

Abstract
A biomimetic approach involving the self-assembly of mineral within the pores of three-dimensional porous polymer scaffolds is a promising strategy to integrate advantages of inorganic and organic phases into a single material for hard tissue engineering. Such a material enhances the ability of progenitor cells to differentiate down an osteoblast lineage in vitro and in vivo, compared with polymer scaffolds. The mechanisms regulating mineral formation in this one-step process, however, are poorly understood, especially the effects of ionic activity products (IP) of the mineralizing solution and incubation time. The aims of this study were to define the structure and composition of mineral formed within the pores of biodegradable polymer scaffolds as a function of IP and time. Three-dimensional poly(lactide-co-glycolide) scaffolds were fabricated by solvent casting/particulate leaching and incubated for 4–16 days in six variants of simulated body fluid whose IPs were varied by adjusting ionic concentrations. Scanning electron microscopy, X-ray diffraction, and Fourier transform infrared spectroscopy demonstrated the formation of carbonated apatite with sub-micrometer sized crystals that grew into spherical globules extending out of the scaffold pore surfaces. As IP increased, more mineral grew on the scaffold pore surfaces, but the apatite became less crystalline and the Ca/P molar ratio decreased from 1.63 ± 0.005 to 1.51 ± 0.002. Since morphology, composition, and structure of mineral are factors that affect cell function, this study demonstrates that the IP of the mineralizing solution is an important modulator of material properties, potentially leading to enhanced control of cell function.
Keywords: biomimetic, mineralization, apatite, ionic activity products, scaffold
INTRODUCTION
Apatites are suitable biomaterials for bone substitution, repair, and regeneration because of their osteoconductive and bioactive properties.1–5 However, the low mechanical properties of apatites prohibit their use as bulk materials in many load-bearing applications. To integrate the biological advantages of these materials into functional biomaterials, apatites have been used as coatings on metals,6–8 ceramics, glasses,9–13 and polymers,14–17 as well as a second phase in polymer/ceramic composites.18,19 Different strategies to deposit surface layers of apatite and create bone-bonding ability have been studied, including plasma spraying,20,21 ion sputtering,22,23 and laser ablation.24,25 These techniques, however, require high temperature treatments under high vacuum, and are not suitable for coating polymer substrates, which melt under those conditions. The high temperatures of those processes also prohibit the incorporation of biologically active factors. Further, hydroxyapatite (HAp) that has been exposed to a heat treatment exhibits high crystallinity, resulting in a structure and properties that differ from bone.26,27
A biomimetic process can provide simple and favorable conditions (e.g., body temperature and atmospheric pressure) for the synthesis of bone-like apatite. In such strategies, apatite self-assembles onto a substrate during incubation in simulated body fluid (SBF), a supersaturated solution, whose ionic concentrations are similar to blood plasma.14,15,28–30 The apatite formed by this method is similar to bone mineral in composition and crystallinity.14,31 This bone-like apatite is therefore expected to provide a better environment for bone cell attachment and proliferation than sintered stoichiometric HAp, since most bone mineral is nonstoichiometric carbonated apatite (CAp).32 Mineral formation via this aqueous process is governed by both surface characteristics of the substrate and incubating parameters, such as solution composition, ionic strength, pH, and temperature, as well as incubation time.33
Hard tissue engineering requires scaffolds to guide and promote controlled cellular growth and differentiation. Poly(lactide-co-glycolide) (PLGA) is one of the polymers used because of its controllable biodegradability and biocompatibility.34 Key characteristics of PLGA scaffolds include sufficient porosity and interconnectivity between the pores, to facilitate cell proliferation, influx of biomolecules and nutrients, efflux of wastes, and vascularization. PLGA scaffolds by themselves, however, exhibit poor osteoconductivity and low mechanical properties.14
To compensate for these design deficiencies, three-dimensional porous PLGA scaffolds have been hybridized with inorganic coatings by mineralization in SBF.14,29,35,36 One of the unique qualities of PLGA is that functional groups that chelate Ca ions can be created by simple hydrolysis, without the need for a separate functionalization step or surface pre-treatment. Therefore, self-assembly of mineral onto PLGA substrates can be achieved via a one-step process, in which samples are both hydrolyzed and mineralized in SBF. Surface-mineralized PLGA exhibits enhanced mechanical properties, yet retains the high porosity of the polymer scaffold.14 In addition, in vitro (cell proliferation, osteoblast differentiation, and cytoskeletal organization) and in vivo (volume fraction of regenerated bone) functions of osteoblast precursor cells are enhanced on mineralized surfaces compared with their polymeric counterparts in both two and three dimensions.35,37
Even though the mineralized layer of these composites is the surface on which interactions with cells occur, there is only limited information on the structure and composition of the mineral formed within the pores of three-dimensional polymer scaffolds as a function of processing parameters. We therefore investigated the regulation of the structure and composition of the mineral formed within the pores of three-dimensional PLGA scaffolds by varying the ionic activity product (IP) of the incubating solution and incubation time. Changes in morphology, chemical composition, and structure of the mineralized surfaces of the scaffolds were analyzed by scanning electron microscopy (SEM), X-ray diffraction (XRD), Fourier transform IR (FTIR), and optical emission spectrometry (OES).
MATERIALS AND METHODS
Synthesis of polymer scaffolds
PLGA with a lactic/glycolic acid ratio of 85/15 (Medisorb, Cincinnati, OH) and chloroform (Sigma-Aldrich, St. Louis, MO) were used for preparing three-dimensional scaffolds by a solvent casting, particulate leaching process, as previously described.14 NaCl (Sigma-Aldrich) particles were sieved in the range of 250–425 μm to dictate the pore size in the scaffolds. The disk-shaped scaffolds were fabricated to a thickness of 3 mm and a diameter of 13 mm.
