

Abstract
Ketamine has important anesthetic, analgesic, and psychotropic actions. It is widely believed that NMDA receptor inhibition accounts for ketamine actions, but there remains a dearth of behavioral evidence to support this hypothesis. Here, we present an alternative, behaviorally relevant molecular substrate for anesthetic effects of ketamine: the HCN1 pacemaker channels that underlie a neuronal hyperpolarization-activated cationic current (I h). Ketamine caused subunit-specific inhibition of recombinant HCN1-containing channels and neuronal I h at clinically relevant concentrations; the channels were more potently inhibited by S-(+)-ketamine than racemic ketamine, consistent with anesthetic actions of the compounds. In cortical pyramidal neurons from wild-type, but not HCN1 knock-out mice, ketamine induced membrane hyperpolarization and enhanced dendritosomatic synaptic coupling; both effects are known to promote cortical synchronization and support slow cortical rhythms, like those accompanying anesthetic-induced hypnosis. Accordingly, we found that the potency for ketamine to provoke a loss-of-righting reflex, a behavioral correlate of hypnosis, was strongly reduced in HCN1 knock-out mice. In addition, hypnotic sensitivity to two other intravenous anesthetics in HCN1 knock-out mice matched effects on HCN1 channels; propofol selectively inhibited HCN1 channels and propofol sensitivity was diminished in HCN1 knock-out mice, whereas etomidate had no effect on HCN1 channels and hypnotic sensitivity to etomidate was unaffected by HCN1 gene deletion. These data advance HCN1 channels as a novel molecular target for ketamine, provide a plausible neuronal mechanism for enhanced cortical synchronization during anesthetic-induced hypnosis and suggest that HCN1 channels might contribute to other unexplained actions of ketamine.
Keywords: anesthesia, Ih, hyperpolarization-activated current, propofol, cortical pyramidal neurons, sleep
Introduction
Ketamine is a clinically important drug commonly used in pediatric and obstetric anesthesia and for perioperative analgesia (Wolff and Winstock, 2006); at subanesthetic doses, ketamine has antidepressant actions (Zarate et al., 2006), and its psychedelic properties make it a popular drug of abuse (Wolff and Winstock, 2006); experimentally, it induces cortically driven rhythms reminiscent of slow oscillations that are a prominent and organizing feature of sleep (Amzica and Steriade, 1998). It is generally believed that these actions of ketamine are due to effects on NMDA receptors because inhibition of those receptors is observed with low micromolar affinity and a stereoselectivity that matches the anesthetic actions of the drug (Franks and Lieb, 1994; Kohrs and Durieux, 1998; Wolff and Winstock, 2006; Sinner and Graf, 2008). However, it is unlikely that inhibition of NMDA receptors can account for the full spectrum of ketamine action. For example, MK-801 is a more potent NMDA receptor inhibitor than ketamine but has little hypnotic action, even at high doses (Daniell, 1990; Kelland et al., 1993; Stabernack et al., 2003; Irifune et al., 2007). Moreover, mice with deletions of the widely expressed NMDA receptor NR2A subunit fail to exhibit specific deficits in ketamine action (Petrenko et al., 2004, 2006; Sato et al., 2005). Other identified substrates for ketamine action are modulated at supraclinical concentrations (e.g., Kv channels) (Schnoebel et al., 2005) and/or they are expressed in brain regions that are unlikely to mediate hypnotic effects (e.g., α6 GABAA receptors in cerebellum) (Hevers et al., 2008). Therefore, we considered the possibility that alternative molecular targets might contribute to ketamine action, focusing on HCN channel subunits that underlie a hyperpolarization-activated pacemaker current (I h) implicated in neuronal rhythmogenesis.
Here, we show that ketamine is a potent inhibitor of cloned HCN1-containing channels and of I h in cortical pyramidal neurons; concordant with its anesthetic actions, we found that inhibition by ketamine is stereoselective and evident at clinically relevant concentrations. Ketamine causes HCN1-dependent membrane hyperpolarization and synaptic enhancement. Most importantly, the ability of ketamine to induce hypnosis is strongly attenuated in HCN1 knock-out mice. These data indicate a prominent contribution from HCN1 channels to anesthetic actions of ketamine; they establish an alternative, behaviorally relevant molecular target for ketamine, supporting HCN1 channels as a viable substrate for anesthetic-induced hypnosis.
Materials and Methods
Recordings from cloned HCN channels in mammalian cells.
HEK 293 cells were transiently transfected with mouse HCN channel constructs (obtained from Drs. B. Santoro and S. A. Siegelbaum, Columbia University, New York, NY), together with eGFP, and whole-cell recordings of mHCN channel currents were obtained as described previously (Chen et al., 2005a). The tandem-linked HCN1–HCN2 construct was made by concatenating mHCN1 and mHCN2 clones, also as described previously (Chen et al., 2005a).
HCN1 knock-out mice.
Mice used in this work were age, sex and weight-matched HCN1 knock-out and B6129SF2/J mice; both represent hybrids of the parental C57BL/6 and 129Sv substrains (Jackson ImmunoResearch Laboratories; Stock #s: 005034 and 101045). Mice homozygous for the targeted mutation are viable, fertile, normal in size and longevity and they do not display any gross physical or behavioral abnormalities (Nolan et al., 2003 2004). For some experiments, we compared HCN1+/+ and HCN1−/− littermates; these animals were generated from heterozygous HCN1+/− mice, obtained by crossing animals from the homozygous HCN1 knock-out line with C57BL/6J mice.
Electrophysiological recordings from mouse cortical pyramidal neurons.
