

Abstract
Mechanical stretch rapidly activates multiple signaling cascades, including phospholipases and kinases, to stimulate protein synthesis and growth. The purpose of this study was to determine whether PLA2 activation contributes to stretch-induced phosphorylation of ERK2 in skeletal muscle myotubes. Myotubes derived from neonatal C57 mice were cultured on silicone membranes and subjected to brief cyclic stretch. Inhibition of PLA2 prevented ERK2 phosphorylation, while inhibition of prostaglandin or leukotriene synthesis did not. ERK2 phosphorylation was also blocked by genistein and PD98059, implicating the canonical raf-MEK-ERK cassette. It appears that PLA2, but not further metabolism of arachidonic acid, is required for stretch-induced activation of ERK2. Exposure to exogenous arachidonic acid had no effect on ERK2 phosphorylation, but exposure to lysophosphatidylcholine, the other metabolite of PLA2, caused a dose-dependent increase in ERK2 phosphorylation. These results suggest that stretch-induced activation of ERK2 may result from an interaction between PLA2 derived lysophosphatidylcholine and membrane receptors.
Keywords: mechanotransduction, MAP kinase, phospholipase a2
Introduction
Mechanical signals are important for the development and maintenance of skeletal muscle function. In vivo, brief periods of forceful exertion are sufficient stimulate protein synthesis over a few hours and to promote increases in muscle mass [1]. In isolated muscle, passive stretch is sufficient to reduce the negative nitrogen balance, and stretch of cultured myotubes dramatically promotes their morphological maturation and development of a contractile matrix [2, 3]. Many signaling processes are involved in this process, but the ultimate mechanosensor(s) are still ambiguous.
Phospholipase A2 (PLA2) enzymes may be mechanically activated [4], because kinetic parameters depend on membrane phase structure and packing defects [5]. PLA2 activity results in the cleavage phospholipids, liberating a free fatty acid, often an arachidonic acid (AA) and lysophospholipid, often a lysophosphatidylcholine (LPC). Cytoplasmic PLA2 activity derives from at least 15 different proteins in three distinct families: the calcium-dependent cPLA2 or Group IV family, the calcium-independent iPLA2 or Group VI family, and the platelet activating factor (PAF-AH) or Group VII and Group VIII families [6, 7]. Inhibition of PLA2 blocks stretch-induced AA release, stretch-induced muscle damage, and, in kidney cells, stretch-induced phosphorylation of ERK 1/2 [8, 9].
Both AA and LPC are biologically active molecules, but AA has shown special importance. AA metabolism is quite complex, as it is subject to oxidative modification and specific metabolism by cyclooxygenase (COX) into prostaglandins (PG), by lipoxygenase (LOX) into leukotrienes, or by cytochrome p450 into epoxyeicosatrienoic acid (EET) [6]. In muscle, attention has focused on PGs, which contribute to stretch- or exercise-stimulated protein synthesis and hypertrophy in vivo and in vitro [1, 2, 10]. In humans, the COX inhibitors ibuprofen and acetaminophen reduce exercise-stimulated protein synthesis in proportion with their ability to prevent PG accretion [1, 11]. Ibuprofen is also an effective inhibitor of passive stretch-stimulated protein synthesis in whole muscle, but meclofenamic acid, which also inhibits PLA2 [12], is even more effective [2].
Signaling through ERK1/2 is also important to hypertrophic signaling [13, 14], and ERK2 is rapidly phosphorylated and activated by mechanical signals [15-17]. ERK2 activity can be increased by stretch-activated PLA2 in kidney cells [8] and contributes to upregulation of COX2 in cardiomyocytes [18]. Its activity is also required for transcription of cyclins, activation of cyclin dependent kinases, and progression through the cell cycle [19]. In addition to its transcriptional role, ERK2 phosphorylates translational regulators and is required for hypertrophy of muscle fibers [13]. In cardiac myocytes, ERK2 contributes to endothelin-induced activation of mTOR, p70S6K, and eIF4E [14], and this mechanism may be active in skeletal myotubes.