Mineralization of polymer scaffolds
SBF with different IPs were prepared by changing ionic concentrations (Table I). Each solution was prepared by dissolving reagent-grade chemicals (NaCl, KCl, MgSO4, MgCl2, NaHCO3, CaCl2, KH2PO4) (Sigma-Aldrich) into deionized water. To maintain thermodynamic conditions conducive to heterogeneous nucleation and control the ionic solubility such that solutions were supersaturated, SBFs were buffered to three different pH values (7.4 for 0.75–1.00 × SBF, 7.0 for 1.25–1.50 × SBF, or 6.8 for 1.75–2.00 × SBF) at 37°C with Tris-HCl (Sigma-Aldrich) and 1N NaOH solution. Tris-HCl solution was buffered to pH 7.4 for control groups. All the SBFs and Tris-HCl solutions were sterilized through 0.22-μm nylon filter before use. Each scaffold was immersed in 60 mL of SBF solution at 37°C, which was renewed every 24 h to ensure constant initial IPs. After 4, 8, 12, and 16 days of incubation, the mineralized samples were gently rinsed in deionized water and dried in a vacuum drier at room temperature.
TABLE I.
Concentrations (mM) of Ions and Ionic Activity Products (IP) of Human Blood Plasma and the Simulated Body Fluids (SBF) Used in this Study
| Na+ | K+ | Mg2+ | Ca2+ | Cl− | log IP | pH | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Blood plasma | 142.0 | 5.0 | 1.5 | 2.5 | 103.0 | 27.0 | 1.0 | 0.5 | −96.3 | 7.2–7.4 | |||
| 0.75 × SBF | 105.8 | 3.8 | 1.1 | 1.9 | 114.0 | 3.2 | 0.8 | 0.4 | −98.3 | 7.4 | |||
| 1.00 × SBF | 141.0 | 5.0 | 1.5 | 2.5 | 152.0 | 4.2 | 1.0 | 0.5 | −96.3 | 7.4 | |||
| 1.25 × SBF | 175.3 | 6.3 | 1.9 | 3.1 | 190.0 | 5.3 | 1.3 | 0.6 | −95.6 | 7.0 | |||
| 1.50 × SBF | 211.5 | 7.5 | 2.3 | 3.8 | 228.0 | 6.3 | 1.5 | 0.8 | −94.3 | 7.0 | |||
| 1.75 × SBF | 246.8 | 8.8 | 2.6 | 4.4 | 266.0 | 7.4 | 1.8 | 0.9 | −93.7 | 6.8 | |||
| 2.00 × SBF | 282.0 | 10.0 | 3.0 | 5.0 | 304.0 | 8.4 | 2.0 | 1.0 | −92.8 | 6.8 |
Characterization of mineral
The mass of each scaffold was determined before incubation and after 4, 8, 12, and 16 days of immersion. Morphological changes of the PLGA pore surface and the mineral formed within the scaffolds were analyzed by SEM (Philips XL30 FEG SEM), operating at 15 kV. Specimens were bisected and sputter-coated with gold before SEM examination. Formation of mineral crystals on the scaffolds was determined by XRD (TF-XRD, Rigaku). To avoid altering the properties of the mineral, the mineral layer was not separated from the surface of the PLGA scaffold. The whole mineralized scaffold was adhered on a sample holder along the X-ray incident direction and scanned in the 2θ range of 5–60° with a scanning step of 0.01° at 2°/min, which is a proper scanning speed for the characterization of apatite formed from SBFs.29,38 Cu Kα radiation was used for the diffraction with a voltage of 40 kV and a current of 100 mA. As references, HAp (Sigma-Aldrich No. 289396) and CAp with 6 wt % of (generous gift from Dr. Michael Morris at the University of Michigan) were scanned under the same conditions.
Compositional analyses via FTIR were carried out on a Perkin Elmer Spectrum BX FTIR in the wave number range of 2000–400 cm−1. PLGA scaffolds with and without mineral were ground into fine particles, mixed with KBr (FTIR grade) at a mass ratio of 100:1, and compressed into a pellet at 6.89 × 104 kPa. To verify the composition of the mineral on the surface of the scaffold, the mineral layer was dissolved in a 1N HCl solution for 24 h and analyzed with OES to obtain molar ratios of calcium and phosphorus. After every 24-h incubation period, the SBF was also sampled, acidified with HNO3, and subjected to the OES to calculate the daily consumption of each element.
Samples sizes and statistical analyses
For quantitatively determining changes in mass and Ca/P ratio, five scaffolds were prepared for each of the SBF groups and control groups at each of the incubation times. For analyzing ion transfer from solution to scaffold, SBFs used for 24-h incubation were collected from five separate containers of each group. Two-way ANOVAs were used to analyze effects of time and IP on mass change and Ca/P ratio. The Holm–Sidak method was chosen for pair-wise multiple comparisons. Differences were considered significant if p < 0.05.
RESULTS
IP determines rate of mineralization
Altering the IP of the mineralizing solutions led to differences in the amount and rate of mineralization during the one-step mineralization process (Figs. 1 and 2), but the minerals formed from solutions of all IP exhibited similar morphologies of sub-micrometer sized flake-like apatite (Fig. 3). During the first 8 days of incubation, in solutions of all IP, most of the surface area of the scaffolds was unaltered and was similar to the pore surfaces of the PLGA scaffolds before incubation [Fig. 1(a,b)]. Mineral deposition, however, was accelerated following day 8. From all solutions, except 0.75 × SBF, mineral accumulated in a radial pattern, which grew into spherical globules extending out of the plane of the pore surfaces by days 12 and 16 [Fig. 1(c,d)]. At a given time, as IP increased, more mineral was observed on the pore surfaces of the scaffolds (Fig. 2). At day 16 of incubation, the entire pore surfaces of the scaffolds incubated in high IP solutions (1.50–2.00 × SBF) were coated with mineral globules that merged to form continuous layers [Fig. 2(d–f)]. On the other hand, scaffolds incubated in lower IP solutions (1.00 × SBF and 1.25 × SBF) were only partially mineralized [Fig. 2(b,c)]. Mineralization was rarely observed on scaffolds incubated at the lowest IP (0.75 × SBF) [Fig. 2(a)].
