrogenase (SDH) (complex II) (+44%) and malate dehydrogenase (MDH) (+54%) are elevated (p < 0.01). These enzymes make up the second half of the TCA cycle and are dehydrogenases but not decarboxylases. Activities of the other TCA cycle enzymes are unchanged. All of the changes in TCA cycle enzyme activities correlate with the Clinical Dementia Rating scale (CDR) shortly before the patient died (p < 0.01). The correlation coefficients range from 0.52 (ICDH) to 0.77 (PDHC). The activity of the KGDHC has been studied in a separate group of patients that have APOEε4 genes, a gene known to predispose to the development of AD. In them, the reduction in KGDHC with AD is even more highly correlated to the decline in CDR (0.77 compared to 0.52).14 The reduction in activity of KGDHC has been reported by a number of groups, and there are no-contravening reports in the literature. The simultaneous changes in multiple enzymes of the TCA cycle suggest a coordinate regulation of the TCA cycle at either the protein or gene level. The results suggest that strategies to improve TCA cycle metabolism might benefit AD patients.
Fig. 1.
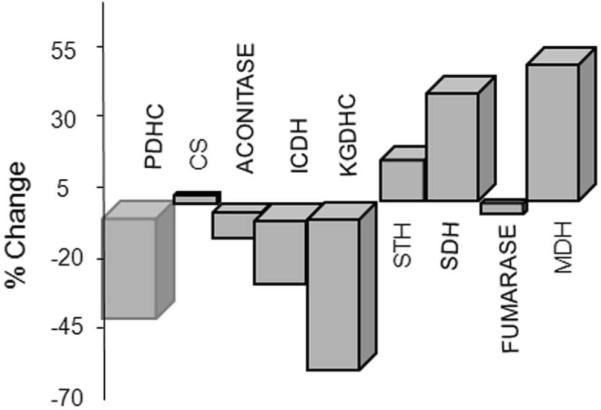
Activities of TCA cycle enzymes change in brains of patients with AD. Activities of PDHC and all of the TCA cycle enzymes were measured in autopsied brains from patients with AD. Data are expressed as percent of control.44
The sensitivity of KGDHC to oxidative stress and the decline in its activity in AD
The underlying basis for the changes in the activities of the TCA cycle enzymes is unknown. Efforts have focused on KGDHC because its activity is among the lowest, and it can be a rate controlling step in the TCA cycle KGDHC is a multi-enzyme complex that consists of an ordered array of multiple copies of three proteins: α-ketoglutarate dehydrogenase (E1k, EC 1.2.4.2), dihydrolipoyl succinyltransferase (E2k, EC 2.3.1.61) and dihydrolipoyl dehydrogenase (E3, EC 1.8.1.4). In the non-genetic forms of AD, the protein levels of the three components are not altered. This suggests that the reduction in AD is related to post-translational modifications. In brains from patients that died bearing the APP670/671 mutation, the protein levels of E1k and E2k are diminished. This could be due to a reduction in protein or to oxidant modification of the antigenic sites that are recognized by the antibodies. The data suggest that the decline KGDHC activities in both genetic and non-genetic forms of AD could be secondary to oxidative stress.
KGDHC is sensitive to a variety of different oxidative stressors. This is true, whether the oxidants are added to the isolated enzyme complex or to cells, or if the oxidant levels in cells or in living mice are altered by inhibiting metabolism or by manipulating antioxidant enzymes such as superoxide dismutase (SOD). KGDHC is inactivated by a variety of oxidants including peroxynitrite, NO,19 hydroxynonenal,20 H2O2 (in mM concentrations),21 chloroamine and sodium hypochlorite (in nM concentrations).22 H2O2 diminishes KGDHC activity in synaptosomes,23 fibroblasts and N2a cells.21, 22 KGDHC in intact mitochondria is inactivated by hydroxynonenal (HNE),24 a marker of oxidative stress that is elevated in brains of both AD patients and animals with impaired brain metabolism due to thiamine-deficiency. Treatment of rat heart mitochondria with HNE selectively inhibits KGDHC and PDHC, while other NADH-linked dehydrogenases and the electron chain complexes are unaffected. The oxidant induced changes can mimic the immunoreactive differences in KGDHC between the genetic and non-genetic forms of AD described above as shown by the selective changes in response to peroxynitrite and NO•. Peroxynitrite and NO inactivate isolated KGDHC and KGDHC in intact cells.25, 26 However, peroxynitrite, but not NO•, diminishes immunoreactivity of E1k and E2k.