Both PLA2 activity and ERK phosphorylation increase rapidly with stretch in myotubes, and both are linked to stretch-induced growth. While some cPLA2s require phosphorylation by ERK for full activation [20], both PGF2α and PGE2 receptors lead to phosphorylation of ERK [21]. PLA2 activity may be directly influenced by mechanical distortion of the cell membrane [4], but ERK phosphorylation is increased by many different stretch-related stimuli [16, 17]. It is not clear whether PLA2 activity and ERK 1/2 represent separate mechanotransduction cascades or links within a single chain.
The experiments in this paper were intended to test the hypothesis that the immediate phosphorylation of ERK2 by stretch is dependent on PLA2 signaling and to test the contribution of specific metabolites. Primary cultured myotubes were subjected to cyclic stretch and treated with inhibitors of AA metabolism. The results suggest that PLA2 activity and LPC, but not a metabolite of AA, promotes rapid ERK2 phosphorylation. PLA2 is an instigator of the cellular response to mechanical stimulation.
Materials and methods
Cell culture
Myoblasts were isolated from neonatal C57 mice by enzymatic dissociation of hindlimb musculature and purified by selective trypsinization and differential adhesion, using procedures reviewed and approved by Georgia Institute of Technology's IACUC and in compliance with the Guide for Care and Use of Laboratory Animals. Briefly, 2-5 day old mouse pups were sacrificed by isoflurane overdose and washed with 70% ethanol. Hindlimb musculature was minced between crossed razor blades and incubated 30 minutes at 37°C in dissociation solution (10 mM phosphate buffered saline (PBS) containing 10 mM CaCl2, 1.5 U/ml collagenase, 2.4 U/ml dispase). Cells were resuspended in growth media (GM, Ham's F-10 containing 20% fetal bovine serum, 2.5 ng/ml bFGF, 100 ug/ml streptomycin and 100 IU/ml penicillin) and myogenic cells were enriched over 8-12 passages by gentle trypsinization and brief preplating. Myogenicity was validated by desmin staining, and only cultures >97% desmin positive were used. Experiments were routinely performed on cells between passage 10 and 22.
For stretch experiments, cells were seeded at 105 cells/cm2 on Matrigel (BD Biosciences, San Jose) coated silicone membranes fixed between stainless steel clamps [22]. Cultures were allowed to adhere and proliferate for 24 hours and were then subjected to an initial 25% stretch coincident with replacement of GM with differentiation media (DM, DMEM containing 2% horse serum and antibiotics). This initial stretch results in cultures forming myotubes aligned with the axis of stretch, and cultures were maintained at this length (125% of seeding length), which serves as a reference for all subsequent length changes. Cultures were maintained in DM until well fused, generally 3-4 days after the media switch.
Culture media was replaced with serum and antibiotic free Ham's F-10 60 minutes prior to mechanical stimulation. Mechanical stimulation consisted of sinusoidal length changes between 85% and 115% of the reference length at 0.3 Hz. Cultures were subjected to 15 stretch cycles (45 s) followed by 15 s rest, and the pattern was repeated for 15 minutes. Parallel static cultures were maintained on elastic membranes, subjected to the 25% stretch with switch to differentiation media, but were not subjected to cyclic stretches.
Interventions
Pharmacological interventions were used to probe the roles of PLA2, COX, LOX, and receptors. Each intervention experiment consisted of a two by two design of mechanical stimulation and drug treatment. Static cultures are designated as “Control,” and vehicle treated cultures as “No drug.” Each treatment was repeated on a minimum of three independent experiments, each with 2-3 repeated wells, resulting in final group sizes (n) around 9. Drugs were suspended in water (BDM, pertussis toxin), DMSO (MA, IBU, BW-B70C, PD146176, genistein, BIM-1), or ethanol (AACOCF3), prior to dilution into serum free F-10 media.
To directly test the effects of PLA2 products, cultures were incubated for 60 minutes in serum free F-10 media, then exposed to AA or lysophosphatidylcholine for 15 mintues. AA (CN Biosciences) was suspended in ethanol at 100 mM, and added to CO2-equilibrated F-10 to give a final concentration of 1-10 μM. Synthetic LPC (1-stearoyl-2-hydroxy-sn-glycero-3-phosphocholine, Avanti Polar Lipids) was dispersed in PBS at 1 mM by vortexing for 60 seconds immediately before use. An aliquot of this dispersion was added to CO2-equilibrated F-10 to give a final concentration of 1-20 μM and mixed by vigorous trituration.