Figure 1.

SEM images of the pore surfaces of (a) a PLGA scaffold before mineralization, and a scaffold mineralized in 2.00 × SBF at (b) day 8, (c) day 12, and (d) day 16. At a given ionic activity product, the amount of mineralization increases with incubation time. Inset in (d) shows flake-like structure of mineral at higher magnification.
Figure 2.

SEM images of scaffold surfaces after 16 day incubation in (a) 0.75 × SBF, (b) 1.00 × SBF, (c) 1.25 × SBF, (d) 1.50 × SBF, (e) 1.75 × SBF, and (f) 2.00 × SBF. At a given incubation time, the amount of mineralization increases with increasing ionic activity product.
Figure 3.

Highly magnified nanoscale structures of the minerals after 16 day incubation in (a) 1.25 × SBF, (b) 1.50 × SBF, (c) 1.75 × SBF, and (d) 2.00 × SBF.
Mass change due to ion transfer varies with IP and incubation time
The amount of mineralization, as determined by percent increase in mass over time, increased significantly with both incubation time (p < 0.001) and IP (p < 0.001) (Fig. 4). No significant increase in mass occurred before day 8 in any IP group. In higher IPs(1.25–2.00 × SBF), significant increases in mass occurred between days 8 and 12, and 12 and 16 (p < 0.05). Within the two lowest IP groups, scaffold mass did not significantly change over 16 days. The percent change in mass between any two IPs, as well as between mineralized samples and controls, had statistical significance at 12 and 16 days (p < 0.01 at both times).
Figure 4.

Mass increases relative to time zero values for scaffolds incubated in deionized water + Tris-HCl (◇), 0.75 × SBF (▲), 1.00 × SBF (●), 1.25 × SBF (■), 1.50 × SBF (△), 1.75 × SBF (○), and 2.00 × SBF (□).
Transfer of Ca and P occurred nonlinearly from the SBF to the surfaces of the scaffolds (Fig. 5), and this supports the nonlinear increases in mass with time. Initial concentrations of Ca and P in 60 mL of 2.00 × SBF were 5.0 and 2.0 mM, respectively (Table I). For the first 4 days, Ca and P atoms were not depleted from SBF. Between days 5 and 9, the amount of atomic depletion increased, which indicates mineral formation resulting from the consumption of Ca and P atoms in SBF. After day 9, consumption of Ca and P ions from solution continued, but at a lower rate than between days 5 and 9.
Figure 5.

Ion concentrations of residual Ca and P in 2.00 × SBF after being used for 24 h.
IP affects crystallinity of the apatite
After 16 days of incubation, all of the mineralized layers had structures of apatite, but different crystallinities, depending on the ionic products of the SBFs (Fig. 6). All XRD results were normalized to the intensity of the broad hump of PLGA at 17°. In all apatites, peaks were observed between 31.4 and 32.2°, corresponding to either (211) of HAp or (112) of CAp apatite, and at 45.4°, corresponding to (203) of HAp.39 Peaks between 31.4 and 32.2° were shifted to higher wavenumber and broadened. Peak shift and broadening result from phase transition and/or strain. As the apatites in this study were formed from static solutions, it is reasonable to assume that peak changes resulted from phase transition rather than strain. However, phase transition between HAp and CAp cannot be clearly determined with XRD, because peak positions of HAp and CAp are very close to each other or overlap.39 It is especially difficult to differentiate diffraction patterns of CAp from HAp when the content is low (Fig. 6, reference peaks).40 The contents of the groups shown in Figure 6 were between 5.42 and 7.23 wt % (calculation method is described in the following FTIR result section). Carbonation, however, is more clearly confirmed by FTIR analysis (Fig. 7).
Figure 6.

X-ray diffraction patterns of the mineralized pore surfaces of the PLGA scaffolds after 16-day incubation in simulated body fluids of different ionic activity product. Apatite formed from low IP SBF (0.75 × SBF) showed sharp peaks, indicating high crystallinity, whereas comparatively broader peaks were exhibited in the apatites formed from high IP SBF (2.00 × SBF) indicating low crystallinity.
Figure 7.

FTIR spectra of the mineralized pore surfaces of PLGA scaffolds as a function of ionic activity product at day 4(a), 8(b), 12(c), and 16(d). Band intensities of phosphate and carbonate increased with ionic activity product and incubation time. At longer incubation times, more type B carbonate formed. Bands within dotted boxes were stacked, enlarged, and inserted to present changes in [insets in Fig. 7(a–d)]. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
Apatite formed from low IP SBF (0.75 × SBF) showed sharp peaks, indicating high crystallinity, whereas comparatively broader peaks were exhibited in the apatites formed from high IP SBFs, indicating low crystallinity. Since full-width-at-half maximum (FWHM) is one of the parameters most widely used as an indicator of apatite crystallinity,41 FWHM of the most intense peak [either (211) of HAp or (112) of CAp] was measured as a relative gage of crystallinity between sample groups. Peak width, however, was not in direct proportion to the IPs of SBFs (FWHM values: 0.75 × SBF, 0.294; 1.0 × SBF, 0.147; 1.25 × SBF, 0.147; 1.5 × SBF, 0.147; 1.75 × SBF, 0.294; 2.0 × SBF, 1.035).