KDGHC activity is also reduced in animal and cell models when oxidative stress increases. Transgenic SOD knockout mice have reduced KGDHC activities in their brains.27 KGDHC is diminished in cells that overexpress monoamine oxidase. Increased substrate (i.e., more ROS) exaggerates the reduction in KGDHC.28 Exposure of CHO cells to hyperoxia inactivates KGDHC to a greater extent than numerous other enzymes including SDH, NADH dehydrogenase, and glycerol phosphate dehydrogenase.29, 30 The results suggest that KGDHC is normally regulated by ROS signals that become “stuck” in neurodegenerative diseases.
The sensitivity of KGDHC to oxidants may be modulated by glutathione, the major antioxidant in the mitochondria. Western blots suggest a direct interaction of purified KGDHC with GSSG.31 Treatment of KGDHC with increasing concentrations of GSSG progressively enhances glutathionylation of E1k, E2k and E3 [Figure 2]. Glutathione causes a slower migration of E1k and E2k. It creates a second band of E3, decreases E3 immunoreactivity, increases E3 glutathionylation [Figure 2].31 Thus, the results are consistent with regulation of KGDHC by glutathione. The glutathionylation of the individual subunits suggests that interaction of each subunit with glutathione may contribute to the response of the complex in different ways. Precise interpretation of the consequences of glutathionylation of KGDHC is limited because the data is limited to Western blot analysis of isolated KGDHC.
Fig. 2.
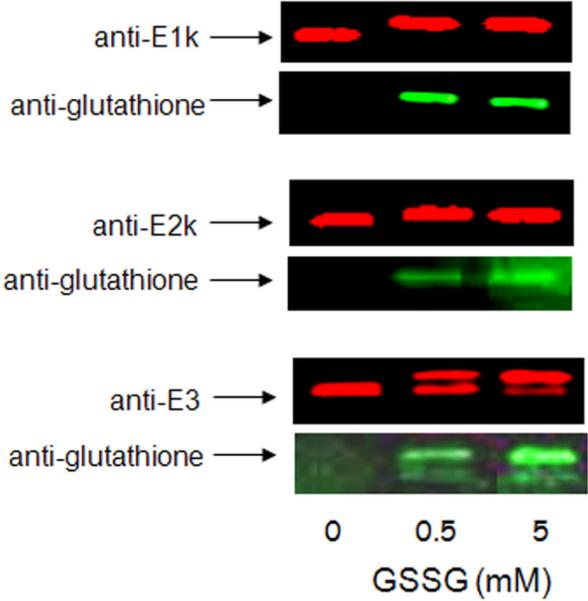
GSSG appears to glutathionylate KGDHC. Purified KGDHC was incubated with GSSG. The samples were run on SDS-PAGE and probed with antibodies to each of the three components of KGDHC and to glutathione. The immunoreactive bands clearly overlapped. Analagous results were obtained with GSH.31
Normally, only a small percent of the total mitochondrial glutathione is present as GSSG. The ratio of GSSG to GSH may change in pathological conditions induced by oxidative stress, and this may promote the glutathionylation of KGDHC. Manipulation of glutathione in cells reveals that the level of glutathione is not as critical as glutathionylation in altering KGDHC activity. Reducing glutathione concentration by about 40% by blocking its synthesis with buthionine sulfoximine (BSO) did not alter the activity of KGDHC [Figure 3]. On the other hand, H2O2 induces a similar change in glutathione concentrations as BSO and reduces KGDHC activity by more than 50%. The change in activity is not exaggerated if BSO and H2O2 are added together. H2O2 is known to reduce glutathione concentrations by promoting movement of glutathione to protein. The fact that much higher concentrations of H2O2 are required to inactivate isolated enzymes than in cells suggest that redox sensitive mechanisms must be involved. Thus, the evidence suggests that redox regulated glutathionylation is one possible mechanism to account for the loss of KGDHC activity with H2O2
Fig. 3.

Lack of correlation of total free cellular glutathione to KGDHC activity in E2k-100 cells. Cells were incubate with either BSO, H2O2 or their combination and total free cellular glutathione was determined.31
Individual components of KGDHC and the response to oxidative stress
Experiments with cells lines with varying levels of E2k reveal that E2k protein levels, complex activity and the response to oxidants do not change in parallel with each other. A reduction in the levels of E2k message and protein to two thirds of normal (E2k67) diminishes KGDHC activity by about one third.32 However, KGDHC activity returns to normal if E2k message and protein are diminished by only one third of normal (E2k30). On the other hand, the response of cells to H2O2 parallels the E2k protein levels [Figure 4].
Fig. 4.
