

Abstract
Retroviral Gag proteins encode small peptide motifs known as late domains that promote the release of virions from infected cells by interacting directly with host cell factors. Three types of retroviral late domains, with core sequences P(T/S)AP, YPXnL, and PPPY, have been identified. HIV-1 encodes a primary P(T/S)AP-type late domain and an apparently secondary late domain sequence of the YPXnL type. The P(T/S)AP and YPXnL motifs interact with the endosomal sorting factors Tsg101 and Alix, respectively. Although biochemical and structural studies support a direct binding between HIV-1 p6 and Alix, the physiological role of Alix in HIV-1 biology remains undefined. To elucidate the function of the p6–Alix interaction in HIV-1 replication, we introduced a series of mutations in the p6 Alix binding site and evaluated the effects on virus particle production and virus replication in a range of cell types, including physiologically relevant primary T cells and macrophages. We also examined the effects of the Alix binding site mutations on virion morphogenesis and single-cycle virus infectivity. We determined that the p6–Alix interaction plays an important role in HIV-1 replication and observed a particularly severe impact of Alix binding site mutations when they were combined with mutational inactivation of the Tsg101 binding site.
Introduction
Retroviral particles are generated by the expression of the viral Gag precursor proteins (Adamson and Freed, 2007; Freed, 1998; Swanstrom and Wills, 1997). These polyprotein precursors contain functional domains responsible for carrying out all the steps necessary for virus-like particle (VLP) production. The matrix (MA) domain directs the association of Gag with the host cell membrane, capsid (CA) and nucleocapsid (NC) domains mediate Gag-Gag multimerization, and motifs known as late domains promote the release of virus particles from the plasma membrane.
Retroviral late domains interact with distinct cellular factors involved in the endosomal sorting of cargo proteins and the biogenesis of vesicles that bud into late endosomes to form multivesicular bodies (MVBs) (Bieniasz, 2006; Demirov and Freed, 2004; Fujii, Hurley, and Freed, 2007; Morita and Sundquist, 2004). At the core of this cellular budding machinery are three multiprotein complexes known as the endosomal sorting complexes required for transport (ESCRT) I, II, and III (Hurley and Emr, 2006). Retroviruses, as well as a variety of other enveloped viruses, have evolved to hijack this cellular budding machinery to facilitate the pinching off of virus particles from the infected cell. Retroviruses encode three main classes of late domain. Pro-Thr/Ser-Ala-Pro (PT/SAP) motifs interact with Tsg101, a component of ESCRT-I. Tyr-Pro-Xn-Leu (YPXnL, where X is a variable residue and n is 1-3) motifs bind the apoptosis-linked gene 2 (ALG-2)-interacting protein (Alix, formerly known as AIP1), a protein that harbors binding sites for both ESCRT-I and ESCRT-III. Pro-Pro-Pro-Tyr (PPPY) motifs interact with members of the Nedd4 family of E3 ubiquitin ligases (Bieniasz, 2006; Demirov and Freed, 2004; Morita and Sundquist, 2004). Alix is composed of three major structural domains (Fujii, Hurley, and Freed, 2007; Gottlinger, 2007; Martin-Serrano and Marsh, 2007): 1) an N-terminal domain that bears homology to yeast BroI. This domain contains a binding site for the ESCRT-III component charged multivesicular body protein 4 (CHMP4). 2) A central domain that adopts a V-like fold (hence the name V domain) that binds YPXnL motifs of HIV-1 and EIAV Gag (Zhai 2008; Fisher 2007; Lee 2007). 3) A proline-rich domain that binds a number of factors, including the ESCRT-I component Tsg101 (Fujii, Hurley, and Freed, 2007; Gottlinger, 2007; Martin-Serrano and Marsh, 2007). Alix functions not only in MVB biogenesis but also in apoptosis, endocytosis, and cytokinesis pathways (Carlton, Agromayor, and Martin-Serrano, 2008; Carlton and Martin-Serrano, 2007; Fujii, Hurley, and Freed, 2007; Gottlinger, 2007; Morita et al., 2007; Odorizzi, 2006; Trioulier et al., 2004; Vito et al., 1999).
Many retroviral Gag proteins bear multiple late domains. HIV-1, for example, encodes two late domain motifs, PT/SAP and YPXnL, in the p6 region of its Gag precursor protein, Pr55Gag. The PT/SAP motif is located between p6 residues 7 and 10. The primary Alix binding sequence is located near the C-terminus of p6, between residues 36 and 44 (36YPLASLRSL44) with p6-Y36, L41, and L44 being particularly critical for Gag–Alix binding (Martin-Serrano et al., 2003; Munshi et al., 2007; Strack et al., 2003; von Schwedler et al., 2003). Alix has also been reported to bind the NC domain of HIV-1 p6, perhaps providing alternative links between Gag and ESCRT-I and ESCRT-III (Dussupt et al., 2009; Popov et al., 2008). It is well established that the PT/SAP motif plays a major role in HIV-1 budding. Deletion of p6 severely impairs HIV-1 particle production (Gottlinger et al., 1991), a defect that maps to the PT/SAP motif (Huang et al., 1995). HIV-1 p6 binds Tsg101 in a PT/SAP-dependent manner (Garrus et al., 2001; VerPlank et al., 2001), and siRNA-mediated depletion of Tsg101 impairs HIV-1 budding (Garrus et al., 2001). Fusion of Tsg101 to the C-terminus of HIV-1 p6 rescues the budding defect imposed by PT/SAP mutation (Martin-Serrano, Zang, and Bieniasz, 2001), and overexpression of the N-terminal, Gag-binding domain of Tsg101 (TSG-5′) blocks HIV-1 budding in a dominant-negative manner (Demirov et al., 2002; Goila-Gaur et al., 2003).
Whereas the role of Tsg101 in HIV-1 release is well established, the function of Alix in HIV-1 budding is less clear. Deletions in p6 that abolish the Alix binding site (Demirov, Orenstein, and Freed, 2002; Gottlinger et al., 1991; Huang et al., 1995) or mutation of one of the key residues (Leu41) in this binding site (Huang et al., 1995; Munshi et al., 2007) do not significantly reduce particle production in HeLa or COS cells. siRNA-mediated depletion of Alix markedly inhibits the release of equine infectious anemia virus (EIAV) but not HIV-1 (Martin-Serrano et al., 2003). In contrast, deletion of the Alix binding site does have a significant effect on particle production in the context of a “minimal” HIV-1 Gag construct from which large regions have been removed (Strack et al., 2003). Overexpression of the Gag-binding, V domain of Alix potently inhibits the release of both EIAV (Chen et al., 2005; Lee et al., 2007; Munshi et al., 2007) and HIV-1 (Lee et al., 2007; Munshi et al., 2007). The Alix V domain disrupts particle budding by binding directly to Gag, as mutations in either p6 or the V domain that abrogate their interaction eliminate the budding defect (Lee et al., 2007; Munshi et al., 2007). Finally, overexpression of full-length Alix (Fisher et al., 2007; Usami, Popov, and Gottlinger, 2007) or its N-terminal Bro1 domain (Dussupt et al., 2009) rescues the defect in particle budding imposed by mutation of the PT/SAP motif. This rescue requires an intact ESCRT-III binding site in the Bro1 domain (Dussupt et al., 2009; Fisher et al., 2007; Usami, Popov, and Gottlinger, 2007). Although Alix depletion or Alix binding site mutations do not appear to significantly inhibit HIV-1 budding, they have been reported to reduce the infectivity of released particles (Fisher et al., 2007; Martin-Serrano and Bieniasz, 2003; Martin-Serrano et al., 2003), perhaps due to defects in virion morphogenesis.
The studies cited above suggest that under some circumstances, Alix may play a positive role in HIV-1 budding and infectivity; however, an analysis of the effects of Alix binding site mutations on HIV-1 release and replication in physiologically relevant cell types has not been reported. To this end, we introduced a panel of mutations into the primary Alix binding site in p6 and analyzed the effects on virus budding in HeLa cells and virus release and replication in T cells and primary monocyte-derived macrophages (MDMs). The results demonstrate a cell-type-dependent role for Alix in HIV-1 replication and also suggest that Tsg101 and Alix function cooperatively in promoting virus replication.