Western blotting
Immediately following treatment, cultures were removed from the stretch apparatus, rinsed with ice cold PBS, and collected by scraping in detergent buffer (1% Triton-X 100, 50 mM tris, 250 mM NaCl, 25 mM EDTA, protease inhibitors (Sigma), 100 mM NaF and 4μg/ml NaVO3) for 30 minutes. Insoluble material was cleared by centrifugation at 16k ×g for 5 min, and soluble protein concentration was assayed by bicinchronic acid assay (Pierce).
To assay phosphorylation of ERK2, Akt, and p70S6 kinase, 4 μg soluble protein was separated by SDS-PAGE through 10% acrylamide mini-gels. Proteins were transferred to nitrocellulose membranes, blocked with 5% nonfat dry milk in Tween-TBS (TTBS) and hybridized with phospho-specific antibody to ERK, Akt, or p70S6k (Cell Signaling). Membranes were rinsed, incubated with horseradish peroxidase conjugated secondary antibody (Jackson, 1:20,000), and detected by enhanced chemiluminescence (Amersham). Bands were visualized by exposure to radiographic film and quantified by scanning densitometry. Membranes were then stripped and re-probed with non-phospho-specific antibodies ERK2 (BD Biosciences), Akt (Cell Signaling) or p70S6k (Santa Cruz Biotechnology). All samples from an experiment were run on the same gel, along with a serum-stimulated positive control, and normalized to the average of that experiment's No Drug-Control samples.
Statistics
All results are reported as mean±S.D. Comparisons within each stretch experiment are made by two-way ANOVA (drug × stretch). Sample size was chosen to provide statistical resolution of one standard deviation with statistical power of 0.8 for interaction effects. Statistical trends were recognized at a threshold of 10%. All intervention experiments follow the 2×2 design, and no post-hoc T-tests were performed, because the interaction term directly tests whether the intervention alters the stretch response. Exogenous lipid treatments were analyzed by 1-way ANOVA followed by post-hoc T-tests using the Bonferroni/Dunn correction for multiple comparisons.
Results and Discussion
Stretch-induced phosphorylation of several proteins was measured by Western blot. In most cases, p70S6 kinase was not significantly phosphorylated on Thr-389 following stretch (p-values between 0.26 and 0.77, data not shown), consistent with previous observations using this stretch model [23]. The single exception to this was in experiments using 100 uM genistein, in which stretch produced a 50±60% increase (p=0.04) increase in the vehicle (DMSO) treated wells (data not shown). This was not seen in other DMSO treated cells and is believed to be a statistical artifact. The activity of pla2g4a may be modulated by ERK 1/2-dependent phosphorylation [20], but its phosphorylation was not altered by cyclic stretch (p-values between 0.64 and 0.68, data not shown). This indicates that the activity of this PLA2 isoform is not influenced by phosphorylation during the course of these experiments. In contrast, pooled across all experiments (n=118, all No Drug, Control/Stretch samples), cyclic stretch produced a 130±130% increase in ERK2 phosphorylation. The extent of phosphorylation varied among experiments (see below), but was always significant at the p<0.01 level, ranging from 70±35% (n=9) to 220±90% (n=9).
Previous experiments [2] have shown than 100 μM meclofenamic acid (MA) prevents stretch induced increase in protein synthesis and PG generation in whole muscle. MA is a COX inhibitor (IC50 0.6-2.5 μM) and a LOX inhibitor (IC50 47-110 μm) [24], but also an inhibitor of PLA2, with an IC50 of 0.05-400 μM, depending on Ca2+ [12]. To determine whether a similar process contributes to stretch-induced ERK2 phosphorylation, myotubes were treated with 30 μM MA prior to stretch. MA completely eliminated the stretch-induced phosphorylation of ERK2 (Drug × stretch p<0.001; fig. 1), involvement of lipid signals, however, the multiple actions of MA at high micromolar concentration make it difficult to decipher what that mechanism might be.
Figure 1.