IP influences chemical composition of apatite over time
Solution IP, as well as incubation time, affected the composition of the mineralized scaffolds (Fig. 7). The absorption bands were characterized based on the studies of synthetic and biological apatites.27,42–45 Band intensities of increased with increasing IP and incubation time [insets in Fig. 7(a–d)]. As the band intensities of are weak at days 4 and 8, it is permissible to use ν3 (1630 and 1551 cm−1) of for assigning IR on calcium phosphates, since the ν1 band (1430 cm−1) and ν4 band (1454 cm−1) are masked by the bands from PLGA (1419 and 1456 cm−1) [Fig. 7(a,b)].45 On days 4 and 8 of incubation, bands at 1630 and 1551 cm−1 (ν3), denoted by hollow squares (□), were more evident on the scaffolds mineralized in higher IP solutions [insets in Fig. 7(a,b)]. As the band at 873 cm−1 (ν3) also overlaps with a band from the PLGA control (867 cm−1), this band was not clearly distinguished at days 4 and 8 [Fig. 7(a,b)], and became evident only in the highest IP (2.00 × SBF) at days 12 and 16 [insets in Fig. 7(c,d)].
bands at 1037 (ν3), 960 (ν1), 602 (ν4), and 563 cm−1 (ν4), denoted by solid circles (●), were detected on scaffolds incubated in higher IP solutions at all times, but showed increases in intensity following 12 and 16 days of incubation in 1.25–2.00 × SBFs [Fig. 7(c,d)], which indicated that groups became involved in mineral formation. At days 12 and 16 of incubation, more vivid bands were present at 1457 (ν3b of type B CAp) and 1419 cm−1 (ν3a of type B CAp) indicating that a larger percentage of the was type B carbonate, in which replaces in the apatite structure [insets in Fig. 7(c,d)]. From the FTIR analysis (Fig. 7), the content was calculated by using a correlation between the ratio of a absorption band at 1420 cm−1 (ν1) to a absorption band at 600 cm−1 (ν4) and known wt %.40 At day 16, the concentration of the apatite was inversely proportional to IP ( wt %: 1.25 × SBF, 7.23; 1.5 × SBF, 6.15; 1.75 × SBF, 5.80; 2.0 × SBF, 5.42).
The Ca/P ratios of the mineral decreased from 1.63 ± 0.005 to 1.51 ± 0.002 with increasing IP (Fig. 8). At both days 12 and 16, there was a significant effect of IP, with a significant decrease in Ca/P occurring between successively greater IPs (p < 0.01). Within each IP, there was no effect of time on Ca/P between days 12 and 16.
Figure 8.
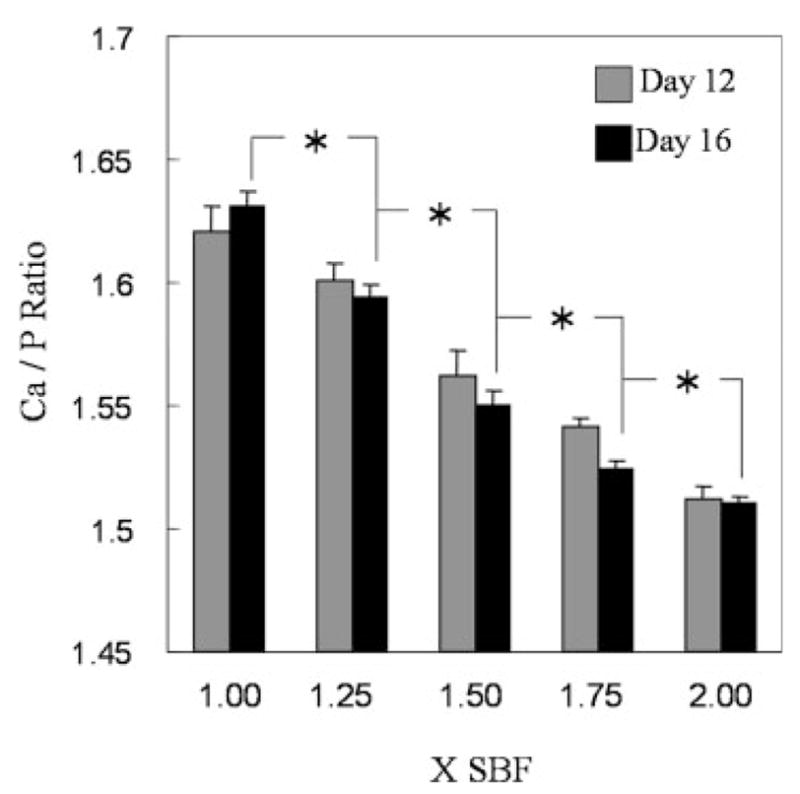
Ca/P ratio of the mineralized layers formed from six different simulated body fluids at days 12 and 16. Asterisks denote significant differences in Ca/P between different ionic activity products at both 12 and 16 days.
DISCUSSION
The formation of mineral on substrates incubated in solutions of different IP is dependent on incubation time, and the rate of mineralization can be accelerated by increasing IP. Mineralization of a polymer substrate has two distinct stages, as does mineralization in vivo.32 Initially, following surface hydrolysis, mineral nucleation occurs on the polymer surface (primary nucleation) and then additional mineral crystals grow on the nucleated mineral (crystal growth). For PLGA substrates, surface hydrolysis, primary nucleation, and growth occur in an efficient, single-step treatment in SBF without the need for a separate functionalization step or surface pretreatment in water or NaOH. For initiating primary nucleation on a PLGA scaffold, a combination of conditions is required; hydroxyl groups and hydrophilic carboxylic acid should be provided from ester bond scission in the carbon backbone of the PLGA, and a large amount of ions should be locally accumulated around the scaffold.