Results
Inhibition of the Alix–p6 interaction is induced by mutations between p6 residues 36 and 44
To define the relevance of the Alix–p6 interaction in the HIV-1 replication cycle, we introduced a series of mutations into the primary Alix binding site in p6 in the context of the full-length HIV-1 molecular clone pNL4-3 (Fig. 1A). Biochemical and structural studies implicated p6 residues Y36, L41, and L44 in Alix binding (Munshi et al., 2007; Strack et al., 2003; von Schwedler et al., 2003; Zhai et al., 2008). We therefore constructed mutants p6-Y36A, Y36S/L44H, and Y36S/L44R. For this study, we also used our previously reported mutants p6-L41R (Huang 1995) and L41A (Munshi et al., 2007) (Fig. 1A). The Y36A and L41A mutations also result in single-amino-acid changes in the overlapping pol open reading frame, in a region referred to as p6* (Fig. 1A).
Fig. 1.
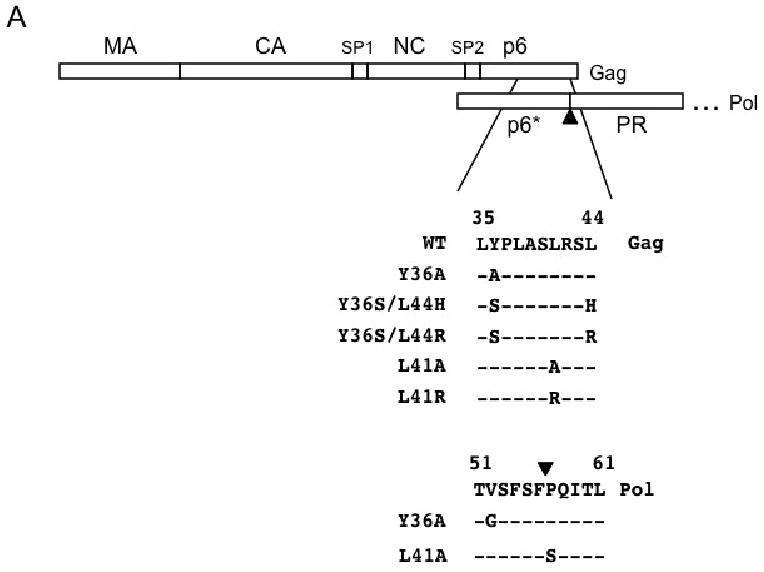
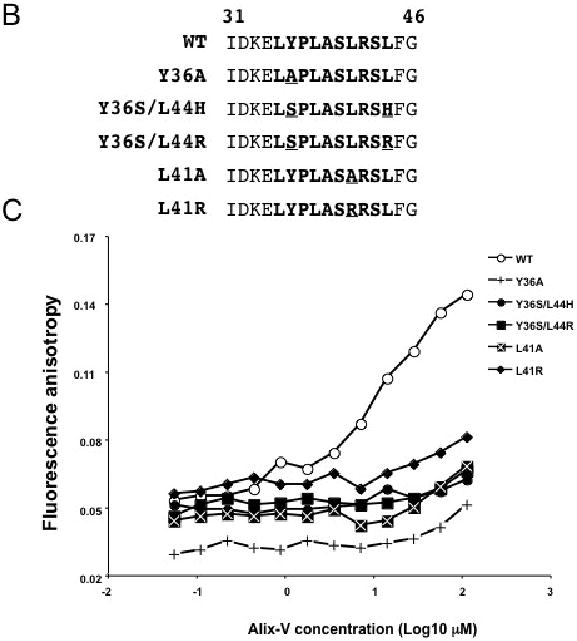
Mutational analysis of the Alix binding site in HIV-1 p6. (A) Organization of Gag and the N-terminal region of Pol, including p6* and the N-terminal portion of PR, are indicated. Amino acid sequence in Gag of WT and p6 mutants is indicated. The Y36A and L41A mutations also introduced single-amino-acid substitutions in p6*, as shown. Arrowhead indicates the cleavage site between p6* and PR. (B) Sequence of the peptides used to measure the effects of Alix binding site mutations on binding to the Alix V domain. The core Alix binding site is shown in bold face text; positions of mutations are underlined. (C) Measurement of binding between the p6-derived peptides and the Alix V domain. 50 nM concentrations of the FITC-labeled peptides were combined with increasing amounts of Alix V domain. Binding was determined by fluorescence anisotropy.
To determine the effect of the YPXnL mutations on Alix binding, we used a fluorescence anisotropy-based assay that measures the binding affinity between the purified Alix V domain and fluorescently labeled peptides derived from p6 residues I31 to G46 (Fig. 1B). A fixed amount of synthetic peptide in solution was incubated with increasing amounts of purified Alix-V. As reported previously (Luttge et al., 2008), the p6-derived peptide containing the wild-type (WT) YPXnL motif bound Alix-V efficiently (Kd ∼15 μM), whereas the mutant peptides exhibited minimal binding (Fig. 1C). These data indicate that the YPXnL mutations block the binding between p6 and Alix.
Mutations in the Alix binding site impose variable and cell-type-dependent effects on Gag processing and virus particle production
Our previous results demonstrated that the L41A and L41R mutations affect Gag processing but do not inhibit particle release in HeLa cells (Munshi et al., 2007). To examine the virus release phenotype of a larger panel of YPXnL mutants in a broader range of cell types, we performed radioimmunoprecipitation assays not only in HeLa cells but also in more physiologically relevant cell types, namely, T cells and MDM. For the HeLa experiments (Fig. 2A), cells were transfected in parallel with WT or p6-mutant molecular clones and were metabolically labeled with [35S]Met/Cys. The levels of cell- and virion-associated HIV-1 proteins were determined by radioimmunoprecipitation of cell and virion lysates with anti-HIV-1 immunoglobulin (HIV-Ig). Virus release efficiency was calculated based on the levels of virion-associated Gag relative to the total (cell plus virion) Gag expressed (see Materials and methods). As reported previously (Munshi et al., 2007), the L41A and L41R mutations did not reduce the efficiency of virus particle production. However, both L41A and L41R increased the ratio of p25 (the CA-SP1 Gag processing intermediate) to mature p24 (CA). This was seen most clearly when the radioimmunoprecipitates were resolved by polyacrylamide gel electrophoresis under conditions that allowed separation of p25 and p24 bands (Fig. 2A, lower gel panel). As reported previously (Huang et al., 1995), the PTAP- mutation severely (by ∼10-fold) reduced particle production, as did the PTAP-/L41A double mutant (Fig. 2A). The Y36S/L44H and Y36S/L44R double mutations did not reduce virus budding but increased the p25 to p24 ratio (Fig. 2A). Interestingly, the Y36A mutant exhibited a phenotype distinct from that of the other Alix binding site mutants; namely, it displayed a marked (∼5-fold) defect in virus release efficiency, coupled with a striking increase in the ratio of Pr55Gag to p24 (Fig. 2A). This phenotype was observed consistently in a number of independent experiments (n = 7). The ratio of p25/p24 to Pr55Gag was 1.7 +/- 0.59 for the WT and 0.09 +/- 0.04 for Y36A. Because the Y36A mutation also introduced a single amino acid change in the overlapping pol-encoded p6* peptide (Fig. 1A), it seemed possible that the defect in Gag processing and particle release could be due to dysregulated PR activity. However, we observed that the Y36A mutation reduced virus particle production by ∼5-fold in the context of a molecular clone encoding an enzymatically inactive PR (data not shown). Thus, the Y36A mutation disrupts HIV-1 particle release independent of PR activity.
Fig. 2.