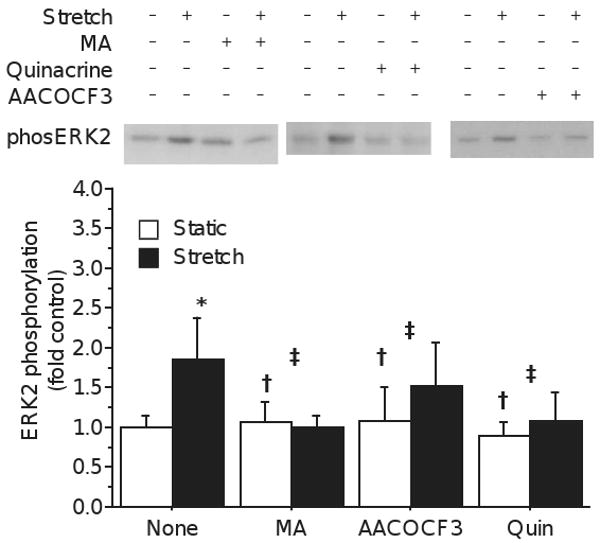
Representative blot showing increased intensity of labeling with phsopho-specific (Thr202/Tyr204) ERK antibody following stretch, and the effect of several PLA2 inhibitors. In the absence of drug, stretch (solid bars) induced significant phosphorylation of ERK2 relative to unstretched controls (open bars). Stretch-induced phosphorylation was blocked by incubation with 30 μM meclofenamic acid (MA), 100 μM AACOCF3 (AACOCF3) or 100 μM quniacrine (quin). Bars represent mean±S.D.; significance notations represent 2-way ANOVA statistics: * main effect of Stretch p<0.05; † main effect of Drug p<0.05; ‡ interaction of Drug × Stretch p<0.05. Note that significance of Stretch is indicated only over the vehicle-treated bars, but refers to the effect of main effect of stretch independent of drug.
To clarify the relative roles of COX and LOX, myotubes were subjected to stretch with broad-spectrum inhibitors of these enzymes. Pre-incubation of myotubes with 100 μM ibuprofen to block COX activity failed to prevent ERK2 phosphorylation (Drug × stretch p=0.65; figure 2), although ibuprofen substantially blocks exercise-induced PG synthesis [1]. The stretch-induced increase in protein synthesis and myoblast proliferation can be mimicked by addition of exogenous PGF2a [25, 26], but the lack of ibuprofen effect in the present study suggests that COX-mediated PG synthesis does not contribute to stretch-induced ERK signaling. Prostanoid signaling following stretch is similar to that seen after ionophore treatment, and NDGA (a non-specific LOX inhibitor) prevents both ionophore and muscular dystrophy induced membrane damage [27, 28]. Inhibition of LOX with a cocktail of 10 μM PD146176, a specific inhibitor of 15-LOX, and 10 μM BW-B 70C, a specific inhibitor of 5-LOX, also failed to prevent ERK2 phosphorylation (Drug × stretch p=0.52; fig. 2). COX and LOX represent two of the major AA metabolic pathways, COX being particularly important for the control of protein turnover and proliferation of myoblasts, but they do not appear to contribute to the rapid phosphorylation of ERK 1/2.
Figure 2.
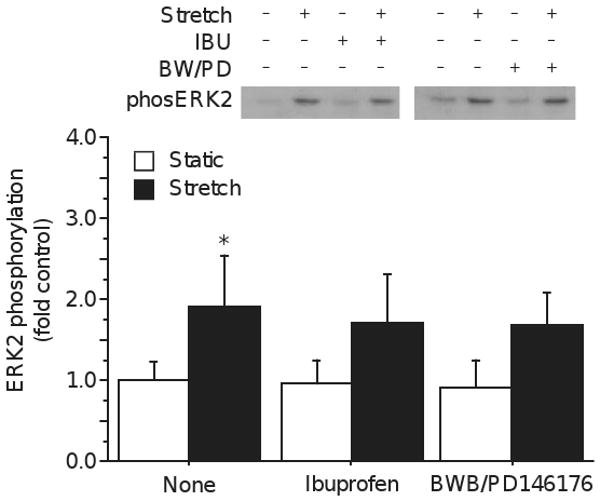
Stretch induced phosphorylation of ERK2 is not blocked by inhibition of cyclooxygenase by ibuprofen (Drug × Stretch p=0.65, power = 0.07) or of lipoxygenase with a cocktail of BW-B70C and PD146176 (Drug × Stretch p=0.52, power = 0.09). (Mean±S.D.; * Stretch p<0.05; † Drug p<0.05; ‡ Drug × Stretch p<0.05)
Generation of AA by phospholipase A2 (PLA2) is a rate-limiting step in the synthesis of prostaglandins and other eicosanoids, so PLA2 inhibitors would be expected to reduce PG production independent of COX activity. To evaluate whether the inhibition of stretch-induced ERK phosphorylation resulted from the inhibition of PLA2, myotube cultures were treated with other PLA2 inhibitors. Treatment with 100 μM quinacrine, an inhibitor of all three PLA2 classes, blocked stretch-induced ERK2 phosphorylation (Drug × Stretch p=0.02, fig. 1). Similarly, 100 μM AACOCF3, an inhibitor of cPLA2 and iPLA2 by a mechanism different than quinacrine [29], also reduced ERK2 phosphorylation (Drug × Stretch p=0.005, fig. 1). Thus, three inhibitors of PLA2 activity prevent the stretch-induced phosphorylation of ERK2, while inhibitors of subsequent AA metabolism do not. Although each of the three inhibitors has non-specific effects, they block PLA2 activity by different mechanisms and are expected to have distinct non-specific effects. The common consequence of reducing stretch-induced ERK2 phosphorylation indicates that PLA2 activity, through one of at least 8 muscle proteins with PLA2 activity [7],