At day 4, the polymer surfaces did not exhibit any morphological changes indicative of primary nucleation and were similar to the nonmineralized control samples. Ion depletion of SBF due to primary nucleation was not observed either during the first 4 days of incubation (Fig. 5). However, there were changes in FTIR spectra for scaffolds incubated in higher IP solutions at day 4 (Fig. 7), indicating some ionic binding to the polymer surface. This finding is, however, likely the result of a highly localized existence of a small amount of nuclei, rather than coverage of a large percentage of the pore surfaces with mineral. These trends imply that IP has little effect on reducing the time for surface hydrolysis, which is in accord with the fact that surface hydrolysis of PLGA occurs by the water molecules in SBF. After surface hydrolysis and mineral nucleation, less energy and time are required to add ions or ion clusters to already existing mineral nuclei than are required to form the initial mineral crystals.32 SBFs with higher IP can provide more accumulated clusters of calcium, , and ions around a scaffold, which eventually lead to differences in the rate of the mineral growth, compared with SBFs with a low IP.
The morphology of the apatite formed from SBF is determined by parameters of the fluid, including Ca/P molar ratio,46 and concentrations of Mg2+, F−, and .47,48 In this study, there were no morphological differences between apatites formed from solutions of different IP. This could be due to the constant Ca/P molar ratio of 2.5/1 and the constant fraction of Mg2+ and in all solutions. Use of three different pH values of SBF, depending on IP, to maintain heterogeneous nucleation, did not lead to changes in mineral morphology. Generally, SBFs with higher IPs (e.g., 5 × SBF) are either buffered to mildly acidic pH values (e.g., 5.8 or lower) and/or contacted with an acidic gas-like carbon dioxide, since increasing IP of SBF is limited by the low solubility of Ca–P salts.36,49–51 Under conditions of high IP and low pH of an SBF, a rapid increase in pH is observed, along with homogeneous nucleation in the solution, which can also lead to changes in apatite morphology with pH.49 However, within the narrower and more physiological pH range used in this study (6.8–7.4), the morphology of the apatite was not affected by pH, at either the macro- or micro-scales (Fig. 3).
All of the apatites were flake-like and composed of sub-micrometer sized structures (Fig. 3). These features are distinct, compared with previous studies, in which micrometer-sized plate-like structures were reported.47–49 Differences in the size and shape of the mineral structure reported by others could be because of compositional differences in SBFs, in which either only reagents containing Ca and P were doubled,31 or lack of crystal growth inhibitors, such as Mg2+ and , resulting in higher Ca–P/inhibitor ratios than that we used.47,48 Inhibitors such as Mg2+ and interfere with the formation of a Ca–P complex and can lead to the formation of a poorly crystallized Ca-deficient apatite.48
All of the minerals synthesized in this study were type B CAps, and their compositions were dependent on IP. Although the Ca/P molar ratios of the SBFs were kept constant at 2.5, the Ca/P molar ratio of the apatite increased from 1.51 to 1.63 as IP decreased, compared with the apatite in bone, which has a Ca/P ratio of 1.57–1.62. The monotonic decrease in Ca/P ratio with increasing IP could be because of the inverse correlation between substitution of and IP. Since substitution of a trivalent ion ( ) by a bivalent ion ( ) should leave a negative residual electric charge in the unit cell of an apatite, one substitution of ion for ion results in a half Ca and one P deficiency, which leads to the increase in the Ca/P molar ratio.40 FTIR data show that relatively more ions were incorporated with ions in apatites incubated in higher IP, which indicates that the substitution of ion for ion in the apatite structure arose more readily in low IP solutions. This interpretation of phase transition between the HAp and the CAp is supported by the calculated concentrations of , which showed wt % is inversely proportional to IP. Ca/P molar ratios achieved in this study make these materials suitable for biological purposes, as calcium phosphate with Ca/P ratios in the range 1.50–1.67 yields the most beneficial tissue responses.52,53
The long period of incubation required for complete mineralization of the pore surfaces in this onestep process was mainly because of the slow surface hydrolysis of the PLGA. To achieve faster hydrolysis, pretreatment of metals8,54 and polymers17,29,31 with water or an alkaline solution, such as NaOH, has been introduced. This alkaline treatment, however, needs to be carried out on polymer substrates without compromising polymer integrity.17,31 Since polymers such as PLGA degrade in a basic solution, the concentration of NaOH and pretreatment time should be carefully determined so that only the surfaces of the PLGA pores become hydrolyzed to induce faster mineralization, but the scaffold itself maintains mechanical stability. Therefore, when designing hydrolysis processes, geometric parameters of the scaffolds, such as porosity and thickness, also need to be considered. Pretreatment in 0.2N NaOH solution for 30 min was suitable for the scaffolds used in this study. After 4 days of incubation in 1 × SBF, the alkaline pretreated scaffolds were mineralized as much as non-pretreated ones were on day 12, and the mass loss of alkaline-treated scaffolds was not significantly different from scaffolds exposed to just deionized water (data not shown). More importantly, this pretreatment did not affect any properties of the apatite (e.g., morphology, composition) formed by the biomimetic process. Another strategy to accelerate mineralization is to use a more concentrated SBF.36,50,51 However, occlusion of pores in more closed cell porous structures, such as scaffolds formed from salt leaching, is a possibility.
We previously developed a system in which three-dimensional porous PLGA scaffolds self-assemble a bone-like mineral extracellular matrix analogue using one type of SBF.14,35,37 The features of this mineral are consistent with biological apatite in terms of its mineral morphology, chemical composition, Ca/P molar ratio, and low crystallinity. Compared with nonmineralized PLGA scaffolds, this hybrid substrate led to significantly enhanced cell adhesion, differentiation, and cytoskeletal organization in vitro.35,37 When bone marrow stromal cells were seeded on mineralized scaffolds and transplanted into mice, a significantly higher volume fraction of regenerated bone was achieved compared with non-mineralized PLGA controls, supporting the hypothesis that a biomimetic microenvironment will enhance cell differentiation and increase bone formation.35 Substrate mineralization also causes expression of different cell adhesion contacts and modifies cell motility.37 For critical-size calvarial defects, apatite-coated PLGA leads to bone regeneration with a high mineral density, whereas uncoated PLGA scaffolds do not.55 In addition, partial dissolution of the apatite can increase Ca and P ion concentrations in the local environment and cause solution-mediated effects on cell function.56 Altering crystallinity and Ca/P ratio of the mineral, as we have demonstrated in these studies, may therefore lead to adjustable dissolution rates, which can control solution-mediated effects, as well as delivery of growth factors or other biomolecules.