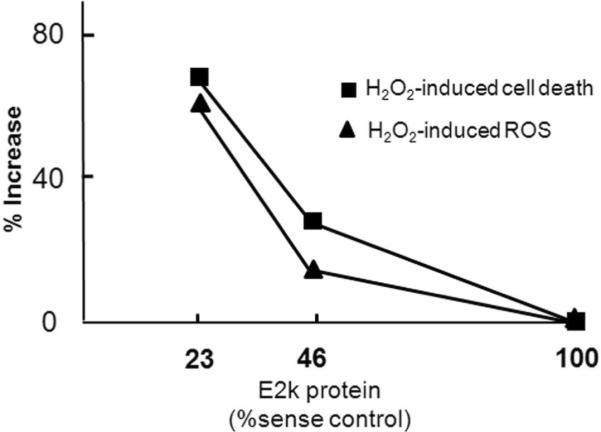
Diminished E2k in HEK cells increases the H2O2 induced ROS and cell death. Cells with various levels of E2k protein subunits were utilized. KGDHC activity declined in the cells with about a 50 percent reduction but returned to normal in the lines with the lowest levels of E2k. The cells were incubated with H2O2 and cell death and fluorescence were determined.32
The H2O2 induced-increase in ROS and cell death is greater in cells with diminished E2k than in controls. E2k likely possesses unique functions related to the ability of cells to respond to oxidants. The individual components of KGDHC also vary in their ability to produce reactive oxygen species. Under reducing conditions KGDHC is the primary source of H2O2 in mitochondria. The ROS originate with E3 33, 34(see Chapter in this volume by Starkov). The results reveal that E2k is important in buffering external ROS and E3 is important in the production of endogenous ROS. The response and role of E1k in oxidative stress has not been examined.
Modification of the interaction of the two TCA cycle enzymes KGDHC and MDH by E2k
Extensive studies suggest co-ordinate gene regulation of the enzymes of glycolysis (see Ratan chapter) and the electron transport chain (see chapter by Scarpulla in this volume). Co-ordinate regulation of the enzymes of the TCA cycle has not been established. MDH activity varies with the E2k levels in cells with variable Ek2 protein levels. In cells with two thirds normal E2k protein and diminished KGDHC activity, MDH activity decreases about 20%. In cells with one third normal E2k, KGDHC activity increases back to nearly normal and MDH activity increases about 20%. Thus, the pattern of changes with varying E2k protein levels is similar for activities of MDH and KGDHC. This pattern differs from what is found in brains from AD patients (i.e., KGDHC activity is reduced and MDH is increased), suggesting that an additional factor such as oxidative stress may be involved in AD.
Experiments to test whether MDH can be modified by the same oxidant-induced changes that alter KGDHC reveal that MDH is only sensitive to oxidants if E2k is diminished [Figure 5].31 H2O2 at concentrations that reduce KGDHC do not alter MDH in cells with a normal complement of E2k and KGDHC activity. However, in cells in which E2k protein levels are diminished by 70%, H2O2 increases the activity of MDH. This occurs even though MDH is already elevated in the E2k deficient lines. The message levels are affected in a similar fashion. These changes are not mediated by glutathione because glutathione levels are not altered by a one hour of H2O2 treatment. Furthermore, diminishing glutathione by a 24 hour treatment with BSO did not alter MDH. Thus, changes in MDH in response to oxidants are modulated by E2k levels, which modify the response of MDH to oxidants at both the message and protein level.s.31
Fig. 5.

E2k modifies the response of MDH activity and message to oxidants. Cells with varying levels of the E2k subunit of KGDHC were incubated with H2O2 and the response of MDH activitity and message levels were determined.31
AD related changes in calcium and oxidative stress
Mitochondria and endoplasmic reticulum (ER) interact closely in the regulation of cellular calcium (see chapter by Rizzutto in this volume). Cellular calcium dynamics are altered in cells from patients with AD.37-40 AD-related changes in calcium regulation are perhaps best studied in cultured cells and the majority of studies have been done in fibroblasts. Such cultured cells from AD patients or from presenilin-1 transgenic mice have exaggerated ER calcium stores35, 36 and diminished mitochondrial calcium uptake.37 Thus, the bombesin (or bradykinin) releasable stores (BRCS) are exaggerated in cells from AD patients. The changes appear disease specific and can be detected in cells before the patients develop AD. To determine if the increase might also be related to oxidant-induced changes, a series of oxidants were screened for their ability to produce AD-like changes in cellular calcium dynamics. Several oxidants induced increases in BRCS. H2O2 was the most effective at exaggerating BRCS without altering cytosolic free calcium.38 The results are consistent with oxidant induced changes being responsible for the AD-related changes in ER calcium in AD cells.