When examining the effect of different venom-derived PLA2s on homogeneous liposomes, Lehtonen and Kinnunen [4] found that osmotic swelling of the liposomes increased activity at lower pressures, but inhibited the activity at higher pressures. They found that the extent of osmotic activation depended strongly on liposome composition, being greatest in liposomes composed of tightly packed, saturated fatty acid tails. Burack and Biltonen [5] suggest that PLA2 is most active at the interface between liquid ordered and liquid disordered phases, such as the boundary of lipid rafts, possibly as a consequence of high local curvature. These results seem to be related to lipid packing stresses, which are reduced by dilatation. Mechanical stretch requires gross rearrangement of the cell membrane, including substantial increase in effective surface area, and may impose sufficient dilatation and mixing to facilitate PLA2 activity. Further, the sensitivity to membrane stress varies across PLA2 enzymes, so different gene products may respond to different degrees of stretch.
PLA2 activity produces a free fatty acid and lysophospholipid. Because AA metabolites appeared not to participate in the phosphorylation of ERK2, static cultures were incubated with exogenous lipids to evaluate the influence of PLA2 products. Exposure for 15 minutes to AA, in concentrations up to 10 μM, did not alter ERK2 phosphorylation (Fig. 3). In contrast, exposure to synthetic LPC produced a dose-dependent increase in ERK2 phosphorylation, reaching statistical significance at 10 μM (Fig. 3). This suggested that the products of PLA2 activity might contribute to separate signaling cascades, with the AA contributing to control of protein synthesis and the LPC contributing to transcriptional regulation through ERK2.
Figure 3.

Phosphorylation of ERK2 was not affected by addition of exogenous AA (1 μM or 10μM), but was increased by addition of 10 or 20 μM lysophosphatidycholine (LPC). (Mean ± S.D.; † significantly different from None, p<0.003)
The presence of lysophospholipids, including LPC and lysophosphatidic acid (LPA) significantly alters local membrane biophysics, and can directly alter channel conductivity and receptor kinetics. LPC collects in cardiac myocytes during ischemia-reperfusion and contributes to calcium toxicity through bepradil and flunarizine sensitive calcium channels [30]. LPC is also capable of activating protein kinase C (PKC) [31], which may be a mechanism to phosphorylate ERK. To determine whether stretch-induced ERK2 phosphorylation depends on PKC, cultures were treated with BIM-1 for 60 minutes prior to stretch. There was a trend for 1 μM BIM-1 to reduce ERK2 phosphorylation overall (p=0.07, fig. 4), and a trend for ERK2 to be less phosphorylated in BIM-1 stretched cultures (160±34% of static vs 205±64% for vehicle-treated stretch, p=0.09), but this did not reach statistical significance. BIM-1 is an inhibitor of conventional PKCs with IC50 in the nanomolar range, well below the applied concentration, and if PKC is involved in the stretch-ERK pathway, it is likely only one of redundant branches.
Figure 4.