Mice of either sex (14–22 d old) were anesthetized (ketamine/xylazine: 200/14 mg/kg, i.m.) and transverse brain slices prepared as described previously (Chen et al., 2005a). Slices were submerged in a recording chamber on a Zeiss Axioskop FS microscope and visualized with Nomarski optics; pyramidal cells were targeted for recording based on location in the slice and characteristic size and shape. For voltage-clamp recording, pipettes (2–4 MΩ) were filled with the following (in mm): 120 KCH3O3S; 4 NaCl; 1 MgCl2; 0.5 CaCl2; 10 HEPES; 10 EGTA; 3 MgATP; 0.3 GTP-Tris, pH 7.2; for current clamp, pipette solution contained the following (in mm): 17.5 KCl; 122.5 potassium gluconate; 10 HEPES; 0.2 EGTA; 9 NaCl; 1 MgCl2; 3 MgATP; 0.3 GTP-Tris, pH 7.2. To record spontaneous and miniature EPSCs, the internal solution contained the following (in mm): 128 CsCH3O3S; 9 NaCl; 2 MgCl2; 10 HEPES; 10 EGTA; 3 MgATP; 0.3 GTP-Tris, pH 7.3.
Most experiments were performed at room temperature (∼24°C), but in some cases, the perfusate was passed through an in-line heater (Warner SH-27) that was adjusted by hand with a DC power supply to raise bath temperature to 33°C, as monitored at the inlet to the recording chamber (Physitemp; TH-5). Recordings were obtained with an Axopatch 200B amplifier in standard bath solution containing the following (in mm): 140 NaCl; 3 KCl; 10 HEPES; 2 CaCl2; 2 MgCl2; 10 glucose; we routinely added tetrodotoxin (TTX, 0.5 μm; Alomone Labs), BaCl2 (200 μm) and bicuculline/strychnine (both at 30 μm; Sigma) to the bath, except where noted. In some experiments, ZD-7288 was used to block I h (50 μm; Tocris Cookson), and CNQX was used to verify that EPSPs were glutamatergic (10 μm; Sigma). Ketamine (Ketaset; Fort Dodge Animal Health), S-(+)-ketamine (Sigma) and etomidate (Bedford Laboratories) were diluted into the bath solution at the indicated concentrations. Propofol (Sigma) was prepared as a 100 mg/ml stock solution in ethanol and diluted in bath solution to the indicated concentrations; propofol was applied to neurons in the slice for ∼5 min, a point at which we found inhibition of I h in cortical neurons was stable and maximal (Chen et al., 2005a)
The properties of HCN channel currents and neuronal I h were determined from whole-cell voltage-clamp experiments, essentially as described previously (Chen et al., 2005a). Under current clamp, we measured input resistance and the magnitude of depolarizing “sag” from voltage response to hyperpolarizing current injection. We examined resonant properties of cortical pyramidal neurons by using an impedance (Z) amplitude profile (ZAP) method (Hutcheon and Yarom, 2000). A sinusoidal current waveform of quadratically increasing frequency (0.5–15 Hz, in 15 s) was injected and a ratio of the Fourier transforms of membrane voltage and input current was used to derive the impedance magnitude (Z) power spectra; these were fitted with an asymmetric peak function to obtain resonant frequency (λmax; input frequency corresponding to the peak impedance) and a Q ratio that describes the degree of resonance (peak impedance divided by impedance at 0.5 Hz). We also measured summation properties of evoked EPSPs. For this, a pipette connected to a stimulator (Grass S48) via a stimulus isolation unit (Grass SIU5) was filled with standard bath solution and placed in the superficial layers of the cortex; synaptic responses were evoked by applying 40 Hz, 5–10 V pulses (corresponding to 2–10 μA) and summation was quantified as the ratio of the amplitudes of the last and first evoked EPSP (EPSP5:EPSP1). To analyze sEPSCs and mEPSCs we used the Mini Analysis Program (Synaptosoft).
Analysis of anesthetic action in mice.
Bolus injections of ketamine and propofol (both 5–30 mg/kg) or etomidate (0.625–10 mg/kg) were administered via the tail vein of each mouse (2–4 months old); injectable propofol (Diprivan) was from Abraxis Pharmaceuticals. Each animal was injected with only a single concentration of the drug on any given day; some animals received multiple doses (on different days, always separated by at least 1 week). Hypnosis was established by loss of righting response (LORR) that occurred within 10 s after completion of the injection and which persisted for at least 10 s thereafter (Garfield and Bukusoglu, 1996); we also measured the latency to regain the righting reflex (Garfield and Bukusoglu, 1996). To obtain EC50 values for hypnosis a series of quantal dose-response curves was constructed and compared statistically using Prism 3.0.
Data acquisition and analysis.
Results are presented as mean ± SEM. Data were analyzed statistically using one-way and two-way ANOVA or Student's t test; post hoc pairwise comparisons used Bonferroni's correction of the t test (Excel and/or SigmaStat). Dose-response data were fitted and analyzed statistically in Prism 3.0 using a logistic equation with three free parameters (slope, EC50 and maximum) and a fixed origin. Differences in mean values were considered significant if p < 0.05.
Results
Ketamine is a potent, subunit-specific, and stereoselective inhibitor of HCN1-containing channels
We examined effects of ketamine on mouse HCN channel subunits expressed in HEK293 cells (Fig. 1 A,B). In cells expressing homomeric HCN1 channels, ketamine (20 μm) caused a hyperpolarizing shift in voltage dependence of activation (ΔV½: −14.7 ± 2.5 mV) that was also associated with a decrease in maximal current amplitude (percentage inhibition: 32.2 ± 5.3%). In contrast, ketamine had little effect on homomeric HCN2 channel currents (ΔV½: −1.6 ± 2.1 mV; percentage inhibition: 6.4 ± 2.4%), even when endogenous allosteric inhibition of HCN2 channels was relieved in the presence of saturating intracellular cAMP (50 μm) (data not shown). Ketamine produced intermediate effects on tandem-linked HCN1–HCN2 heteromeric channels; it caused a hyperpolarizing shift in V½ of −9.5 ± 0.8 mV and decreased maximal current by 28.9 ± 3.8%. These data indicate that HCN2 homomeric channels are relatively unaffected by ketamine, but HCN1-containing channels are strongly inhibited by ketamine in either homomeric or heteromeric configurations.