The cellular consequences of reduced KGDHC
To determine the consequences of the diminished KGDHC activities, the reductions have been modeled in cells and animal models by drug, dietary and genetic methods. Impairing KGDHC activity alters metabolism. Specific KGDHC inhibitors [phosphonoethyl ester of succinyl phosphonate (PESP) and the carboxy ethyl ester of succinyl phosphonate (CESP)] alter [1-13C]glucose and [U-13C]glutamate metabolism in intact cerebellar granule neurons. Both inhibitors lead to a decreased formation of [4-13C]glutamate from [1-13C]glucose, a reduction in label in glutamate derived from [1-13C]glucose/ [U-13C]glutamate and a decline in the levels of γ-aminobutyric acid (GABA), aspartate, and alanine. These findings are consistent with the hypothesis that decreased KGDHC activity leads to increased α-ketoglutarate formation which leads to enhanced transamination of α-ketoglutarate with valine, leucine, and GABA and to a new equilibrium position of the aspartate aminotransferase reaction. Overall, the findings suggest that some carbon derived from α-ketoglutarate may bypass the block in the TCA cycle at KGDHC by means of the GABA shunt and/or conversion of valine to succinate.39 Thus, these metabolic measures show that impairing flux through KGDHC alters the pathways of metabolism but does not necessarily compromise cellular bioenergetics.
The key position of KGDHC in mitochondrial function suggests that inhibition of KGDHC will have profound effects not only on metabolism but also on other cell functions. Experiments with a relatively selective inhibitor of KGDHC, α-keto-β-methyl valerate (KMV), show that inhibitors of KGDHC release cytochrome C from the mitochondria to the cytosol, activate caspases and alter ER calcium stores before they cause a decline the mitochondrial membrane potentials (a measure of mitochondrial function).40 Thus, the coupling of KGDHC to cell functions appears to occur in multiple ways in addition to its traditional role in production of NADH equivalents.
The consequences of reduced KGDHC in brain in vivo
Both dietary and genetic methods have been used in mice to model the changes in KGDHC that occur in the brains of patients that died with AD. Dihydrolipoamide dehydrogenase (Dld) encodes the E3 subunit of KGDHC and PDHC. Although homozygous (Dld-/-) mice die in utero, the heterozygous (Dld+/-) mice appear normal and the activities of KGDHC and PDHC are about one half normal. Malondialdehyde, a common measure of oxidative stress, is elevated in the brains of these mice and neurotoxins increase malondialdehyde more in the Dld+/- mice than controls. The Dld+/- mice are more vulnerable to toxin models of PD and HD: 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), malonate and 3-nitropropionic acid (3-NP). Studies of isolated brain mitochondria treated with 3-NP showed that both succinate-supported respiration and membrane potentials are suppressed to a greater extent in Dld+/- mice.41 Thus, even partial deficiency of PDHC and KGDHC can have profound effects on brain function and the brain's ability to respond to challenges.41
Preliminary results with a transgenic, heterozygous mouse model of E2k deficiency (DLST+/-) reveal the consequences of the reduction in gene expression of E2k on brain biochemistry, memory and other behaviors. In these mice, E2k mRNA (-47%), E2k protein (-46%) and KGDHC activity (-37%) are reduced in whole brain homogenates. DLST+/- mice display several behavioral alterations. Rotorod performance at high speeds declines 40%. Learning of a discriminative fear conditioning task is also disrupted. These results show that a reduction in KGDHC activity can impair memory in the emotional domain, a finding that may be relevant to the disturbances in emotional memory that are common in AD patients. The DLST+/- mice may be a potentially useful animal model for developing and screening new therapies to treat AD and other related disorders with impaired cognition associated with lowered metabolism.42
Thiamine (vitamin B1) deficiency (TD) models the mild impairment of oxidative metabolism that accompanies AD. TD produces memory deficits and cholinergic dysfunction, characteristic features of AD, in people and in animals [for references see 43]. All three of the major thiamine-dependent enzymes in brain are altered in AD. These include PDHC, KGDHC 44 and transketolase.45 TD reduces the activities of thiamine-dependent enzymes throughout the brain, but produces a time-dependent neuronal loss only in select brain regions. The changes in the brain region with neuronal loss are similar to those in AD and include glial activation, inflammation, abnormalities in oxidative metabolism, oxidative stress and clusters of degenerating neurons.43 While TD increases mRNA levels for eNOS (3.7 fold), IL-1β (43 fold), IL-6 (44 fold) and TNF-α (64 fold) in regions of neuronal loss, the only one of these mRNA levels that increase in areas of cortex without neuronal loss is TNF-α (22 fold).46 The results demonstrate that TD induces quantitative, distinct changes in inflammatory responses and oxidative stress in vulnerable and non-vulnerable regions that may underlie the selective vulnerability characteristic of this condition [for references see 43].