In the present study, we demonstrated that the IP of an SBF is an important and useful factor to modulate properties of bone-like apatite coated on three-dimensional PLGA scaffolds. This approach showed that material properties of the apatite, such as morphology, crystallinity, content, and Ca/P ratio can be controlled in a predictable manner by adjusting only the IP of a mineralizing solution. It is, however, still unclear what physical–chemical properties of apatite are required to optimize cell functions.57 One consensus finding could be that the enhanced biological responses can be achieved only when a material provides similar properties to bone apatite.52,53,56 Apatites formed from the solutions with six different IPs of this study presented features similar to bone apatite in terms of the apatite phase (CAp), the Ca/P ratio (1.57–1.62), and the content (5.42–7.23 wt %). By changing IPs and time of incubation in mineralizing solutions it therefore becomes more feasible to manipulate the chemical and structural properties of the mineral coating on the surface of a polymer scaffold, and this is expected to be able to further modulate biological functions.
Acknowledgments
We thank the University of Michigan Electron Microbeam Analysis Laboratory (EMAL) for use of the SEM.
Contract grant sponsor: NIH; contract grant numbers: R01 DE 013380, R01 DE 015411
Contract grant sponsor: NSF; contract grant number: EAR-9628196
References
- 1.Hench LL, Wilson J. Surface-active biomaterials. Science. 1984;226:630–636. doi: 10.1126/science.6093253. [DOI] [PubMed] [Google Scholar]
- 2.Hench LL. Bioceramics: From concept to clinic. J Am Ceram Soc. 1991;74:1487–1510. [Google Scholar]
- 3.Joris SJ, Amberg CH. The nature of deficiency in nonstoichiometric hydroxyapatites. I. Catalytic activity of calcium and strontium hydroxyapatites. J Phys Chem. 1971;75:3167–3171. [Google Scholar]
- 4.Jarcho M. Calcium phosphate ceramics as hard tissue prosthetics. Clin Orthop. 1981;157:259–278. [PubMed] [Google Scholar]
- 5.Heimke G. Advanced ceramics for biomedical applications. Angew Chem Int Ed Engl. 1989;28:111–116. [Google Scholar]
- 6.Ducheyne P, Radin S, Heughebaert M, Heughebaert JC. Calcium phosphate ceramics coatings on porous titanium: Effect of structure and composition on electrophoretic deposition, vacuum sintering and in vitro dissolution. Biomaterials. 1990;11:244–253. doi: 10.1016/0142-9612(90)90005-b. [DOI] [PubMed] [Google Scholar]
- 7.Kohn DH. Metals in medical applications. Curr Opin Solid State Mater Sci. 1998;3:309–316. [Google Scholar]
- 8.Godley R, Starosvetsky D, Gotman I. Bonelike apatite formation on niobium metal treated in aqueous NaOH. J Mater Sci Mater Med. 2004;15:1073–1077. doi: 10.1023/B:JMSM.0000046388.07961.81. [DOI] [PubMed] [Google Scholar]
- 9.Vallet-Regi M, Romero AM, Ragel CV, LeGeros RZ. XRD, SEM-EDS, and FTIR studies of in vitro growth of an apatite-like layer on sol–gel glasses. J Biomed Mater Res. 1999;44:416–421. doi: 10.1002/(sici)1097-4636(19990315)44:4<416::aid-jbm7>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 10.Miller CA, Kokubo T, Reaney IM, Hatton PV, James PF. Formation of apatite layers on modified canasite glass-ceramics in simulated body fluid. J Biomed Mater Res. 2002;59:473–480. doi: 10.1002/jbm.10018. [DOI] [PubMed] [Google Scholar]
- 11.Ylänen H, Karlsson K, Itala A, Aro HT. Effect of immersion in SBF on porous bioactive bodies made by sintering bioactive glass microspheres. J Non-Crystal Solids. 2000;275:107–115. [Google Scholar]
- 12.Metzger DS, Driskell TD, Paulsrud JR. Tricalcium phosphate ceramic—A resorbable bone implant: Review and current status. J Am Dent Assoc. 1982;105:1035–1048. doi: 10.14219/jada.archive.1982.0408. [DOI] [PubMed] [Google Scholar]
- 13.Brovarone CV, Verne E, Krajewski A, Ravaglioli A. Graded coating on ceramic substrates for biomedical applications. J Euro Ceram Soc. 2001;21:2855–2862. [Google Scholar]
- 14.Murphy WL, Kohn DH, Mooney DJ. Growth of continuous bonelike mineral within porous poly(lactide-co-glycolide) scaffolds in vitro. J Biomed Mater Res. 2000;50:50–58. doi: 10.1002/(sici)1097-4636(200004)50:1<50::aid-jbm8>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 15.Luong LN, Hong SI, Patel RJ, Outslay ME, Kohn DH. Spatial control of protein within biomimetically nucleated mineral. Biomaterials. 2006;27:1175–1186. doi: 10.1016/j.biomaterials.2005.07.043. [DOI] [PubMed] [Google Scholar]
- 16.Abe Y, Kokubo T, Yamamuro T. Apatite coating on ceramics, metals and polymers utilizing a biological process. J Mater Sci Mater Med. 1990;1:233–238. [Google Scholar]