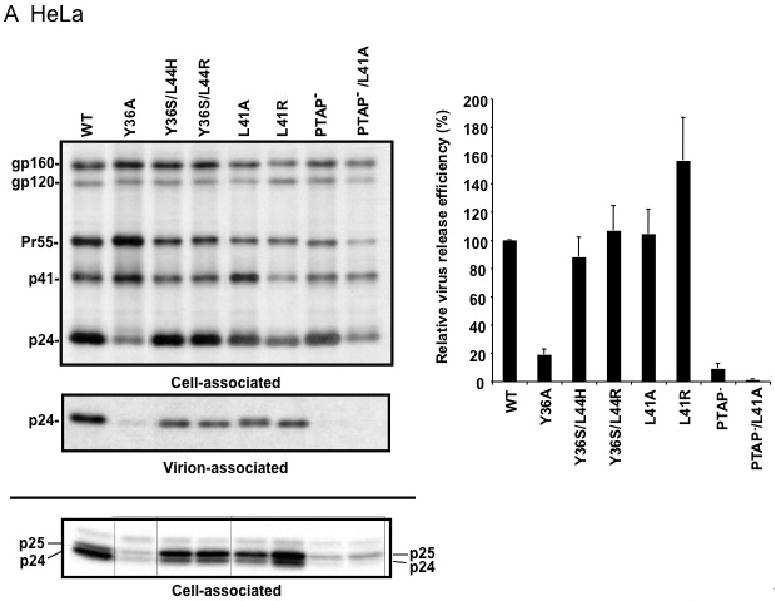
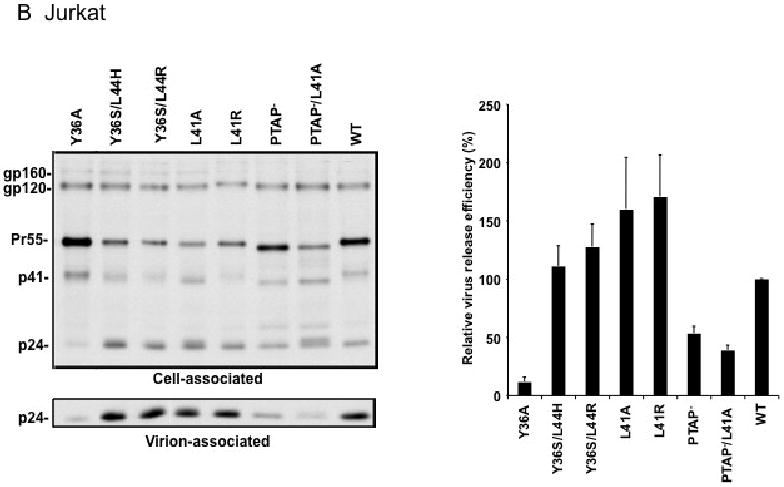
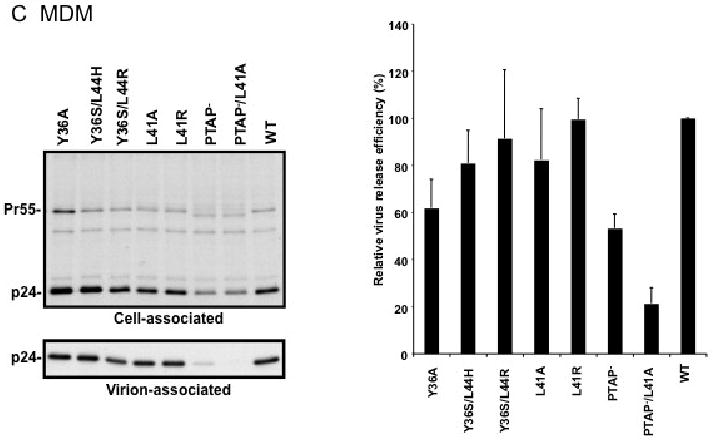
Effect of p6 mutations on HIV-1 particle production in HeLa cells (A), Jurkat T cells (B), and primary MDM (C). HeLa cells were transiently transfected with WT pNL4-3 or p6-mutant derivatives. Jurkat cells were infected with virus stocks obtained by transfecting HeLa cells with WT or mutant pNL4-3 molecular clones, pCMVNLGagPolRRE, and pHCMV-G (see Materials and methods). Primary MDMs were infected with virus stocks obtained by transfecting HeLa cells with pNL(AD8)R- molecular clones encoding WT or mutant p6, pCMVNLGagPolRRE, and pHCMV-G. Transfected or infected cells were metabolically labeled with [35S]Met/Cys, and virions were pelleted by ultracentrifugation. Cell and virion lysates were immunoprecipitated with HIV-Ig and analyzed by SDS-PAGE, followed by phosphorimager analysis. Virus release efficiencies were calculated as the amount of radiolabeled, virion-associated Gag as a fraction of the total (cell plus virion) radiolabeled Gag protein detected, +/- SE. n = 7 (A) or 3 (B and C). In the lower portion of panel (A), is shown a gel run under conditions that optimize the separation of CA-SP1 (p25) and CA (p24). Positions of the Env glycoprotein precursor gp160, the mature surface Env glycoprotein gp120, the Gag precursor Pr55Gag (Pr55), the Gag processing intermediates p41Gag (p41) and CA-SP1 (p25), and CA (p24) are shown.
We next analyzed the effect of the Alix binding site mutations on particle release in more physiologically relevant cell types. WT and mutant virions pseudotyped with vesicular stomatitis virus G glycoprotein (VSV-G) (see Materials and methods) were used to infect the Jurkat T-cell line and virus release efficiency was measured by radioimmunoprecipitation. As in HeLa cells, the L41A, L41R, Y36S/L44R, Y36S/L44H mutations had no significant effect on virus release, whereas the Y36A mutation severely reduced (by ∼7-fold) particle production (Fig. 2B). We also consistently observed an increase in the ratio of Pr55Gag to p24 in cells expressing the Y36A mutant. The PTAP- mutant showed a less pronounced virus release impairment in Jurkat (2-fold) than in HeLa cells (10-fold), consistent with our earlier findings (Demirov, Orenstein, and Freed, 2002). We also examined the effect of the Alix binding site mutations on virus release in primary MDMs (Fig. 2C). Interestingly, not only the L41A, L41R, Y36S/L44R, and Y36S/L44H mutants but also the Y36A mutant showed levels of virus release that were reduced by no more than ∼40%. Likewise, the PTAP- mutant displayed only a modest impairment in particle production in this primary cell type. However, combining mutations in the Tsg101 (PTAP-) and Alix (L41A) binding sites led to a severe (5-fold) defect in particle production (Fig. 2C).
Mutations in the Alix binding site impair HIV-1 replication, particularly when coupled with disruption of the PTAP motif
To evaluate the effect of Alix binding site mutations on virus replication kinetics, we transfected two T-cell lines, Jurkat and CEM(12D7), with full-length pNL4-3 derivatives bearing the Alix binding site mutations, the PTAP- mutations, or combined PTAP- and L41A mutations. Virus replication was monitored by measuring the levels of reverse transcriptase (RT) activity in the medium over time. As we demonstrated previously (Demirov, Orenstein, and Freed, 2002), the PTAP- mutant replicated with some delay relative to WT in Jurkat T cells (Fig. 3A) but showed no detectable replication in CEM(12D7) (Fig. 3B). In Jurkat cells, the Alix binding site mutants displayed significant delays relative to WT, with peak RT levels occurring ∼1-2 weeks later than observed with the WT. In CEM(12D7), replication of the Alix binding site mutants peaked 4-6 days later than WT. Interestingly, combining the PTAP- and L41 mutations abolished virus replication in both cell types (Fig. 3A and B).
Fig. 3.