Diminishing thiamine-dependent enzymes alters brain pathology in Tg19959 transgenic “Alzheimer” mice that are genetically engineered to over express a double mutant form of the amyloid precursor protein (APP) [Figure 6].47 TD exacerbates amyloid plaque pathology in these transgenic mice and enlarges the area occupied by plaques in cortex, hippocampus and thalamus by 50%, 200% and 200%, respectively. TD increases Aβ1-42 levels by about three-fold, β-CTF (C99) levels by 33% and β-secretase (BACE1) protein levels by 43%. TD induces inflammation in areas of plaque formation. Thus, the induction of mild impairment of oxidative metabolism, oxidative stress and inflammation by TD alters metabolism of APP and/or Aβ47
Fig. 6.

Thiamine deficiency accelerates accumulation of compact plaques. Mice that were plaque competent but did not yet have plaques were made thiamine deficient for 10 days. APP processing was altered and plaque accumulation was accelerated.47
Difficulties in selecting antioxidants to protect KGDHC or BRCS
If the changes in KGDHC and the other TCA cycle enzymes are key links in the pathophysiology of AD, approaches that either block the inactivation or re-activate the protein may be beneficial. The data described above suggest that antioxidants would be beneficial. However, the appropriate way to select the right antioxidant is not established, and the selection of antioxidant is not simple even for well-studied oxidants such as H2O2. One reason for the complexity is that every antioxidant is an oxidant under different conditions. If cells are treated with Ginkgo biloba48 or resveratrol49 in the absence of added oxidant, ROS production increases. If oxidant is added externally to the same cells, Ginkgo biloba or resveratrol serve as antioxidants.48, 49 Although various ROS and their reduction by antioxidants can be estimated in cells with different fluorescent probes,6 this technique does not necessarily predict that a target of interest, such as KGDHC, is protected. Trolox blocks the H2O2-induced increase in dichloro-difluorofluorescein signal that is highly correlated to the loss of KGDHC activity. Nevertheless, under the same conditions the trolox does not protect KGDHC from H2O2-induced inactivation [Figure 7]. By contrast, chronic addition of trolox greatly increases KGDHC activity.21 Thus, precise methods for selective antioxidant therapies to reactivate KGDHC that will translate to animals and to patients require further development.
Fig. 7.

Trolox diminishes H2O2-induced ROS but does not protect KGDHC. Cells were incubated with concentrations of Trolox that diminished H2O2 induced increases in cDCF. These concentrations of Trolox did not protect KGDHC from H2O221
Choosing an antioxidant to normalize calcium metabolism in AD models or patients is equally difficult. Measures of BRCS also reveal these problems. Since BRCS increase with AD, a reasonable goal is to reduce BRCS. Chronic treatment with trolox more than doubles BRCS, while N-acetyl cysteine (NAC) and DMSO diminish BRCS. However, the H2O2 induced increase in BRCS is diminished by Trolox, exaggerated by NAC and not affected by DMSO.21 α-Keto-β-methyl-valerate (KMV) is an effective antioxidant for reversing the effects of H2O2 on BRCS. Similar concentrations of KMV diminish the BRCS in cells from AD patients to those of controls.38 Thus, compounds selected to block the effects of H2O2 induced ROS on BRCS make AD cells more like controls [Figure 8]. Although the high concentrations that are required suggest that KMV is a poor compound to test in vivo, the results show that compounds that block oxidant induced changes in BRCS can reverse AD associated changes.
Fig. 8.

KMV diminishes bombesin-releasable calcium stores (BRCS) in oxidant treated fibroblasts and in AD fibroblasts. H2O2 increases BRCS. The increases can be diminished by KMV. The AD related increases in BRCS can also be diminished by KMV. Bars with different symbols are significantly different from each other.38
Studies in animal models of AD suggest that antioxidant strategies may be beneficial in preventing plaque formation. A cross of plaque competent mice with mice bearing disrupted iNOS alleles reduces the levels of protein tyrosine nitration products, lowers the concentration of amyloid-β-peptide and diminishes the cerebral amyloid plaque burden.50 Vitamin E diminishes Aβ levels and amyloid deposition in young plaque competent mice, but not in old mice.51 The problem is that vitamin E does little for human AD; it has been extensively studied without reproducible success. Preliminary studies indicate the antioxidant resveratrol (trans-3, 5, 4- trihydroxystilbene), a polyphenol, can also diminish plaque formation but only in limited regions. Resveratrol possesses antioxidant, neuroprotective and anticancer properties. Chronic consumption of resveratrol (0.2% in diet) increases the well-being of mice fed a high calorie diet.52 This treatment paradigm decreases plaque numbers and percent area occupied by plaques in medial cortex (-48%) and striatum (-89%). Resveratrol also decreases glutathione and increases levels of its precursor cysteine.53 The combination of resveratrol with compounds that enhance mitochondrial functions may be even more effective.