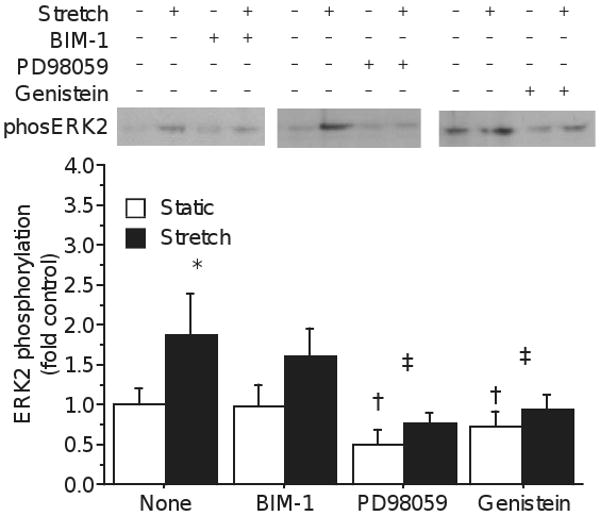
Although BIM-1 treatment appears to reduce stretch-induced ERK2 phosphorylation, statistical tests were inconclusive (Drug × Stretch p = 0.12, power = 0.32), suggesting that PKC is, at best, not a potent mediator of stretch. However, stretch-induced phosphorylation of ERK2 was blocked by both genistein (Drug × Stretch p=0.01), and PD98059 (Drug × Stretch p=0.02), indicating that both tyrosine kinase and MEK activity are required. (Mean±S.D.; * Stretch p<0.05; † Drug p<0.05; ‡ Drug × Stretch p<0.05)
To further examine the pathway leading to ERK2 phosphorylation, cultures were treated with MEK and tyrosine kinase inhibitors. Treatment with the MEK inhibitor PD98059 reduced ERK2 phosphorylation in static culture to 50±20% of vehicle treated and significantly prevented the phosphorylation of ERK2 by stretch (Drug × Stretch p=0.02; fig. 4), demonstrating that phosphorylation of ERK2 is through the canonical MEK cascade. ERK is classically phosphorylated downstream of tyrosine kinases (TKs), including receptor tyrosine kinases, like FGF receptors, and non-receptor tyrosine kinases, like Src. To test the involvement of TKs, cultures were preincubated for 60 minutes with 100 μM genistein, a broad spectrum TK inhibitor. Genistein potently reduced both the ERK phosphorylation in resting cells (p<0.01; fig. 4), and the stretch-induced increase (Drug × Stretch p=0.01). The phospho-ERK antibody recognizes the dual, Thr-185/Tyr-187 phosphorylated form of ERK2, and both genistein and PD98059 may reduce ERK2 phosphorylation through inhibition of MEK.
In summary, brief, cyclical stretch results in the rapid phosphorylation of ERK2 in primary myotubes by a mechanism dependent on PLA2 activity and protein kinases, but not COX or LOX. ERK2 phosphorylation was similarly increased by exogenous LPC, but not by AA, and this is consistent with mechanical membrane deformation facilitating PLA2 activity, with PLA2-generated LPC interacting with membrane protein to activate a tyrosine kinase, leading to activation of MEK1/2 and ERK1/2. Activation of PLA2 may promote the subsequent upregulation of AA metabolizing enzymes, such as COX2, to increase a myotube's capacity for generating growth-promoting prostaglandins, allowing synergy across daily or semi-daily activity bouts.