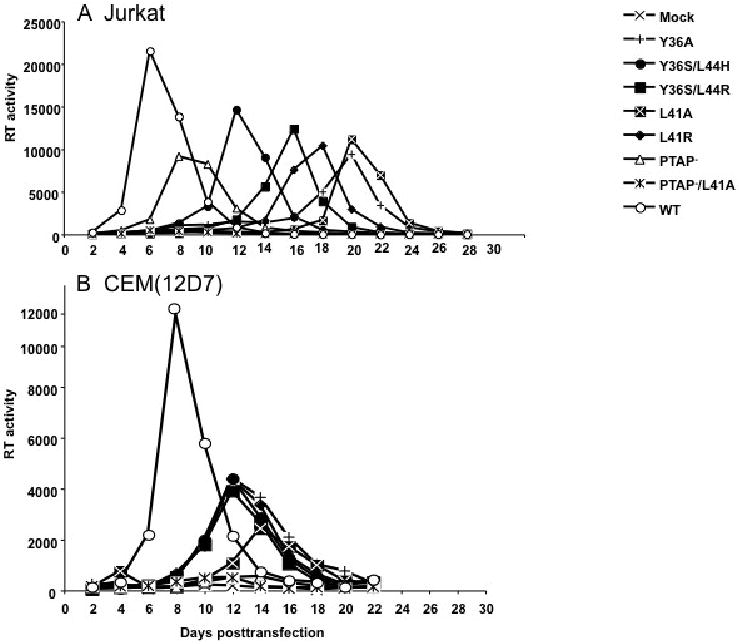
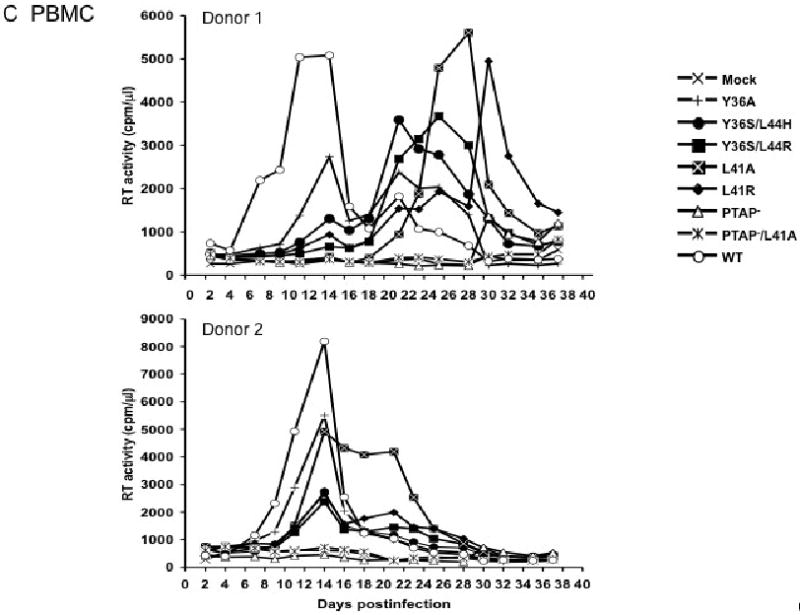
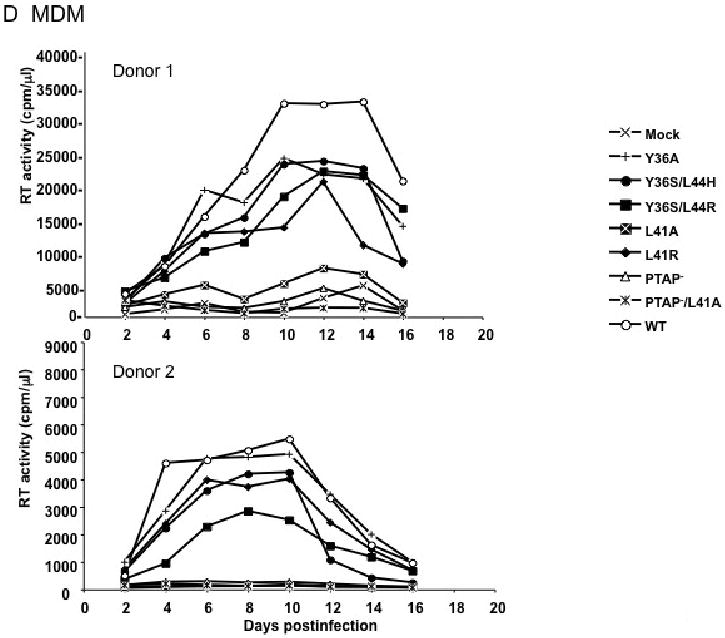
Effect of p6 mutations on virus replication in the Jurkat (A), or CEM(12D7) (B) T-cell lines, PBMC (C), and primary MDM (D). Jurkat and CEM(12D7) T-cell lines were transfected with WT or p6-mutant pNL4-3 molecular clones. Cells were split and culture supernatants collected for RT analysis every 2-3 days. PBMCs were infected with virus stocks obtained by transfecting HeLa cells with WT or the indicated mutant pNL4-3 molecular clones, pCMVNLGagPolRRE, and pHCMV-G. MDMs were infected with virus stocks obtained by transfecting HeLa cells with pNL(AD8)R- molecular clones encoding WT or mutant p6, pCMVNLGagPolRRE, and pHCMV-G. Culture supernatants were reserved every 2 days for RT analysis.
To elucidate the function of the YPXnL motif in HIV-1 replication in primary CD4+ lymphocytes, we prepared VSV-G-pseudotyped virus stocks and used them to infect human peripheral blood mononuclear cells (PBMCs) from several different donors (Fig. 3C). In general, the Alix binding site mutants displayed moderate to severe defects in virus replication; however, donor-to-donor variability was observed. For example, in donor 1, all the Alix binding site mutants replicated with a significant delay relative to the WT. In donor 2, the Alix binding site mutants produced peak RT levels that were approximately 2-5-fold lower than those detected in WT-infected cultures. These data demonstrate that an intact Alix binding site promotes, but is not essential for, HIV-1 replication in primary human PBMCs. The PTAP- and PTAP-/L41A mutants showed no detectable replication in these experiments, although in some assays measurable replication of PTAP- was observed (data not shown), consistent with our earlier report (Demirov, Orenstein, and Freed, 2002).
Finally, we tested the phenotypes of the Alix binding site mutants in primary MDM. The YPXnL Alix-binding motif overlaps with the region of p6 that interacts with the viral accessory protein Vpr (Kondo and Gottlinger, 1996), and Vpr has been shown to be important for HIV-1 replication in MDM (Balliet et al., 1994; Connor et al., 1995; Heinzinger et al., 1994). We therefore performed these experiments in the context of a molecular clone that is both macrophage-tropic and lacks an intact Vpr gene [pNL(AD8)R-; (Freed, Englund, and Martin, 1995)]. In cells obtained from two independent donors, we observed that several of the Alix binding site mutants (Y36A, Y36S/L44H, Y36S/L44R, and L41R) displayed only modest reductions in peak RT values (Fig. 3D). In contrast, the L41A mutant showed severely defective virus replication. As we reported previously (Demirov, Orenstein, and Freed, 2002) replication of the PTAP- mutant was completely blocked, as was that of PTAP-/L41A (Fig. 3D).
Morphology of Alix binding site mutants
The biochemical analysis presented above indicated that some of the Alix binding site mutants, Y36A in particular, induced significant defects in Gag processing, especially in HeLa and T cells. This raised the possibility that defects in virion morphogenesis might be observed. To evaluate this question, we performed thin-section transmission electron microscopy (EM) of HeLa cells transfected with WT or mutant molecular clones (Fig. 4A). To quantify these data, 200 virions per sample were examined and classified into one of several categories: released mature, released immature, released aberrant, budding aberrant, and budding (Fig. 4B). For the WT, approximately 70% of particles were classified as released mature, with small numbers of budding and immature virions detected. As expected (Demirov, Orenstein, and Freed, 2002; Huang et al., 1995), PTAP- and PTAP-/L41A mutants produced very few mature virions and increased numbers, relative to WT, of immature particles and tethered budding structures. While Y36S/L44H, Y36S/L44R, L41A, and L41R mutants produced significant numbers of mature virions (∼50% of the total) they also showed an increase in the number of immature particles. The most striking phenotype for the Alix binding site mutants was that of Y36A, which produced only ∼25% mature virions and a large number (∼30%) of aberrant particles. These data demonstrate rather modest effects of most Alix binding site mutations on virion morphology, but more severe effects in the case of the Y36A mutant. Despite the increased numbers of aberrant particles observed for Y36A, it is important to note that this mutant displayed a significantly higher percentage of mature particles than did PTAP- and PTAP-/L41A (Fig. 4B).