Figure 1.

Ketamine selectively inhibits currents from HCN1 subunit-containing channels. A, Sample currents from HEK293 cells expressing mHCN1, mHCN2, and mHCN1–mHCN2 channel constructs evoked by hyperpolarizing voltage steps from −38 to −118 mV, before and during exposure to ketamine (20 μm); conditioning voltage steps were followed by a step to −88 mV for tail current analysis. B, Summary data showing averaged (±SEM) shift in half activation potential (V½; top) and current inhibition (percentage from control; bottom) evoked by ketamine for each of the indicated HCN channel constructs. *p < 0.05 by paired t test for ketamine versus control (n = 5, 5, and 8 for mHCN1, mHCN2, and mHCN1–mHCN2). C, Averaged values for shift in V½ (left) and amplitude inhibition (right) of heteromeric mHCN1–mHCN2 currents at different concentrations of racemic ketamine (black squares) or its S-(+)-enantiomer (gray triangles). For the shift in V½, calculated EC50 values were, respectively, 8.2 ± 1.2 μm and 4.1 ± 1.2 μm, with corresponding maximal values of −11.9 ± 0.7 mV and −14.5 ± 0.9 mV; for amplitude inhibition, EC50 values were 15.6 ± 1.2 μm and 7.4 ± 1.3 μm, with corresponding maximal values of 44.5 ± 3.0% and 49.3 ± 3.7% inhibition. The insets show normalized fits to highlight differences in potency between ketamine and S-(+)-ketamine. For both ΔV½ and percentage inhibition, EC50 values were significantly different; also, the maximum shift in V½ was significantly greater for S-(+)-ketamine (p < 0.05 by t test, n ≥ 8 cells at each concentration).
Effects of ketamine on heteromeric HCN1–HCN2 channels were determined over a range of concentrations encompassing those achieved clinically (Fig. 1 C). By fitting the shift in V½ (−ΔV½) and inhibition of maximal current amplitude to logistic functions, we estimated half-maximal (EC50) concentrations of ketamine of ∼8 and ∼16 μm, with corresponding maximal values of approximately −12 mV and ∼45% inhibition. These EC50 values are clearly within a clinically relevant range; anesthesia is maintained in humans at free plasma ketamine concentrations of ∼5–10 μm (Idvall et al., 1979; White et al., 1980) and in rats at ∼20 μm (Cohen et al., 1973).
Anesthetic actions of ketamine are stereoselective, with S-(+)-ketamine approximately twice as potent as racemic ketamine (Franks and Lieb, 1994; Kohrs and Durieux, 1998); a similar stereospecificity is observed for ketamine inhibition of NMDA receptors. Thus, viable alternative candidate molecular targets for ketamine action should also satisfy this important stereoselectivity requirement. Indeed, we found a significant left-shift in EC50 for effects of S-(+)-ketamine on V½ (∼4 vs 8 μm) and current amplitude (∼7 vs 16 μm), indicating that S-(+)-ketamine is ∼2-fold more potent as a blocker of HCN1–HCN2 channels than racemic ketamine (Fig. 1 C). In addition to the increase in potency, S-(+)-ketamine is also more efficacious than ketamine in inducing a shift in gating, with maximal ΔV½ of approximately −14 mV. Thus, ketamine inhibits HCN1-containing channels at clinically relevant concentrations and with a stereoselectivity similar to that observed for its anesthetic actions (Franks and Lieb, 1994; Kohrs and Durieux, 1998).
Ketamine inhibits I h and modulates electroresponsive properties in cortical pyramidal neurons
We used HCN1 knock-out mice to verify subunit-specific effects of ketamine on HCN channels in a native setting relevant to anesthetic action and to determine functional consequences on neuronal excitability. Ketamine produces robust slow cortical oscillations in vivo that are likely initiated in layer 5 pyramidal neurons (Amzica and Steriade, 1998), which express both HCN1 and HCN2 subunits (Santoro et al., 2000). In brain slices of somatosensory cortex from wild-type mice (Fig. 2 A,B), ketamine inhibited a relatively fast-activating I h in layer 5 cortical pyramidal neurons via both a hyperpolarizing shift in voltage dependence of activation (ΔV½: −9.7 ± 1.8 mV) and a decrease in maximal current amplitude (percentage inhibition: 29.6 ± 7.1%); in pyramidal neurons from HCN1 knock-out mice (Fig. 2 C,D), ketamine had no effect on the smaller and slower residual I h carried by the remaining HCN2 homomeric channels (see Chen et al., 2008 for further details on effects of HCN1 deletion on basic I h properties in cortical pyramidal neurons).
Figure 2.