- 17.Chen J, Chu B, Hsiao BS. Mineralization of hydroxyapatite in electrospun nanofibrous poly(L-lactic acid) scaffolds. J Biomed Mater Res A. 2006;79:307–17. doi: 10.1002/jbm.a.30799. [DOI] [PubMed] [Google Scholar]
- 18.Marra KG, Szem JW, Kumta PN, DiMilla PA, Weiss LE. In vitro analysis of biodegradable polymer blend/hydroxyl apatite composite for bone tissue engineering. J Biomed Mater Res. 1999;47:324–335. doi: 10.1002/(sici)1097-4636(19991205)47:3<324::aid-jbm6>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 19.Shikinami Y, Okuno M. Bioresorbable devices made of forged composites of hydroxyapatite particles and poly-L-lactide. I. Basic characteristics. Biomaterials. 1999;20:859–877. doi: 10.1016/s0142-9612(98)00241-5. [DOI] [PubMed] [Google Scholar]
- 20.Weaver DT, Van Aken DC, Smith JD. The role of bulk nucleation in the formation of crystalline cordierite coatings produced by air plasma spraying. Mater Sci Eng. 2003;1–2:96–102. [Google Scholar]
- 21.Ding SJ, Huang TH, Kao CT. Immersion behavior of plasma-sprayed modified hydroxyapatite coatings after heat treatment. Surf Coat Technol. 2003;165:248–257. [Google Scholar]
- 22.Ong JL, Bessho K, Carnes DL. Bone response to plasma-sprayed hydroxyapatite and radiofrequency-sputtered calcium phosphate implants in vivo. Int J Oral Maxillofac Implants. 2002;17:581–586. [PubMed] [Google Scholar]
- 23.Burke EM, Lucas LC. Dissolution kinetics of calcium phosphate coatings. Implant Dent. 1998;7:323–330. doi: 10.1097/00008505-199807040-00011. [DOI] [PubMed] [Google Scholar]
- 24.Huang CM, Xu Y, Zangvil A, Kriven WM, Xiong F. Laser ablated coatings on ceramic fibers for ceramic matrix composites. Mater Sci Eng. 1995;191:249–256. [Google Scholar]
- 25.Clèries L, Martínez E, Ferńndez-Pradas JM, Sardin G, Esteve J, Morenza JL. Mechanical properties of calcium phosphate coatings deposited by laser ablation. Biomaterials. 2000;21:967–971. doi: 10.1016/s0142-9612(99)00240-9. [DOI] [PubMed] [Google Scholar]
- 26.Durucan C, Brown PW. Low temperature formation of calcium deficient hydroxyapatite-PLA/PLGA composites. J Biomed Mater Res. 2000;51:717–725. doi: 10.1002/1097-4636(20000915)51:4<717::aid-jbm21>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 27.Xu G, Aksay IA, Groves JT. Continuous crystalline carbonate apatite thin films. A biomimetic approach. J Am Chem Soc. 2001;123:2196–2203. doi: 10.1021/ja002537i. [DOI] [PubMed] [Google Scholar]
- 28.Nakamura T, Yamamuro T, Higashi S, Kokubo T, Ito S. A new glass–ceramic for bone replacement: Evaluation of its bonding ability to bone tissue. J Biomed Mater Res. 1985;19:685–698. doi: 10.1002/jbm.820190608. [DOI] [PubMed] [Google Scholar]
- 29.Yuan X, Mak AT, Li J. Formation of bone-like apatite on poly(L-lactic acid) fibers by a biomimetic process. J Biomed Mater Res. 2001;57:140–150. doi: 10.1002/1097-4636(200110)57:1<140::aid-jbm1153>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 30.Kim HM, Kishimoto K, Miyaji F, Kokubo T, Yao T, Suetsugu Y, Tanaka J, Nakamura T. Composition and structure of the apatite formed on PET substrates in SBF modified with various ionic activity products. J Biomed Mater Res. 1999;46:228–235. doi: 10.1002/(sici)1097-4636(199908)46:2<228::aid-jbm12>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 31.Murphy W, Mooney DJ. Bioinspired growth of crystalline carbonate apatite on biodegradable polymer substrata. J Am Chem Soc. 2002;124:1910–1917. doi: 10.1021/ja012433n. [DOI] [PubMed] [Google Scholar]
- 32.Bostrom MP, Boskey A, Kaufman JK, Einhorn TA. Orthopaedic basic science: Formation and function of bone. Rosemont, IL: American Academy of Orthopaedic Surgeons; 2000. pp. 320–334. [Google Scholar]
- 33.Nancollas GH, Wu W. Biomineralization mechanism: A kinetics and interfacial energy approach. J Crystal Growth. 2000;211:137–142. [Google Scholar]
- 34.Thomson RC, Yaszemski MJ, Mikos AG. Polymer scaffold processing. In: Lanza RP, Langer R, Chick WL, editors. Principles of Tissue Engineering. Austin, Texas: R.G. Landes Co., Academic Press; 1997. pp. 263–272. [Google Scholar]
- 35.Kohn DH, Shin K, Hong SI, Jayasuriya AC, Leonova EV, Rosello RA, Krebsbach PH. Self-assembled mineral scaffolds as model systems for biomineralization and tissue engineering. In: Landis WJ, Sodek J, editors. Proceedings of the Eighth International Conference of the Chemistry and Biology of Mineralized Tissue; Toronto: University of Toronto Press; 2005. pp. 216–219. [Google Scholar]
- 36.Chen W, Mak A, Li J, Wang M, Shum A. Formation of apatite on poly(α-hydroxy acid) in an accelerated biomimetic process. J Biomed Mater Res B. 2005;73:68–76. doi: 10.1002/jbm.b.30178. [DOI] [PubMed] [Google Scholar]