Fig. 4.
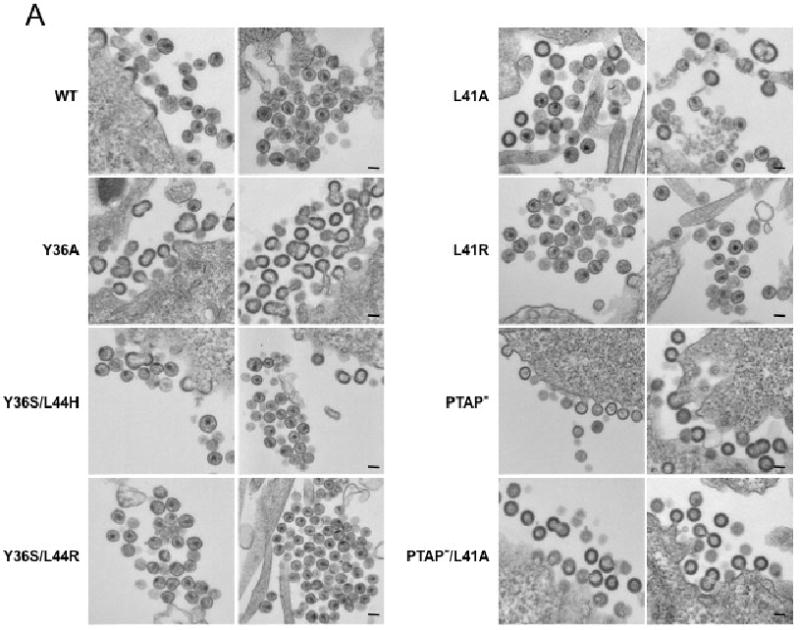
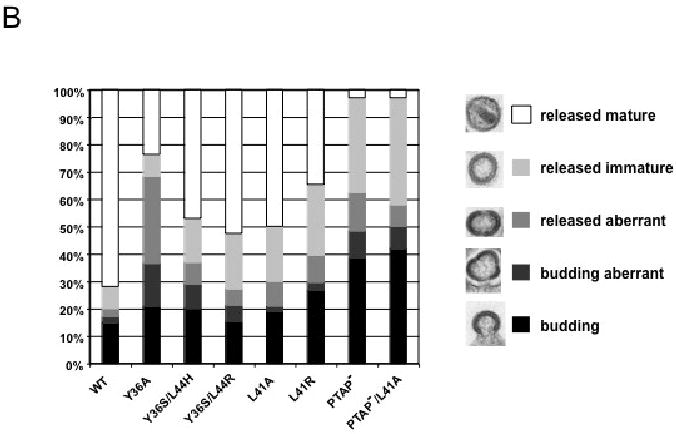
Effect of p6 mutations on virion morphogenesis. (A) HeLa cells were transiently transfected with WT pNL4-3 or derivatives containing the indicated p6 mutations. Transfected cells were fixed 24 hr posttransfection and processed for transmission EM. Scale bar indicates 100 nm. (B) Quantification of virion morphology. >200 virions/sample were examined and categorized as budding, budding aberrant, released aberrant, released immature, and released mature.
Single-cycle infectivity of Alix binding site mutants
Because the analyses presented in Fig. 3 measure virus replication in multiple rounds of infection, we wished to determine the effect of the Alix binding site mutations on single-cycle virus infectivity. For this purpose, we used the CD4+, CCR5+, CXCR4+ HeLa-derived indicator cell line TZM-bl, which harbors integrated luciferase and lacZ reporter genes under transcriptional control of the HIV-1 LTR (Wei et al., 2002). WT and mutant virus stocks were generated in HeLa cells, normalized for RT activity, and used to infect TZM-bl cells. We observed that the Alix binding site mutations reduced infectivity by approximately two-fold (Fig. 5) under a range of input inocula (data not shown). In contrast, mutation of the PTAP motif very severely disrupted virus infectivity. These results demonstrate that mutational inactivation of the Alix binding site modestly inhibits single-round HIV-1 infectivity.
Fig. 5.
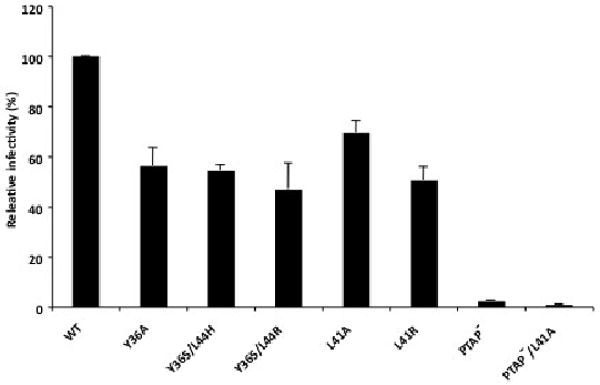
Effect of Alix binding site mutations on single-cycle infectivity. Virus stocks were prepared in HeLa cells, normalized for RT activity, and used to infect TZM-bl cells. Luciferase activity was measured two days postinfection. n >3, +/- SD.
Discussion
The results of this study clearly demonstrate a role for the Alix binding site of p6 in HIV-1 replication. Although all of the mutations reported here severely disrupt p6–Alix binding, the phenotypes of the individual mutants are not identical. In particular, the Y36A mutation imposes a Gag processing defect in HeLa and Jurkat cells that is not evident with the other mutants; this defect is characterized by an increase in the ratio of Pr55Gag to p25/p24. The Y36A mutation also impairs particle budding in HeLa cells, as visualized by the accumulation of defective tethered virions at the plasma membrane.
In addition to the substitution in p6, the Y36A mutation also introduces a Val-to-Gly change in codon 52 of pol, in the p6* domain upstream of the PR coding region. It is possible that this substitution in p6* contributes to the defects in Gag processing and particle budding observed with the Y36A mutant. However, in primary MDM this mutation does not impose a Gag processing defect, and indeed, Y36A replicates in this primary cell type with kinetics similar to those of the WT. We also observed that the virus release defect imposed by Y36A was maintained in the context of an inactive PR. This latter result indicates that the release defect imposed by Y36A is not a result of dysregulation of PR activity. Finally, a recent study demonstrated that substitution of residue 52 of p6* had no effect on HIV-1 replication, even in highly sensitive virus competition assays (Leiherer, Ludwig, and Wagner, 2009). These observations all suggest that it is the Y36A mutation in p6, not the V52G change in p6*, that is the cause of the defects observed for Y36A. The L41A mutation also introduces a coding change in pol, in contrast to L41R, which does not. The phenotypes of these two mutants are similar in terms of virus release efficiency, virion morphology, replication in T-cell lines and primary T cells; however, the L41A mutant appears to be more impaired in primary MDMs.
It is noteworthy that the particle tethering phenotype of Y36A closely resembles that induced by overexpression of the Alix V domain (Lee et al., 2007; Munshi et al., 2007). Because several of the Alix binding site mutants produce virus particles efficiently in HeLa cells, these results suggest that the Y36A mutation, and the overexpressed Alix V domain, act to disrupt particle budding by mechanisms that entail more than simply blocking the p6–Alix interaction. The defect induced by Y36A or by Alix V domain overexpression could, for example, be caused by global changes in p6 folding which could potentially influence the upstream interaction with Tsg101. Because all of the Alix binding site mutants appear to be severely defective for p6-Alix interaction (Fig. 1C) yet exhibit some striking differences in terms of replication capacity and virion morphology, it is possible that some of the mutations (Y36A in particular) may impose defects in addition to impaired Alix binding. However, despite their altered morphology, Y36A virions exhibit reductions in infectivity of only ∼2-fold. This is likely due to the fact that the Y36A mutant still generates a significant fraction of mature virions (∼2-3-fold reduced relative to WT as opposed to ∼10-20-fold reduced for PTAP-; Fig. 4B).