Ketamine inhibits I h in cortical pyramidal neurons from wild-type mice, but not from HCN1 knock-out mice. A, C, Voltage-clamp recordings of I h in cortical pyramidal neurons from wild-type and HCN1 knock-out mice, under control conditions (top) and during exposure to ketamine (20 μm, bottom). B, D, Averaged steady-state I–V (top) and voltage dependence of I h activation (bottom) under control conditions (squares) and in the presence of ketamine (20 μm, triangles) in cortical pyramidal neurons from wild-type and HCN1−/− mice. Ketamine induced an approximately −10 mV shift in V½ of I h activation and an approximately 30% decrease in I h amplitude (at −128 mV) in cortical neurons from wild-type mice but had no effect on I h in HCN1 knock-out mice (ΔV½ approximately −2 mV; ∼5% inhibition). *p < 0.05 by paired t test; n = 5 each for wild type and HCN1 knock-outs. Vm, Membrane voltage.
Under current clamp, ketamine diminished two other electroresponsive properties associated with neuronal I h: voltage-dependent sag and resonance. The I h-dependent depolarizing sag was reduced in the presence of 20 μm ketamine (supplemental Fig. S1, available at www.jneurosci.org as supplemental material) (by ∼40% at −90 mV; from 3.5 ± 1.2 mV to 2.2 ± 1.0 mV, n = 5, p < 0.05). Likewise, as shown in Figure 3, resonance properties assayed using a ZAP protocol (Hutcheon and Yarom, 2000; Nolan et al., 2007) in cortical pyramidal neurons from wild-type mice were attenuated by ketamine and essentially eliminated by supramaximal concentration of ZD-7288, an HCN channel blocker (50 μm). For example, resonant frequency (λmax) decreased from 1.5 ± 0.2 to 1.1 ± 0.2 to 0.5 ± 0.0 (p < 0.05, n = 5) and, concurrently, the associated Q value, a ratio of the peak impedance to the impedance at 0.5 Hz, decreased from 1.5 ± 0.1 in control to 1.2 ± 0.1 in ketamine to 1.0 ± 0.0 in ZD-7288 (p < 0.05, n = 5). By this same analysis, we found no evidence for resonance in neurons from HCN1 knock-out mice (Fig. 3 B,C), indicating that resonant properties modulated by ketamine in cells from wild-type mice were conferred by HCN1 channels (Nolan et al., 2007).
Figure 3.

Ketamine decreases resonant properties of cortical pyramidal neurons in wild-type but not in HCN1 knock-out mice. A, B, Sample voltage records (top) obtained during injection of a sinusoidal current waveform (bottom) with frequency increasing quadratically from 0.5 to 15 Hz (in 15 s) in cortical pyramidal neurons from wild-type (A) and HCN1 knock-out (B) mice; after control records were obtained, cells were exposed sequentially to ketamine (20 μm) and ZD-7288 (50 μm). In the cell from a wild-type mouse, note that the greatest voltage response is observed at a frequency above 0.5 Hz (blue arrow), but this is shifted toward lower frequencies by ketamine (red arrow) and ZD-7288. C, A Fourier transform was applied to the voltage and current waveforms and the ratio used to obtain impedance power as a function of stimulus frequency; those relationships were fitted with an asymmetric peak function (overlaid lines) for each cell to obtain averaged values (±SEM) for the Q ratio, Zmax and λmax (insets), which represent, respectively, the ratio of peak impedance to that at 0.5 Hz, the peak impedance, and the frequency at peak impedance. As expected for inhibition of I h, ketamine and ZD-7288 increased impedance magnitude (Zmax) and altered resonant properties in cortical pyramidal cells from wild-type mice by shifting the Q value and λmax; in neurons from HCN1 knock-out mice, only Zmax was affected and only by ZD-7288, presumably reflecting effects of the drug on residual HCN2 channels that do not contribute substantially to cell resonant properties. *,† p < 0.05 from control and ketamine conditions, by two-way RM-ANOVA, n = 5 each for wild type and HCN1 knock-outs.
HCN channels generate an instantaneous component of current in cortical pyramidal neurons in addition to the well known voltage- and time-dependent I h (Nolan et al., 2004, 2007; Chen et al., 2005a, 2008). Accordingly, we found that ketamine also inhibited a constitutive inward current near resting membrane potential in cells from wild-type mice (∼13 pA at −70 mV; from −22.7 ± 3.2 pA to −9.9 ± 3.9 pA, n = 5, p < 0.05); this current was due to I h because it was not seen in neurons from HCN1 knock-out mice. Correspondingly, ketamine caused membrane hyperpolarization and increased input resistance (R N) in cortical pyramidal cells from wild-type but not HCN1 knock-out mice (Fig. 4 A,B). Thus, ketamine inhibits voltage-dependent and constitutive components of I h in cortical neurons, with the expected effects on electroresponsive properties; consistent with its preferential action on HCN1 subunits, these actions of ketamine were not observed in HCN1 knock-out mice.
Figure 4.