- 37.Leonova EV, Pennington KE, Krebsbach PH, Kohn DH. Substrate mineralization stimulates focal adhesion contact redistribution and cell motility of bone marrow stromal cells. J Biomed Mater Res A. 2006;79:263–270. doi: 10.1002/jbm.a.30786. [DOI] [PubMed] [Google Scholar]
- 38.Kokubo T, Takadama H. How useful is SBF in predicting in vivo bone bioactivity? Biomaterials. 2006;27:2907–2915. doi: 10.1016/j.biomaterials.2006.01.017. [DOI] [PubMed] [Google Scholar]
- 39.Powder Diffraction File: Inorganic Phases. Swarthmore, PA: JCPDS International Centre for Diffraction Data; 1987. [Google Scholar]
- 40.LeGeros RZ. Formation and Stability of Synthetic Apatites: Effect of Some Elements. In: Myers HM, editor. Calcium Phosphates in Oral Biology and Medicine. California: Karger; 1999. pp. 82–107. [Google Scholar]
- 41.Baig AA, Fox JA, Young RA, Wang Z, Hsu J, Higuchi WI, Chhettry A, Zhuang H, Otsuka M. Relationships among carbonated apatite solubility, crystallite size and microstrain parameters. Calcif Tissue Int. 1999;64:437–449. doi: 10.1007/pl00005826. [DOI] [PubMed] [Google Scholar]
- 42.Chang MC, Tanaka J. FT-IR study for hydroxyapatite/collagen nanocomposite cross-linked by glutaraldehyde. Biomaterials. 2002;23:4811–4818. doi: 10.1016/s0142-9612(02)00232-6. [DOI] [PubMed] [Google Scholar]
- 43.Rehman I, Bonfield W. Characterization of hydroxyapatite and carbonated apatite by photo acoustic FTIR spectroscopy. J Mater Sci Mater Med. 1997;8:1–4. doi: 10.1023/a:1018570213546. [DOI] [PubMed] [Google Scholar]
- 44.Koutsopoulos S. Synthesis and characterization of hydroxyapatite crystals: A review study on the analytical methods. J Biomed Mater Res. 2002;62:600–612. doi: 10.1002/jbm.10280. [DOI] [PubMed] [Google Scholar]
- 45.Rey C, Collins B, Goehl T, Dickson R, Glimcher MJ. The carbonate environment in bone mineral: A resolution-enhanced Fourier transform infrared spectroscopy study. Calcif Tissue Int. 1989;45:157–164. doi: 10.1007/BF02556059. [DOI] [PubMed] [Google Scholar]
- 46.Cheng PT, Pritzker KPH. Solution of Ca/P ratio affects calcium phosphate crystal phases. Calcif Tissues Int. 1983;35:596–601. doi: 10.1007/BF02405100. [DOI] [PubMed] [Google Scholar]
- 47.Chou YF, Huang W, Dunn JC, Miller TA, Wu BM. The effect of biomimetic apatite structure on osteoblast viability, proliferation, and gene expression. Biomaterials. 2005;26:285–295. doi: 10.1016/j.biomaterials.2004.02.030. [DOI] [PubMed] [Google Scholar]
- 48.Barrère F, Layrolle P, van Blitterswijk CA, de Groot K. Biomimetic calcium phosphate coatings on Ti6Al4V: A crystal growth study of octacalcium phosphate and inhibition by Mg2+ and . Bone. 1999;25:107S–111S. doi: 10.1016/s8756-3282(99)00145-3. [DOI] [PubMed] [Google Scholar]
- 49.Chou YF, Chiou WA, Xu Y, Dunn JC, Wu BM. The effect of pH on the structural evolution of accelerated biomimetic apatite. Biomaterials. 2004;25:5323–5331. doi: 10.1016/j.biomaterials.2003.12.037. [DOI] [PubMed] [Google Scholar]
- 50.BarrÈre F, van Blitterswijk CA, de Groot K, Layrolle P. Influence of ionic strength and carbonate on the Ca–P coating formation from SBFx5 solution. Biomaterials. 2002;23:1921–1930. doi: 10.1016/s0142-9612(01)00318-0. [DOI] [PubMed] [Google Scholar]
- 51.BarrÈre F, van Blitterswijk CA, de Groot K, Layrolle P. Nucleation of biomimetic Ca–P coatings on Ti6Al4V from a SBFx5 solution: Influence of magnesium. Biomaterials. 2002;23:2211–2220. doi: 10.1016/s0142-9612(01)00354-4. [DOI] [PubMed] [Google Scholar]
- 52.Kohn DH. Bioceramics. In: Kutz M, editor. Biomedical Engineers Handbook. New York: McGraw-Hill; 2002. pp. 13.1–13.24. [Google Scholar]
- 53.Posner AS. The mineral of bone. Clin Orthop Relat Res. 1985;200:87–99. [PubMed] [Google Scholar]
- 54.Miyazaki T, Kim HM, Kokubo T, Miyaji F, Kato H, Nakamura T. Effect of thermal treatment on apatite-forming ability of NaOH-treated tantalum metal. J Mater Sci Mater Med. 2001;12:683–687. doi: 10.1023/a:1011260224120. [DOI] [PubMed] [Google Scholar]
- 55.Cowan CM, Shi Y-Y, Aalami OO, Chou Y-F, Mari C, Thomas R, Quarto N, Chontag CH, Wu B, Longaker MT. Adipose-derived adult stromal cells heal critical-size mouse calvarial defects. Nat Biotechnol. 2004;25:560–567. doi: 10.1038/nbt958. [DOI] [PubMed] [Google Scholar]
- 56.LeGeros RZ. Properties of osteoconductive biomaterials: Calcium phosphates. Clin Orthop Relat Res. 2002;395:81–98. doi: 10.1097/00003086-200202000-00009. [DOI] [PubMed] [Google Scholar]
- 57.Suzuki T, Hukkanen M, Ohashi R, Yokogawa Y, Nishizawa K, Nagata F, Buttery L, Polak J. Growth and adhesion of osteoblast-like cells derived from neonatal rat calvaria on calcium phosphate ceramics. J Biosci Bioeng. 2000;89:18–26. doi: 10.1016/s1389-1723(00)88045-7. [DOI] [PubMed] [Google Scholar]