Acknowledgments
This work was supported by NIH grant AR48884 to Grace K. Pavlath. The NIH had no role in the design, performance or interpretation of the study.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Trappe TA, White F, Lambert CP, Cesar D, Hellerstein M, Evans WJ. Effect of ibuprofen and acetaminophen on postexercise muscle protein synthesis. Am J Physiol. 2002;282:E551–556. doi: 10.1152/ajpendo.00352.2001. [DOI] [PubMed] [Google Scholar]
- 2.Palmer RM, Reeds PJ, Atkinson T, Smith RH. The influence of changes in tension on protein synthesis and prostaglandin release in isolated rabbit muscles. Biochem J. 1983;214:1011–1014. doi: 10.1042/bj2141011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.De Deyne PG. Formation of sarcomeres in developing myotubes: role of mechanical stretch and contractile activation. Am J Physiol. 2000;279:C1801–1811. doi: 10.1152/ajpcell.2000.279.6.C1801. [DOI] [PubMed] [Google Scholar]
- 4.Lehtonen JY, Kinnunen PK. Phospholipase A2 as a mechanosensor. Biophys J. 1995;68:1888–1894. doi: 10.1016/S0006-3495(95)80366-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Burack WR, Biltonen RL. Lipid bilayer heterogeneities and modulation of phospholipase A2 activity. Chem Phys Lipids. 1994;73:209–222. doi: 10.1016/0009-3084(94)90182-1. [DOI] [PubMed] [Google Scholar]
- 6.Kudo I, Murakami M. Phospholipase A2 enzymes. Prostaglandins Other Lipid Mediat. 2002;68-69:3–58. doi: 10.1016/s0090-6980(02)00020-5. [DOI] [PubMed] [Google Scholar]
- 7.Poulsen KA, Pedersen SF, Kolko M, Lambert IH. Induction of group VIA phospholipase A2 activity during in vitro ischemia in C2C12 myotubes is associated with changes in the level of its splice variants. Am J Physiol. 2007;293:C1605–1615. doi: 10.1152/ajpcell.00012.2007. [DOI] [PubMed] [Google Scholar]
- 8.Alexander LD, Alagarsamy S, Douglas JG. Cyclic stretch-induced cPLA2 mediates ERK 1/2 signaling in rabbit proximal tubule cells. Kidney Int. 2004;65:551–563. doi: 10.1111/j.1523-1755.2004.00405.x. [DOI] [PubMed] [Google Scholar]
- 9.Willems ME, Stauber WT. Attenuation of stretch-induced histopathologic changes of skeletal muscles by quinacrine. Muscle Nerve. 2003;27:65–71. doi: 10.1002/mus.10281. [DOI] [PubMed] [Google Scholar]
- 10.Vandenburgh HH, Hatfaludy S, Sohar I, Shansky J. Stretch-induced prostaglandins and protein turnover in cultured skeletal muscle. Am J Physiol. 1990;259:C232–240. doi: 10.1152/ajpcell.1990.259.2.C232. [DOI] [PubMed] [Google Scholar]
- 11.Trappe TA, Fluckey JD, White F, Lambert CP, Evans WJ. Skeletal muscle PGF(2)(alpha) and PGE(2) in response to eccentric resistance exercise: influence of ibuprofen acetaminophen. J Clin Endocrinol Metab. 2001;86:5067–5070. doi: 10.1210/jcem.86.10.7928. [DOI] [PubMed] [Google Scholar]
- 12.Franson RC, Eisen D, Jesse R, Lanni C. Inhibition of highly purified mammalian phospholipases A2 by non-steroidal anti-inflammatory agents. Modulation by calcium ions. Biochem J. 1980;186:633–636. doi: 10.1042/bj1860633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Haddad F, Adams GR. Inhibition of MAP/ERK kinase prevents IGF-I-induced hypertrophy in rat muscles. J Appl Physiol. 2004;96:203–210. doi: 10.1152/japplphysiol.00856.2003. [DOI] [PubMed] [Google Scholar]
- 14.Wang L, Proud CG. Ras/Erk signaling is essential for activation of protein synthesis by Gq protein-coupled receptor agonists in adult cardiomyocytes. Circ Res. 2002;91:821–829. doi: 10.1161/01.res.0000041029.97988.e9. [DOI] [PubMed] [Google Scholar]
- 15.Martineau LC, Gardiner PF. Insight into skeletal muscle mechanotransduction: MAPK activation is quantitatively related to tension. J Appl Physiol. 2001;91:693–702. doi: 10.1152/jappl.2001.91.2.693. [DOI] [PubMed] [Google Scholar]