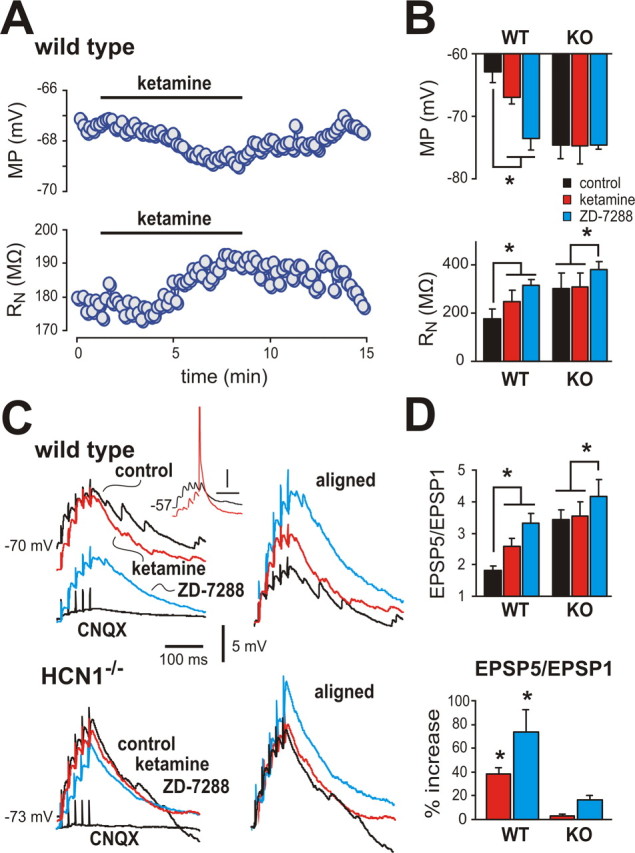
Ketamine causes membrane hyperpolarization, increases R N, and enhances EPSP temporal summation in cortical pyramidal neurons from wild-type mice, but not from HCN1 knock-out mice. A, Effect of ketamine (20 μm) on membrane potential (MP, top) and input resistance (R N, bottom) in a cortical pyramidal neuron from a wild-type mouse. B, Averaged data (±SEM) illustrating effects of ketamine and the I h blocker, ZD-7288 (50 μm) on MP (top) and R N (bottom) in cortical pyramidal neurons from wild-type and HCN1 knock-out mice (n = 5 each). C, Sample voltage traces show EPSP recordings (left) obtained from cortical pyramidal neuron from wild-type (top) and HCN1 knock-out (bottom) mice in response to 40 Hz stimulation under control conditions, and during exposure to ketamine and ZD-7288. Right, EPSPs were aligned to initial membrane potential and normalized to the amplitude of the first EPSP in the train to highlight drug effects on temporal summation. Inset, Effects of ketamine from a more depolarized starting potential illustrate increased excitability resulting from enhanced EPSP summation despite ketamine-induced membrane hyperpolarization (spike is truncated; time scale represents 50 ms). D, Averaged EPSP summation ratio (EPSP5/EPSP1) for wild-type mice and HCN1 knock-out mice (n = 6 and 5, respectively) under the indicated conditions (top); averaged percentage increase of EPSP summation induced by ketamine and ZD-7288 (bottom). Ketamine enhanced EPSP summation in cortical neurons from wild-type animals, but not from HCN1 knock-out mice. *p < 0.05 by two-way RM-ANOVA or in D, by paired t test versus control. WT, Wild type; KO, knock-out.
Ketamine enhances dendritosomatic synaptic transfer in cortical pyramidal neurons
In pyramidal neurons, HCN1 subunits contribute to dendritic I h, which provides an electrotonic shunt leading to sublinear summation of EPSPs; inhibition of I h, therefore, promotes enhanced EPSP summation (Magee, 2000). Accordingly, we found that ketamine enhanced summation of bicuculline-resistant, CNQX-sensitive excitatory synaptic inputs measured in pyramidal neurons following 40 Hz extracellular stimulation of the superficial cortex (Fig. 4 C,D); this is clearly evident in the aligned and normalized traces, where the amplitude of the final evoked EPSP (EPSP5) relative to the first (EPSP1) was substantially increased by ketamine (averaged EPSP5:EPSP1 ratio increased by ∼33%; from 2.1 ± 0.2 to 2.8 ± 0.3, n = 6, p < 0.05) and further enhanced by the I h blocker, ZD-7288 (by ∼67%; to 3.4 ± 0.3, p < 0.05). Although most of our experiments were performed at room temperature, the effects of ketamine on EPSP summation were identical at temperatures closer to those encountered physiologically (i.e., EPSP5:EPSP1 ratio increased in ketamine by ∼34% at 33°C; p < 0.05, n = 4). The enhanced EPSP summation evoked by ketamine in wild-type mice could be attributed definitively to inhibition of I h since it was absent in HCN1 knock-out mice (Fig. 4 C,D); a residual effect of ZD-7288 in knock-out mice reflects inhibition of remaining HCN2 channels. Note that despite membrane hyperpolarization by ketamine, the concomitant increase in EPSP summation can lead to spike discharge (Fig. 4 C, inset). The frequency and amplitude of spontaneous and miniature EPSCs (sEPSCs and mEPSCs) were not different in pyramidal neurons from wild-type and HCN1 knock-out mice, and ketamine had no effect on either sEPSC or mEPSC properties in these mouse lines (supplemental Fig. S2, available at www.jneurosci.org as supplemental material) (n = 5 each). This suggests that ketamine-induced enhancement of synaptic summation was independent of any presynaptic actions and primarily postsynaptic, as expected. Thus, in addition to ketamine-induced membrane hyperpolarization and increased R N of cortical pyramidal neurons, ketamine also promotes more effective dendritosomatic information transfer in cortical pyramidal neurons.
It is important to note that the experiments depicted in Figure 4 C were performed after blocking GABAA receptors with bicuculline. In the absence of bicuculline, however, we observed a sublinear summation of evoked EPSPs in pyramidal neurons from HCN1 knock-out mice that was essentially identical to that seen in neurons from wild-type mice (supplemental Fig. S3, available at www.jneurosci.org as supplemental material); the summation ratio was 2.0 ± 0.1 in wild-type mice and 2.1 ± 0.1 in HCN1 knock-outs (n = 10 each). In neurons from HCN1 knock-out mice the enhancement of EPSP summation by bicuculline was ∼3-fold greater than observed in cells from wild-type mice (supplemental Fig. S3, available at www.jneurosci.org as supplemental material); bicuculline increased EPSP5:EPSP1 by ∼12.9 ± 5.7% in control animals and by 37.3 ± 5.8% in HCN1 knock-outs (n = 6 and 5, respectively; p < 0.05). Thus, despite the loss of HCN1 channels in knock-out mice, a compensatory GABAergic mechanism provides normal dendritosomatic coupling in pyramidal neurons. Interestingly, it was recently reported that upregulated GABAA current serves to preserve relatively normal excitability in hippocampal pyramidal neurons of mice with genetic deletion of Kv4.2 (Andrásfalvy et al., 2008), a channel that also exhibits preferential distal dendritic localization (Magee, 2000). In sum, these data indicate that ketamine is unable to enhance EPSP summation in cortical pyramidal neurons from HCN1 knock-out mice, but that baseline EPSP summation in knock-out animals is otherwise quantitatively indistinguishable from that seen in wild-type cells.