Mutations in the Alix binding site resulted in significant delays in virus replication in a range of cell types (Fig. 3). Viruses collected at the delayed RT peaks demonstrated enhanced replication kinetics upon repassage in fresh Jurkat T cells relative to the original mutants. Sequencing of the putative revertant isolates revealed several potential compensatory mutations (Fujii and Freed, unpublished). Y36A acquired a p6-L41I mutation. Several of the mutants acquired single-amino-acid substitutions in the gp120 and gp41 Env glycoproteins and also truncations in the Vpu open reading frame. Whether these p6, Env, and Vpu mutations are able to rescue the replication defects imposed by the Alix binding site changes, and the mechanism by which they might do so, are currently under investigation.
With the exception of Y36A, the virus release defect in HeLa cells imposed by Alix binding site mutations is considerably less severe than that observed with PT/SAP mutations that disrupt the p6–Tsg101 interaction. As reported earlier for L41A and L41R (Munshi et al., 2007), the only indication of a budding defect for most of the mutants in HeLa cells is an increase in the levels of the Gag processing intermediate p25 (CA-SP1) and p41 (MA-CA) (Fig. 2A and data not shown). Because the processing of CA-SP1 to CA occurs late in the Gag processing cascade, CA-SP1 accumulation frequently accompanies a budding block (Demirov, Orenstein, and Freed, 2002; Garrus et al., 2001; Gottlinger et al., 1991). Although the release defect imposed by PT/SAP mutations is more severe than that observed for Alix binding site substitutions, the latter class of mutants displayed significant and cell-type-dependent defects in virus replication. For example, in Jurkat T cells, mutation of Y36, L41, and L44 had a more severe effect on virus replication kinetics than did the PTAP- mutation. The situation was reversed in CEM(12D7) cells in which, as reported previously (Demirov, Orenstein, and Freed, 2002; Huang et al., 1995), the PTAP- mutant showed no detectable replication whereas the Alix binding site mutants were only modestly delayed. Significantly from a biological relevance point of view, in primary MDM the Alix binding site mutations were relatively well tolerated whereas the PTAP- mutations completely blocked replication. Regardless of the relative impact on virus replication of mutations in the Tsg101 and Alix binding sites, combining these two sets of mutations (e.g., PTAP-/L41A) consistently resulted in a highly defective phenotype, independent of host cell type. One possible explanation for the differences in severity of Alix binding site mutations is that the target cell types analyzed here express different levels of Alix and Tsg101. For example, cell types that express high levels of Alix and low levels of Tsg101 would be predicted to be more dependent on Alix for the establishment of a productive spreading infection; conversely, very high levels of Alix expression could promote Tsg101-independent release, as has been observed (Fisher et al., 2007; Usami, Popov, and Gottlinger, 2007). However, we observed that the Jurkat T-cell line expresses higher levels of Tsg101 than does CEM(12D7) and that both T-cell lines express comparable levels of Alix (Fujii and Freed, unpublished). These results suggest that differences in Tsg101 and Alix expression levels are not sufficient to explain the cell-type-dependent phenotypes observed here. Further studies will be required to clarify this issue, and in a broader sense, to elucidate the basis for the late domain redundancy so often observed in retroviral Gag proteins. This is not only an interesting question from the perspective of understanding virus budding but also is critical in determining whether successful antiviral strategies that target virus release will need to simultaneously disrupt Gag interactions with multiple late domain partners.
Materials and methods
Cells
HeLa cells were cultured in Dulbecco's-modified Eagle medium supplemented with 10% fetal bovine serum (FBS) as described previously (Freed and Martin, 1994). The Jurkat and CEM(12D7) T-cell lines were maintained in RPMI-1640 supplemented with 10% FBS as previously described (Demirov, Orenstein, and Freed, 2002; Freed and Martin, 1994). Human PBMC and primary MDM were prepared and cultured as reported (Demirov, Orenstein, and Freed, 2002; Freed, Englund, and Martin, 1995; Gousset et al., 2008). TZM-bl cells (Wei et al., 2002) were obtained from J. Kappes though the NIH AIDS Research and Reference Reagent Program.
Plasmids, mutagenesis, and DNA cloning
The full-length HIV-1 molecular clones pNL4-3 (Adachi et al., 1986), the macrophage-tropic derivative pNL(AD8) (Freed, Englund, and Martin, 1995), and the Vpr-defective pNL(AD8) [pNL(AD8)R-; (Freed, Englund, and Martin, 1995)) were used for this study. The PTAP-, L41R (Huang et al., 1995) and L41A (Munshi et al., 2007) mutants have been described previously. The remaining mutants were constructed by PCR-based mutagenesis methods as reported (Goila-Gaur et al., 2003). Primer sequences will be made available upon request. The pNL(AD8)R-versions of the p6 mutants were constructed by introducing the PflMI-BamHI fragment (pNL4-3 nucleotide 5304-8466) from pNL(AD8)R- into the pNL4-3 derivatives containing the Alix binding site mutations. Plasmid DNA was purified with the plasmid purification maxi prep kit (QIAGEN, Valencia, CA).
Transfections, preparation of virus stocks, and infections
HeLa cell transfections were performed with the Lipofectamin 2000 reagent (Invitrogen, Carlsbad, CA) according to the manufacturer's instructions. Jurkat and CEM(12D7) T cells were transfected with DEAE-dextran as previously reported (Freed et al., 1994). For preparing VSV-G-pseudotyped virus stocks, HeLa cells were cotransfected with WT or mutant molecular clones, a VSV-G expression vector [pHCMV-G (Yee, Friedmann, and Burns, 1994)] and the WT HIV-1 GagPol expression plasmid pCMVNLGagPolRRE (Ono and Freed, 2001). Transfected-cell supernatants were passed through a 0.45-μm-pore-size filter, normalized for RT activity (Freed and Martin, 1994), and used in infections as indicated. Infections of T-cell lines, PBMC, and MDM were performed as described (Demirov, Orenstein, and Freed, 2002). Virus stocks were normalized for RT activity prior to infections. Infectivity assays in TZM-bl cells were performed as reported (Waheed et al., 2006).
Radioimmunoprecipitation and Western blot analysis
The quantitative radioimmunoprecipitation virus release assay has been described in detail previously (Demirov, Orenstein, and Freed, 2002; Waheed, Ono, and Freed, 2009). Briefly, transfected or infected cells were metabolically labeled with [35S]Met/Cys. After approximately 16 h of metabolic labeling, virions were pelleted by ultracentrifugation. Cell and virus lysates were immunoprecipitated with HIV immunoglobulin (HIV-Ig) obtained from the NIH AIDS Research and Reference Reagent Program and subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), followed by fluorography. Virus release efficiency was calculated as the amount of virion p24 divided by total Gag (cell Pr55Gag + cell p24 + virion p24). For resolution of p25 (CA-SP1) and p24 (CA) bands, samples were separated by SDS-PAGE on 12% gels for 4-5 hrs at 200V.
EM analysis
Transfected HeLa cells were fixed in 2% glutaraldehyde-100 mM sodium cacodylate solution. The methods for transmission EM were described previously (Freed et al., 1994).
Fluorescence anisotropy
The methods used for purification of the Alix V domain have been described (Luttge et al., 2008). Custom peptides were synthesized and amino terminally labeled with fluorescein isothiocyanate (FITC; EZBiolabs). FITC-conjugated peptides at 50 nM were incubated with 1 to 100 μM concentrations of purified Alix V domain. Methods for detection of protein–peptide interactions by fluorescence anisotropy have been described previously (Liu et al., 2006).