Summary
These cell and animal models support the hypothesis that mitochondrial changes are central to the pathophysiology of AD. The findings suggest that many of the changes in AD may be secondary to mitochondrial abnormalities including deficits in key enzymes of the TCA cycle. The cellular and molecular changes in these models provide insight into the pathophysiology of AD and offer models in which to test possible therapeutic strategies and particular drugs including the strategies proposed in the chapters by Blass (on stimulating metabolism in presence of an antioxidant), Ratan (on altering cascades of gene responses) and Beal (driving mitochondrial function by specific small molecules).
ACKNOWLEDGEMENTS
This work was supported by NIH grants AG14600, AG11921 and AG14930 and the Burke Medical research Institute.
ABBREVIATIONS
- AD
Alzheimer's disease
- APP
Amyloid precursor protein
- BRCS
Bombesin (or bradykinin) releasable calcium stores
- CDR
Clinical dementia rating
- CS
Citrate synthase (EC 4.1.3.7)
- ICDH
Isocitric acid dehydrogenase (EC 1.1.1.41)
- KGDHC
α-Ketoglutaric acid dehydrogenase complex (KGDHC; EC 1.2.4.2, EC 2.3.1.61, EC 1.6.4.3)
- KMV
α-Keto-β-methyl valerate
- MDH
Malate dehydrogenase (EC 1.1.1.37)
- NAC
N-Acetylcysteine
- PDHC
Pyruvate dehydrogenase complex (EC 1.2.4.1, EC 2.3.1.12, EC 1.6.4.3)
- ROS
Reactive oxygen species
- SDH
Succinate dehydrogenase (EC 1.3.99.1)
- STH
Succinate thiokinase (EC 6.2.1.4)
- TCA
Tricarboxylic acid
REFERENCES
- 1.Reiman EM, et al. 2005Correlations between apolipoprotein E epsilon4 gene dose and brain-imaging measurements of regional hypometabolism Proc Natl Acad Sci U S A 1028299-302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Reiman EM, et al. 2004Functional brain abnormalities in young adults at genetic risk for late-onset Alzheimer's dementia Proc Natl Acad Sci U S A 101284-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Buckner RL, et al. 2005Molecular, structural, and functional characterization of Alzheimer's disease: evidence for a relationship between default activity, amyloid, and memory J Neurosci 257709-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mosconi L, et al. 2004MCI conversion to dementia and the APOE genotype: a prediction study with FDG-PET Neurology 632332-40 [DOI] [PubMed] [Google Scholar]
- 5.Gibson GE, et al. 1981Brain dysfunction in mild to moderate hypoxia Am J Med 701247-54 [DOI] [PubMed] [Google Scholar]
- 6.Brookes PS.2006Mitochondrial production of oxidants and their role in the regulation of cellular processes . Springer; New York: 2006. New York. [Google Scholar]
- 7.Zhu X, et al. 2007Causes of oxidative stress in Alzheimer disease Cell Mol Life Sci 642202-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mancuso C, et al. 2007Mitochondrial dysfunction, free radical generation and cellular stress response in neurodegenerative disorders Front Biosci 121107-23 [DOI] [PubMed] [Google Scholar]
- 9.Pappolla MA, et al. 1998Evidence of oxidative stress and in vivo neurotoxicity of beta-amyloid in a transgenic mouse model of Alzheimer's disease: a chronic oxidative paradigm for testing antioxidant therapies in vivo Am J Pathol 152871-7 [PMC free article] [PubMed] [Google Scholar]
- 10.Calingasan NY, Uchida K, Gibson GE.1999Protein-bound acrolein: a novel marker of oxidative stress in Alzheimer's disease J Neurochem 72751-6 [DOI] [PubMed] [Google Scholar]
- 11.Pratico D, et al. 200412/15-lipoxygenase is increased in Alzheimer's disease: possible involvement in brain oxidative stress Am J Pathol 1641655-62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yao Y, et al. 2005Elevation of 12/15 lipoxygenase products in AD and mild cognitive impairment Ann Neurol 58623-6 [DOI] [PubMed] [Google Scholar]
- 13.Ojaimi J, et al. 1999Mitochondrial respiratory chain activity in the human brain as a function of age Mech Ageing Dev 11139-47 [DOI] [PubMed] [Google Scholar]
- 14.Gibson GE, et al. 2000Mitochondrial damage in Alzheimer's disease varies with apolipoprotein E genotype Ann Neurol 48297-303 [PubMed] [Google Scholar]