HCN1 knock-out mice are markedly less sensitive to hypnotic actions of ketamine
We reasoned that genetic deletion of this target for ketamine action would reduce anesthetic sensitivity in HCN1 knock-out mice. We were particularly interested in hypnotic actions of the drug since membrane hyperpolarization and enhanced synaptic efficacy in cortical neurons are believed to be important for cortical synchronization associated with sleep-like hypnotic states (Bazhenov et al., 1998; Hill and Tononi, 2005). Indeed, we discovered a strikingly diminished sensitivity of HCN1 knock-out mice to the hypnotic actions of ketamine (Fig. 5); by comparison to control mice, the dose-response relationship for ketamine to evoke a loss-of-righting reflex (LORR) in HCN1 knock-out mice was shifted toward higher concentrations (Fig. 5 A), with nearly a doubling of the EC50 (control: 7.3 ± 0.6 mg/kg; HCN1 knock-out: 13.3 ± 0.6 mg/kg, n = 11 and 15, p < 0.001). Also consistent with decreased sensitivity to ketamine, we found that duration of the LORR was substantially diminished at each effective ketamine dose in HCN1 knock-outs (Fig. 5 B). The LORR obtained at higher drug concentrations likely reflects engagement of different neural mechanisms and distinct molecular targets from those responsible for lower-dose hypnotic actions, as is thought to occur during progression through different planes of anesthesia (Antkowiak, 2002).
Figure 5.

HCN1 knock-out mice are less sensitive to hypnotic actions of ketamine. Mice were injected with incrementing concentrations of ketamine (5–30 mg/kg, i.v.) and the percentage of wild-type and HCN1 knock-out animals that failed to right themselves (LORR) was determined as a measure of hypnosis. HCN1 knock-out mice were less sensitive to hypnotic effects of ketamine, as indicated by the increased EC50 for ketamine-induced LORR (A) and the reduced duration of the LORR (B). *p < 0.05 by two-way RM-ANOVA, n = 11 and 13. The inverted triangle and diamond in A represent data from HCN1+/+ (n = 12) and HCN1−/− (n = 8) littermates obtained from heterozygote HCN1+/− mice following a backcross of HCN1 knock-outs to C57BL/6J mice.
The wild-type and HCN1 knock-out mouse lines used for the behavioral studies are on a similar mixed genetic background (C57BL/6 and 129Sv); however, the 129Sv substrains are not identical and some genetic drift might be expected since homozygous knock-out and wild-type lines were maintained separately. Therefore, we also tested effects of ketamine on LORR in HCN1+/+ and HCN1−/− littermates derived from heterozygotes produced by crossing the knock-out line with C57BL/6J mice. As with the lines maintained as homozygotes, hypnotic effects of ketamine were clearly different in these HCN1+/+ and HCN1−/− littermates; at a 10 mg/kg dose, 75% of HCN1+/+ mice were unresponsive, whereas only 12.5% of the HCN1−/− littermates lost the righting reflex (Fig. 5 A), and LORR duration was shorter in the HCN1 knock-outs (140 ± 27 s vs 19 ± 19 s; n = 12 and 8, p < 0.005). These concordant data from HCN1+/+ and HCN1−/− mice obtained in different mouse lines suggest that the reduced sensitivity to hypnotic actions of ketamine in HCN1 knock-outs is not related to genetic background.
The decreased sensitivity to ketamine-induced hypnosis was also not a nonspecific effect of HCN1 gene deletion. Etomidate is a different class of intravenous anesthetic drug that has no effect on HCN1 channels (Fig. 6 A), and both control and HCN1 knock-out mice were identical in sensitivity to hypnotic effects of etomidate (Fig. 6 B) (EC50 for both control and HCN1 knock-out: 1.3 ± 0.1 mg/kg, n = 10 and 11, p > 0.99). Together, these data suggest that the diminished ability of ketamine to provoke hypnosis in HCN1 knock-out mice can indeed be attributed to deletion of the HCN1 subunit, and is unlikely to be accounted for by differences in genetic background or nonspecific changes that generally affect anesthetic sensitivity.
Figure 6.

Etomidate does not inhibit HCN1 channel currents and hypnotic effects of etomidate are unaffected in HCN1 knock-out mice. A, Sample currents from HEK293 cells expressing mHCN1 channels (n = 5) before and during exposure to etomidate (left, 10 μm); averaged steady-state I–V (right, top) and voltage dependence of I h activation (right, bottom) under control conditions (squares) and in the presence of etomidate (triangles). B, Mice were tested for LORR following injection with different concentrations of etomidate (0.625–10 mg/kg, i.v.); hypnotic effects of etomidate were identical in wild-type and HCN1 knock-out animals, determined as either EC50 (left) or duration of LORR (right). NS, by two-way RM-ANOVA, n = 10 and 12. Vm, Membrane voltage.