- 16.Kumar A, Chaudhry I, Reid MB, Boriek AM. Distinct signaling pathways are activated in response to mechanical stress applied axially and transversely to skeletal muscle fibers. J Biol Chem. 2002;277:46493–46503. doi: 10.1074/jbc.M203654200. [DOI] [PubMed] [Google Scholar]
- 17.Wretman C, Lionikas A, Widegren U, Lannergren J, Westerblad H, Henriksson J. Effects of concentric and eccentric contractions on phosphorylation of MAPK(erk1/2) and MAPK(p38) in isolated rat skeletal muscle. J Physiol (Lond) 2001;535:155–164. doi: 10.1111/j.1469-7793.2001.00155.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Adderley SR, Fitzgerald DJ. Oxidative damage of cardiomyocytes is limited by extracellular regulated kinases 1/2-mediated induction of cyclooxygenase-2. J Biol Chem. 1999;274:5038–5046. doi: 10.1074/jbc.274.8.5038. [DOI] [PubMed] [Google Scholar]
- 19.Meloche S, Pouyssegur J. The ERK1/2 mitogen-activated protein kinase pathway as a master regulator of the G1- to S-phase transition. Oncogene. 2007;26:3227–3239. doi: 10.1038/sj.onc.1210414. [DOI] [PubMed] [Google Scholar]
- 20.Lin LL, Wartmann M, Lin AY, Knopf JL, Seth A, Davis RJ. cPLA2 is phosphorylated and activated by MAP kinase. Cell. 1993;72:269–278. doi: 10.1016/0092-8674(93)90666-e. [DOI] [PubMed] [Google Scholar]
- 21.Bos CL, Richel DJ, Ritsema T, Peppelenbosch MP, Versteeg HH. Prostanoids and prostanoid receptors in signal transduction. Int J Biochem Cell Biol. 2004;36:1187–1205. doi: 10.1016/j.biocel.2003.08.006. [DOI] [PubMed] [Google Scholar]
- 22.Burkholder TJ. Permeability of C2C12 myotube membranes is influenced by stretch velocity. Biochem Biophys Res Commun. 2003;305:266–270. doi: 10.1016/s0006-291x(03)00756-3. [DOI] [PubMed] [Google Scholar]
- 23.Hornberger TA, Armstrong DD, Koh TJ, Burkholder TJ, Esser KA. Intracellular signaling specificity in response to uniaxial vs. multiaxial stretch: implications for mechanotransduction. Am J Physiol. 2005;288:C185–194. doi: 10.1152/ajpcell.00207.2004. [DOI] [PubMed] [Google Scholar]
- 24.Boctor AM, Eickholt M, Pugsley TA. Meclofenamate sodium is an inhibitor of both the 5-lipoxygenase and cyclooxygenase pathways of the arachidonic acid cascade in vitro. Prostaglandins Leukot Med. 1986;23:229–238. doi: 10.1016/0262-1746(86)90190-3. [DOI] [PubMed] [Google Scholar]
- 25.Smith RH, Palmer RM, Reeds PJ. Protein synthesis in isolated rabbit forelimb muscles. The possible role of metabolites of arachidonic acid in the response to intermittent stretching. Biochem J. 1983;214:153–161. doi: 10.1042/bj2140153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Otis JS, Burkholder TJ, Pavlath GK. Stretch-induced myoblast proliferation is dependent on the COX2 pathway. Exp Cell Res. 2005;310:417–425. doi: 10.1016/j.yexcr.2005.08.009. [DOI] [PubMed] [Google Scholar]
- 27.Jackson MJ, Wagenmakers AJ, Edwards RH. Effect of inhibitors of arachidonic acid metabolism on efflux of intracellular enzymes from skeletal muscle following experimental damage. Biochem J. 1987;241:403–407. doi: 10.1042/bj2410403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gati I, Danielsson O, Betmark T, Ernerudh J, Ollinger K, Dizdar N. Effects of inhibitors of the arachidonic acid cascade on primary muscle culture from a Duchenne muscular dystrophy patient. Prostaglandins Leukot Essent Fatty Acids. 2007;77:217–223. doi: 10.1016/j.plefa.2007.10.002. [DOI] [PubMed] [Google Scholar]
- 29.Blackwell GJ, Flower RJ. Inhibition of phospholipase. Br Med Bull. 1983;39:260–264. doi: 10.1093/oxfordjournals.bmb.a071830. [DOI] [PubMed] [Google Scholar]
- 30.Chen M, Xiao CY, Hashizume H, Abiko Y. Differential effects of Ca2+ channel blockers on Ca2+ overload induced by lysophosphatidylcholine in cardiomyocytes. Eur J Pharmacol. 1997;333:261–268. doi: 10.1016/s0014-2999(97)01138-2. [DOI] [PubMed] [Google Scholar]
- 31.Prokazova NV, Zvezdina ND, Korotaeva AA. Effect of lysophosphatidylcholine on transmembrane signal transduction. Biokhimiya. 1998;63:31–37. [PubMed] [Google Scholar]