- 15.Li F, et al. 2004Increased plaque burden in brains of APP mutant MnSOD heterozygous knockout mice J Neurochem 891308-12 [DOI] [PubMed] [Google Scholar]
- 16.Velliquette RA, O'Connor T, Vassar R.2005Energy inhibition elevates beta-secretase levels and activity and is potentially amyloidogenic in APP transgenic mice: possible early events in Alzheimer's disease pathogenesis J Neurosci 2510874-83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xiong K, et al. 2007Mitochondrial respiratory inhibition and oxidative stress elevate beta-secretase (BACE1) proteins and activity in vivo in the rat retina Exp Brain Res 181435-46 [DOI] [PubMed] [Google Scholar]
- 18.Huang X, et al. 1999The A beta peptide of Alzheimer's disease directly produces hydrogen peroxide through metal ion reduction Biochemistry 387609-16 [DOI] [PubMed] [Google Scholar]
- 19.Park LC, et al. 1999Metabolic impairment induces oxidative stress, compromises inflammatory responses, and inactivates a key mitochondrial enzyme in microglia J Neurochem 721948-58 [DOI] [PubMed] [Google Scholar]
- 20.Humphries KM, Szweda LI.1998Selective inactivation of alpha-ketoglutarate dehydrogenase and pyruvate dehydrogenase: reaction of lipoic acid with 4-hydroxy-2-nonenal Biochemistry 3715835-41 [DOI] [PubMed] [Google Scholar]
- 21.Gibson GE, et al. 2002Oxidative stress increases internal calcium stores and reduces a key mitochondrial enzyme Biochim Biophys Acta 1586177-89 [DOI] [PubMed] [Google Scholar]
- 22.Jeitner TM, Xu H, Gibson GE.2005Inhibition of the alpha-ketoglutarate dehydrogenase complex by the myeloperoxidase products, hypochlorous acid and mono-N-chloramine J Neurochem 92302-10 [DOI] [PubMed] [Google Scholar]
- 23.Chinopoulos C, Tretter L, Adam-Vizi V.1999Depolarization of in situ mitochondria due to hydrogen peroxide-induced oxidative stress in nerve terminals: inhibition of alpha-ketoglutarate dehydrogenase J Neurochem 73220-8 [DOI] [PubMed] [Google Scholar]
- 24.Humphries KM, Yoo Y, Szweda LI.1998Inhibition of NADH-linked mitochondrial respiration by 4-hydroxy-2-nonenal Biochemistry 37552-7 [DOI] [PubMed] [Google Scholar]
- 25.Joffe GT, Parks JK, Parker WD., Jr.1998Secondary inhibition of 2-ketoglutarate dehydrogenase complex by MPTP Neuroreport 92781-3 [DOI] [PubMed] [Google Scholar]
- 26.Park LC, et al. 2000Metabolic impairment elicits brain cell type-selective changes in oxidative stress and cell death in culture J Neurochem 74114-24 [DOI] [PubMed] [Google Scholar]
- 27.Hinerfeld D, et al. 2004Endogenous mitochondrial oxidative stress: neurodegeneration, proteomic analysis, specific respiratory chain defects, and efficacious antioxidant therapy in superoxide dismutase 2 null mice J Neurochem 88657-67 [DOI] [PubMed] [Google Scholar]
- 28.Kumar MJ, Nicholls DG, Andersen JK.2003Oxidative alpha-ketoglutarate dehydrogenase inhibition via subtle elevations in monoamine oxidase B levels results in loss of spare respiratory capacity: implications for Parkinson's disease J Biol Chem 27846432-9 [DOI] [PubMed] [Google Scholar]
- 29.Schoonen WG, et al. 1990Hyperoxia-induced clonogenic killing of HeLa cells associated with respiratory failure and selective inactivation of Krebs cycle enzymes Mutat Res 237173-81 [DOI] [PubMed] [Google Scholar]
- 30.Schoonen WG, et al. 1990Respiratory failure and stimulation of glycolysis in Chinese hamster ovary cells exposed to normobaric hyperoxia J Biol Chem 2651118-24 [PubMed] [Google Scholar]
- 31.Shi Q, et al. 2008Novel functions of the alpha-ketoglutarate dehydrogenase complex may mediate diverse oxidant-induced changes in mitochondrial enzymes associated with Alzheimer's disease Biochim Biophys Acta 1782229-38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shi Q, et al. 2005Reduction in the E2k subunit of the alpha-ketoglutarate dehydrogenase complex has effects independent of complex activity J Biol Chem 28010888-96 [DOI] [PubMed] [Google Scholar]
- 33.Starkov AA, et al. 2004Mitochondrial alpha-ketoglutarate dehydrogenase complex generates reactive oxygen species J Neurosci 247779-88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tretter L, Adam-Vizi V.2004Generation of reactive oxygen species in the reaction catalyzed by alpha-ketoglutarate dehydrogenase J Neurosci 247771-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huang HM, et al. 1994Use of cultured fibroblasts in elucidating the pathophysiology and diagnosis of Alzheimer's disease Ann N Y Acad Sci 747225-44 [DOI] [PubMed] [Google Scholar]