HCN1 knock-out mice are less sensitive to hypnotic actions of propofol, another subunit-selective HCN1 channel blocker
Propofol is a GABAA receptor agonist and an intravenous anesthetic that is structurally unrelated to ketamine (Rudolph and Antkowiak, 2004); however, like ketamine, it also selectively inhibits cloned HCN1-containing channels by shifting V½ of I h activation and decreasing maximal current amplitude (Cacheaux et al., 2005; Chen et al., 2005a; Lyashchenko et al., 2007). Consistent with this HCN1 subunit selectivity, propofol (5 μm) caused a hyperpolarizing shift in V½ of I h activation and a decrease in maximal current amplitude in cortical pyramidal neurons from control mice (Fig. 7 A,B), but neither effect was observed on the residual I h in cells from HCN1 knock-out mice (Fig. 7 C,D). In addition, we also found that propofol caused membrane hyperpolarization (from −66.1 ± 3.1 mV to −69.8 ± 3.8 mV), increased input resistance (225.7 ± 31.0 MΩ to 272.3 ± 31.1 MΩ) and diminished I h-dependent depolarizing sag (3.2 ± 0.4 mV to 0.7 ± 0.4 mV) in pyramidal neurons from control mice (p < 0.05 and n = 6 for all); these effects of propofol were not observed in HCN1 knock-out mice (supplemental Fig. S4, available at www.jneurosci.org as supplemental material). Importantly, also, propofol enhanced synaptic summation in cortical pyramidal neurons from control mice, but not from HCN1 knock-out animals (Fig. 7 E,F). These data verify in a native context the subunit selectivity described earlier for propofol inhibition of HCN channel currents in heterologous systems (Cacheaux et al., 2005; Chen et al., 2005a; Lyashchenko et al., 2007), and they are strikingly similar to results obtained for ketamine. Notably, and again similar to our findings with ketamine, HCN1 knock-out mice were less sensitive to hypnotic effects of propofol; as shown in Figure 7 G, the EC50 for propofol in a LORR assay was shifted by ∼40% in HCN1 knock-outs (control: 7.2 ± 0.6 mg/kg; HCN1 knock-out: 10.6 ± 0.8 mg/kg, n = 10 and 13, p < 0.001), with sleep times significantly decreased relative to control mice. So, this structurally and pharmacologically distinct compound has effects similar to ketamine on cloned HCN1-containing channels and on cortical neuronal I h and, as for ketamine, the hypnotic effects of propofol are diminished in HCN1 knock-out mice.
Figure 7.
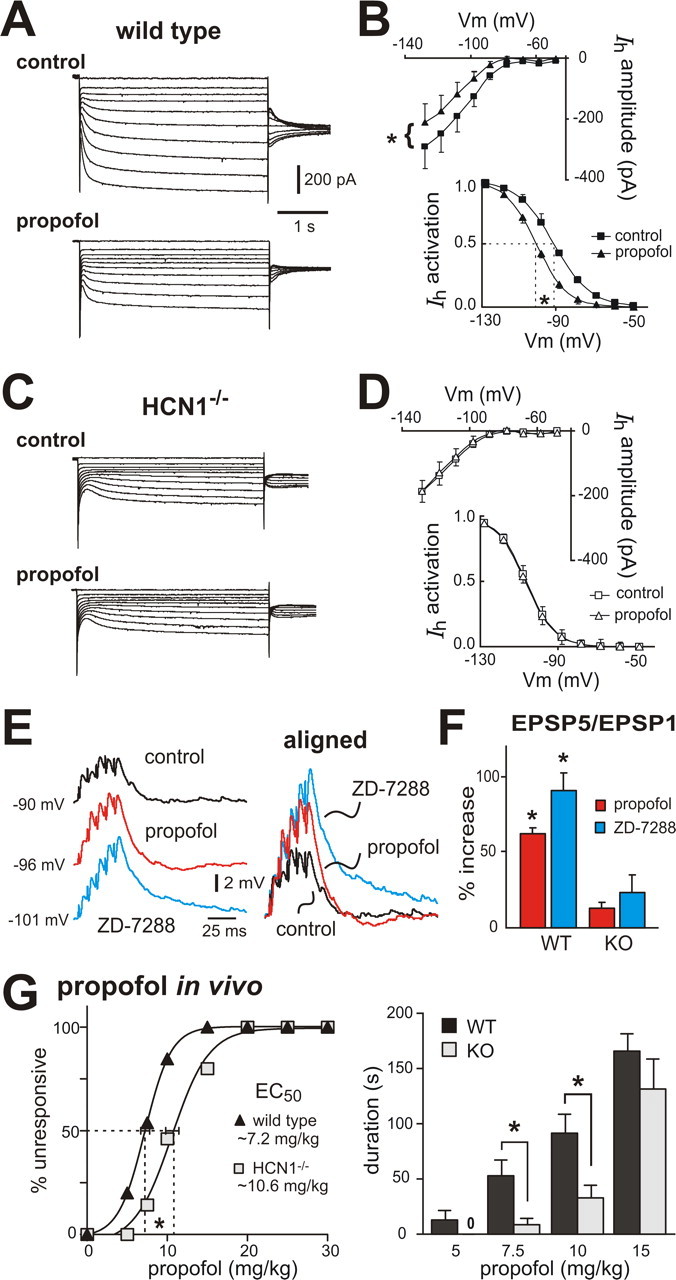
Propofol-induced inhibition of neuronal I h and hypnosis are diminished in HCN1 knock-out mice. A, C, Voltage-clamp recordings of I h in cortical pyramidal neurons from wild-type (A) and HCN1−/− (C) mice under control conditions (top) and during exposure to propofol (5 μm, bottom). B, D, Averaged steady-state I–V (top) and voltage dependence of I h activation (bottom) under control conditions (squares) and in the presence of propofol (triangles) in cortical pyramidal neurons from wild-type (B) and HCN1−/− (D) mice. Propofol induced an approximately −10 mV shift in V½ of I h activation and an approximately 30% decrease in I h amplitude in cortical neurons from wild-type mice (p < 0.01, paired t test, n = 7) but had no effect on I h in HCN1−/− mice (ΔV½ ∼−0.4 mV; ∼0.1% inhibition, n = 6). E, Sample voltage traces show EPSP recordings (left) obtained from cortical pyramidal neuron from wild-type mouse under control conditions, and during exposure to propofol and ZD-7288. EPSPs were aligned to initial membrane potential and normalized to the amplitude of the first EPSP in the train (right) to highlight drug effects on EPSP temporal summation. F, Averaged percentage increase of EPSP summation induced by propofol and ZD-7288 in cortical neurons from wild-type animals and HCN1−/− mice (*p < 0.05 vs control, n = 4 each). G, Mice were tested for LORR after injection with incrementing concentrations of propofol (5–30 mg/kg, i.v.). HCN1 knock-out mice were less sensitive to hypnotic effects of propofol, as indicated by the increased EC50 for propofol-induced LORR (left) and the reduced duration of the LORR (right). *p < 0.05 by two-way RM-ANOVA, n = 10 and 13. Vm, Membrane voltage.