Acknowledgments
We thank members of the Freed laboratory for helpful discussion and critical review of the manuscript. Purification of Alix-V protein was performed by the Protein Expression Laboratory, SAIC, NCI-Frederick. HIV-Ig was obtained through the NIH AIDS Research and Reference Reagent Program. This research was supported by the Intramural Research Program of the Center for Cancer Research, National Cancer Institute, NIH, and by the Intramural AIDS Targeted Antiviral Program. This project was funded in part with federal funds from the National Cancer Institute, NIH, under contract N01-CO-12400. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adachi A, Gendelman HE, Koenig S, Folks T, Willey R, Rabson A, Martin MA. Production of acquired immunodeficiency syndrome-associated retrovirus in human and nonhuman cells transfected with an infectious molecular clone. J Virol. 1986;59(2):284–91. doi: 10.1128/jvi.59.2.284-291.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adamson CS, Freed EO. Human immunodeficiency virus type 1 assembly, release, and maturation. Adv Pharmacol. 2007;55:347–87. doi: 10.1016/S1054-3589(07)55010-6. [DOI] [PubMed] [Google Scholar]
- Balliet JW, Kolson DL, Eiger G, Kim FM, McGann KA, Srinivasan A, Collman R. Distinct effects in primary macrophages and lymphocytes of the human immunodeficiency virus type 1 accessory genes vpr, vpu, and nef: mutational analysis of a primary HIV-1 isolate. Virology. 1994;200(2):623–31. doi: 10.1006/viro.1994.1225. [DOI] [PubMed] [Google Scholar]
- Bieniasz PD. Late budding domains and host proteins in enveloped virus release. Virology. 2006;344(1):55–63. doi: 10.1016/j.virol.2005.09.044. [DOI] [PubMed] [Google Scholar]
- Carlton JG, Agromayor M, Martin-Serrano J. Differential requirements for Alix and ESCRT-III in cytokinesis and HIV-1 release. Proc Natl Acad Sci U S A. 2008;105(30):10541–6. doi: 10.1073/pnas.0802008105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlton JG, Martin-Serrano J. Parallels between cytokinesis and retroviral budding: a role for the ESCRT machinery. Science. 2007;316(5833):1908–12. doi: 10.1126/science.1143422. [DOI] [PubMed] [Google Scholar]
- Chen C, Vincent O, Jin J, Weisz OA, Montelaro RC. Functions of early (AP-2) and late (AIP1/ALIX) endocytic proteins in equine infectious anemia virus budding. J Biol Chem. 2005;280(49):40474–80. doi: 10.1074/jbc.M509317200. [DOI] [PubMed] [Google Scholar]
- Connor RI, Chen BK, Choe S, Landau NR. Vpr is required for efficient replication of human immunodeficiency virus type-1 in mononuclear phagocytes. Virology. 1995;206(2):935–44. doi: 10.1006/viro.1995.1016. [DOI] [PubMed] [Google Scholar]
- Demirov DG, Freed EO. Retrovirus budding. Virus Res. 2004;106(2):87–102. doi: 10.1016/j.virusres.2004.08.007. [DOI] [PubMed] [Google Scholar]
- Demirov DG, Ono A, Orenstein JM, Freed EO. Overexpression of the N-terminal domain of TSG101 inhibits HIV-1 budding by blocking late domain function. Proc Natl Acad Sci U S A. 2002;99(2):955–60. doi: 10.1073/pnas.032511899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demirov DG, Orenstein JM, Freed EO. The late domain of human immunodeficiency virus type 1 p6 promotes virus release in a cell type-dependent manner. J Virol. 2002;76(1):105–117. doi: 10.1128/JVI.76.1.105-117.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dussupt V, Javid MP, Abou-Jaoude G, Jadwin JA, de La Cruz J, Nagashima K, Bouamr F. The nucleocapsid region of HIV-1 Gag cooperates with the PTAP and LYPXnL late domains to recruit the cellular machinery necessary for viral budding. PLoS Pathog. 2009;5(3):e1000339. doi: 10.1371/journal.ppat.1000339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher RD, Chung HY, Zhai Q, Robinson H, Sundquist WI, Hill CP. Structural and biochemical studies of ALIX/AIP1 and its role in retrovirus budding. Cell. 2007;128(5):841–52. doi: 10.1016/j.cell.2007.01.035. [DOI] [PubMed] [Google Scholar]
- Freed EO. HIV-1 gag proteins: diverse functions in the virus life cycle. Virology. 1998;251(1):1–15. doi: 10.1006/viro.1998.9398. [DOI] [PubMed] [Google Scholar]
- Freed EO, Englund G, Martin MA. Role of the basic domain of human immunodeficiency virus type 1 matrix in macrophage infection. J Virol. 1995;69(6):3949–3954. doi: 10.1128/jvi.69.6.3949-3954.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freed EO, Martin MA. Evidence for a functional interaction between the V1/V2 and C4 domains of human immunodeficiency virus type 1 envelope glycoprotein gp120. J Virol. 1994;68(4):2503–2512. doi: 10.1128/jvi.68.4.2503-2512.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freed EO, Orenstein JM, Buckler-White AJ, Martin MA. Single amino acid changes in the human immunodeficiency virus type 1 matrix protein block virus particle production. J Virol. 1994;68(8):5311–5320. doi: 10.1128/jvi.68.8.5311-5320.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii K, Hurley JH, Freed EO. Beyond Tsg101: the role of Alix in ‘ESCRTing’ HIV-1. Nat Rev Microbiol. 2007;5(12):912–6. doi: 10.1038/nrmicro1790. [DOI] [PubMed] [Google Scholar]
- Garrus JE, von Schwedler UK, Pornillos OW, Morham SG, Zavitz KH, Wang HE, Wettstein DA, Stray KM, Cote M, Rich RL, Myszka DG, Sundquist WI. Tsg101 and the vacuolar protein sorting pathway are essential for HIV-1 budding. Cell. 2001;107(1):55–65. doi: 10.1016/s0092-8674(01)00506-2. [DOI] [PubMed] [Google Scholar]
- Goila-Gaur R, Demirov DG, Orenstein JM, Ono A, Freed EO. Defects in human immunodeficiency virus budding and endosomal sorting induced by TSG101 overexpression. J Virol. 2003;77(11):6507–19. doi: 10.1128/JVI.77.11.6507-6519.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottlinger HG. How HIV-1 hijacks ALIX. Nat Struct Mol Biol. 2007;14(4):254–6. doi: 10.1038/nsmb0407-254. [DOI] [PubMed] [Google Scholar]
- Gottlinger HG, Dorfman T, Sodroski JG, Haseltine WA. Effect of mutations affecting the p6 gag protein on human immunodeficiency virus particle release. Proc Natl Acad Sci U S A. 1991;88(8):3195–9. doi: 10.1073/pnas.88.8.3195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gousset K, Ablan SD, Coren LV, Ono A, Soheilian F, Nagashima K, Ott DE, Freed EO. Real-time visualization of HIV-1 GAG trafficking in infected macrophages. PLoS Pathog. 2008;4(3):e1000015. doi: 10.1371/journal.ppat.1000015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinzinger NK, Bukinsky MI, Haggerty SA, Ragland AM, Kewalramani V, Lee MA, Gendelman HE, Ratner L, Stevenson M, Emerman M. The Vpr protein of human immunodeficiency virus type 1 influences nuclear localization of viral nucleic acids in nondividing host cells. Proc Natl Acad Sci U S A. 1994;91(15):7311–5. doi: 10.1073/pnas.91.15.7311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang M, Orenstein JM, Martin MA, Freed EO. p6Gag is required for particle production from full-length human immunodeficiency virus type 1 molecular clones expressing protease. J Virol. 1995;69(11):6810–8. doi: 10.1128/jvi.69.11.6810-6818.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurley JH, Emr SD. The ESCRT complexes: structure and mechanism of a membrane-trafficking network. Annu Rev Biophys Biomol Struct. 2006;35:277–98. doi: 10.1146/annurev.biophys.35.040405.102126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo E, Gottlinger HG. A conserved LXXLF sequence is the major determinant in p6gag required for the incorporation of human immunodeficiency virus type 1 Vpr. J Virol. 1996;70(1):159–64. doi: 10.1128/jvi.70.1.159-164.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Joshi A, Nagashima K, Freed EO, Hurley JH. Structural basis for viral late-domain binding to Alix. Nat Struct Mol Biol. 2007;14(3):194–9. doi: 10.1038/nsmb1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leiherer A, Ludwig C, Wagner R. Influence of extended mutations of the HIV-1 transframe protein p6 on Nef-dependent viral replication and infectivity in vitro. Virology. 2009 doi: 10.1016/j.virol.2009.01.042. [DOI] [PubMed] [Google Scholar]