- 36.Ito E, et al. 1994Internal Ca2+ mobilization is altered in fibroblasts from patients with Alzheimer disease Proc Natl Acad Sci U S A 91534-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Richardson JS.1993Free Radicals in the Genesis of Alzheimer's Diseasea Annals of the New York Academy of Sciences 69573-76 [DOI] [PubMed] [Google Scholar]
- 38.Huang HM, et al. 2004alpha-keto-beta-methyl-n-valeric acid diminishes reactive oxygen species and alters endoplasmic reticulum Ca(2+) stores Free Radic Biol Med 371779-89 [DOI] [PubMed] [Google Scholar]
- 39.Santos SS, et al. 2006Inhibitors of the alpha-ketoglutarate dehydrogenase complex alter [1-13C]glucose and [U-13C]glutamate metabolism in cerebellar granule neurons J Neurosci Res 83450-8 [DOI] [PubMed] [Google Scholar]
- 40.Huang HM, et al. 2003Inhibition of the alpha-ketoglutarate dehydrogenase complex alters mitochondrial function and cellular calcium regulation Biochim Biophys Acta 1637119-26 [DOI] [PubMed] [Google Scholar]
- 41.Klivenyi P, et al. 2004Mice deficient in dihydrolipoamide dehydrogenase show increased vulnerability to MPTP, malonate and 3-nitropropionic acid neurotoxicity J Neurochem 881352-60 [DOI] [PubMed] [Google Scholar]
- 42.Shi Q, et al. Targeted disruption of the E2k subunit of the mitochondrial α-ketoglutarate dehydrogenase complex diminishes complex activity and impairs memory. Soc. Neuroscience. 2007;37:156–13. [Google Scholar]
- 43.Ke ZJ, Gibson GE.2004Selective response of various brain cell types during neurodegeneration induced by mild impairment of oxidative metabolism Neurochem Int 45361-9 [DOI] [PubMed] [Google Scholar]
- 44.Bubber P, et al. 2005Mitochondrial abnormalities in Alzheimer brain: mechanistic implications Ann Neurol 57695-703 [DOI] [PubMed] [Google Scholar]
- 45.Gibson GE, et al. 1988Reduced activities of thiamine-dependent enzymes in the brains and peripheral tissues of patients with Alzheimer's disease Arch Neurol 45836-40 [DOI] [PubMed] [Google Scholar]
- 46.Karuppagounder SS, et al. 2007Changes in inflammatory processes associated with selective vulnerability following mild impairment of oxidative metabolism Neurobiol Dis 26353-62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Karuppagounder SS, et al. 2008Thiamine deficiency induces oxidative stress and exacerbates the plaque pathology in Alzheimer's mouse model Neurobiology of Aging (in press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gibson GE, Zhang H.2000Effects of Ginkgo biloba (EGb761) on metabolism of reactive oxygen species in fibroblasts from Alzheimer's Disease patients and controls. . . In Advances in Ginkgo Biloba Extract Research Vol. Vol 8. ginkgo biloba Extract (EGB 761) as a neuroprotective agent from basic studies to clinical trials . Christen Y, Ed.: 108-121IPSEN Foundation; Marseille [Google Scholar]
- 49.Galfi P, et al. 2005Divergent effects of resveratrol, a polyphenolic phytostilbene, on free radical levels and type of cell death induced by the histone deacetylase inhibitors butyrate and trichostatin A J Steroid Biochem Mol Biol 9439-47 [DOI] [PubMed] [Google Scholar]
- 50.Nathan C, et al. 2005Protection from Alzheimer's-like disease in the mouse by genetic ablation of inducible nitric oxide synthase J Exp Med 2021163-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sung S, et al. 2004Early vitamin E supplementation in young but not aged mice reduces Abeta levels and amyloid deposition in a transgenic model of Alzheimer's disease Faseb J 18323-5 [DOI] [PubMed] [Google Scholar]
- 52.Baur JA, et al. 2006Resveratrol improves health and survival of mice on a high-calorie diet Nature 444337-42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Karuppagounder SS, et al. 2007Chronic consumption of resveratrol reduces plaque pathology in a transgenic model of Alzheimer's disease. . Soc. Neuroscience 37157–17. [DOI] [PMC free article] [PubMed] [Google Scholar]