- Liu F, Stephen AG, Adamson CS, Gousset K, Aman MJ, Freed EO, Fisher RJ, Burke TR., Jr Hydrazone- and hydrazide-containing N-substituted glycines as peptoid surrogates for expedited library synthesis: application to the preparation of Tsg101-directed HIV-1 budding antagonists. Org Lett. 2006;8(22):5165–8. doi: 10.1021/ol0622211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luttge BG, Shehu-Xhilaga M, Demirov DG, Adamson CS, Soheilian F, Nagashima K, Stephen AG, Fisher RJ, Freed EO. Molecular characterization of feline immunodeficiency virus budding. J Virol. 2008;82(5):2106–19. doi: 10.1128/JVI.02337-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Serrano J, Bieniasz PD. A bipartite late-budding domain in human immunodeficiency virus type 1. J Virol. 2003;77(22):12373–7. doi: 10.1128/JVI.77.22.12373-12377.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Serrano J, Marsh M. ALIX catches HIV. Cell Host Microbe. 2007;1(1):5–7. doi: 10.1016/j.chom.2007.02.006. [DOI] [PubMed] [Google Scholar]
- Martin-Serrano J, Yarovoy A, Perez-Caballero D, Bieniasz PD. Divergent retroviral late-budding domains recruit vacuolar protein sorting factors by using alternative adaptor proteins. Proc Natl Acad Sci U S A. 2003;100(21):12414–9. doi: 10.1073/pnas.2133846100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Serrano J, Zang T, Bieniasz PD. HIV-1 and Ebola virus encode small peptide motifs that recruit Tsg101 to sites of particle assembly to facilitate egress. Nat Med. 2001;7(12):1313–9. doi: 10.1038/nm1201-1313. [DOI] [PubMed] [Google Scholar]
- Morita E, Sandrin V, Chung HY, Morham SG, Gygi SP, Rodesch CK, Sundquist WI. Human ESCRT and ALIX proteins interact with proteins of the midbody and function in cytokinesis. Embo J. 2007;26(19):4215–27. doi: 10.1038/sj.emboj.7601850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita E, Sundquist WI. Retrovirus budding. Annu Rev Cell Dev Biol. 2004;20:395–425. doi: 10.1146/annurev.cellbio.20.010403.102350. [DOI] [PubMed] [Google Scholar]
- Munshi UM, Kim J, Nagashima K, Hurley JH, Freed EO. An Alix fragment potently inhibits HIV-1 budding: characterization of binding to retroviral YPXL late domains. J Biol Chem. 2007;282(6):3847–55. doi: 10.1074/jbc.M607489200. [DOI] [PubMed] [Google Scholar]
- Odorizzi G. The multiple personalities of Alix. J Cell Sci. 2006;119(Pt 15):3025–3032. doi: 10.1242/jcs.03072. [DOI] [PubMed] [Google Scholar]
- Ono A, Freed EO. Plasma membrane rafts play a critical role in HIV-1 assembly and release. Proc Natl Acad Sci U S A. 2001;98(24):13925–13930. doi: 10.1073/pnas.241320298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popov S, Popova E, Inoue M, Gottlinger HG. Human immunodeficiency virus type 1 Gag engages the Bro1 domain of ALIX/AIP1 through the nucleocapsid. J Virol. 2008;82(3):1389–98. doi: 10.1128/JVI.01912-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strack B, Calistri A, Craig S, Popova E, Gottlinger HG. AIP1/ALIX is a binding partner for HIV-1 p6 and EIAV p9 functioning in virus budding. Cell. 2003;114(6):689–99. doi: 10.1016/s0092-8674(03)00653-6. [DOI] [PubMed] [Google Scholar]
- Swanstrom R, Wills JW. Synthesis, Assembly, and Processing of Viral Proteins. In: Coffin JM, Hughes SH, Varmus HE, editors. Retroviruses. Cold Spring Harbor Laboratory Press; New York: 1997. pp. 263–334. [PubMed] [Google Scholar]
- Trioulier Y, Torch S, Blot B, Cristina N, Chatellard-Causse C, Verna JM, Sadoul R. Alix, a protein regulating endosomal trafficking, is involved in neuronal death. J Biol Chem. 2004;279(3):2046–2052. doi: 10.1074/jbc.M309243200. [DOI] [PubMed] [Google Scholar]
- Usami Y, Popov S, Gottlinger HG. Potent rescue of human immunodeficiency virus type 1 late domain mutants by ALIX/AIP1 depends on its CHMP4 binding site. J Virol. 2007;81(12):6614–22. doi: 10.1128/JVI.00314-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VerPlank L, Bouamr F, LaGrassa TJ, Agresta B, Kikonyogo A, Leis J, Carter CA. Tsg101, a homologue of ubiquitin-conjugating (E2) enzymes, binds the L domain in HIV type 1 Pr55(Gag) Proc Natl Acad Sci U S A. 2001;98(14):7724–9. doi: 10.1073/pnas.131059198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vito P, Pellegrini L, Guiet C, D'Adamio L. Cloning of AIP1, a novel protein that associates with the apoptosis-linked gene ALG-2 in a Ca2+-dependent reaction. J Biol Chem. 1999;274(3):1533–1540. doi: 10.1074/jbc.274.3.1533. [DOI] [PubMed] [Google Scholar]
- von Schwedler UK, Stuchell M, Muller B, Ward DM, Chung HY, Morita E, Wang HE, Davis T, He GP, Cimbora DM, Scott A, Krausslich HG, Kaplan J, Morham SG, Sundquist WI. The protein network of HIV budding. Cell. 2003;114(6):701–13. doi: 10.1016/s0092-8674(03)00714-1. [DOI] [PubMed] [Google Scholar]
- Waheed AA, Ablan SD, Mankowski MK, Cummins JE, Ptak RG, Schaffner CP, Freed EO. Inhibition of HIV-1 replication by amphotericin B methyl ester: selection for resistant variants. J Biol Chem. 2006;281(39):28699–711. doi: 10.1074/jbc.M603609200. [DOI] [PubMed] [Google Scholar]
- Waheed AA, Ono A, Freed EO. Methods for the study of HIV-1 assembly. Methods Mol Biol. 2009;485:163–84. doi: 10.1007/978-1-59745-170-3_12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei X, Decker JM, Liu H, Zhang Z, Arani RB, Kilby JM, Saag MS, Wu X, Shaw GM, Kappes JC. Emergence of resistant human immunodeficiency virus type 1 in patients receiving fusion inhibitor (T-20) monotherapy. Antimicrob Agents Chemother. 2002;46(6):1896–905. doi: 10.1128/AAC.46.6.1896-1905.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yee JK, Friedmann T, Burns JC. Generation of high-titer pseudotyped retroviral vectors with very broad host range. Methods Cell Biol. 1994;43(Pt A):99–112. doi: 10.1016/s0091-679x(08)60600-7. [DOI] [PubMed] [Google Scholar]
- Zhai Q, Fisher RD, Chung HY, Myszka DG, Sundquist WI, Hill CP. Structural and functional studies of ALIX interactions with YPX(n)L late domains of HIV-1 and EIAV. Nat Struct Mol Biol. 2008;15(1):43–9. doi: 10.1038/nsmb1319. [DOI] [PubMed] [Google Scholar]