Discussion
The results of experiments reported here have implications for understanding both the molecular neuropharmacology of ketamine action and the molecular basis of anesthetic-induced hypnosis. First, we show that HCN1 channels are a behaviorally relevant molecular substrate for ketamine; this result is especially surprising since ketamine is a drug whose actions have been universally associated with inhibition of NMDA receptors (Franks and Lieb, 1994; Sinner and Graf, 2008). These results raise the possibility that effects on HCN1 channels may be relevant for other unexplained aspects of ketamine action (e.g., its analgesic, psychedelic, or antidepressant effects) and they urge caution in the reflexive association of ketamine action with NMDA receptors. Second, we find a reduced sensitivity to hypnotic actions of ketamine and propofol, but not etomidate, in HCN1 knock-out mice; these behavioral results correlate well with inhibitory effects of these distinctly different anesthetic compounds on HCN1 channels, supporting a role for HCN1 channel inhibition in anesthetic-induced hypnosis. Furthermore, we demonstrate that inhibition of I h in cortical neurons by anesthetics alters electroresponsive properties in ways known to promote synchronized oscillatory behavior (Bazhenov et al., 1998; Hill and Tononi, 2005), providing a plausible neural mechanism by which HCN1 channel modulation could contribute to hypnotic actions of those drugs. In sum, these studies reveal HCN1 channels as heretofore unrecognized molecular targets for ketamine and legitimize these channels as clinically relevant substrates for hypnotic actions of anesthetics.
HCN1 channels are a behaviorally relevant molecular target for ketamine
As mentioned, the dramatic decrease in ketamine sensitivity of HCN1 knock-out mice (∼2-fold) was particularly surprising since NMDA receptors are widely believed to represent the molecular target for ketamine action. The case for NMDA receptor involvement in mediating anesthetic actions of ketamine rests largely on observations that ketamine inhibits NMDA receptors with a concentration dependence and stereoselectivity that are concordant with clinical effects of the drug (Franks and Lieb, 1994; Kohrs and Durieux, 1998). Whereas such in vitro results provide a strong correlative argument to implicate NMDA receptors, we find that the same case can be made for HCN1 channels: EC50 values obtained for ketamine inhibition of HCN1 channels are within a clinically relevant range and HCN1 channels are more potently inhibited by S-(+)-ketamine than racemic ketamine. Although concentrations required for inhibition of HCN1 channels (EC50: 8–16 μm) are somewhat higher than for inhibition of NMDA receptor channels (EC50: 1–5 μm) (Orser et al., 1997; Yamakura et al., 2000), it is worth recalling that MK-801 fails to cause hypnosis even though it is more potent than ketamine as an NMDA receptor antagonist (Daniell, 1990; Kelland et al., 1993; Stabernack et al., 2003; Irifune et al., 2007). Notably, and unlike ketamine, MK-801 has no effect on HCN1-containing channels (supplemental Fig. S5, available at www.jneurosci.org as supplemental material). Finally, there are currently no in vivo data from NMDA receptor knock-out mice to support a specific role for these channels in any ketamine-induced anesthetic outcome (Petrenko et al., 2004; Sato et al., 2005), whereas we find a striking deficit in hypnotic sensitivity to ketamine in HCN1 knock-out mice. Clearly, neither these arguments nor our data rule out a contribution from NMDA receptor inhibition to anesthetic actions of ketamine, which may yet be demonstrated. However, these studies demonstrate that HCN1 channels satisfy established in vitro criteria for a suitable ketamine target while also providing corroborating in vivo support for HCN1 contributions to hypnosis, a clinically relevant behavioral outcome.
A common concern in studies with conventional knock-out animals is the possibility of compensatory cellular mechanisms that complicates interpretation of behavioral data. In our studies, however, an upregulated GABAA current in the HCN1 knock-out mice actually provided a fortuitous set of circumstances; it compensated for the loss of I h to yield essentially normal synaptic summation properties under control, undrugged conditions, while still providing a context in which drug effects were absent (i.e., ketamine and propofol had no effect on I h or synaptic summation in the HCN1 knock-out mice). In addition, I h was markedly diminished in cortical neurons from HCN1 knock-out mice, suggesting little, if any, compensatory upregulation of other HCN subunits (see also Nolan et al., 2003, 2004). Indeed, expression of HCN2 mRNA in cortex of juvenile and adult mice is not different between wild-type and HCN1 knock-out mice, and HCN3 and HCN4 expression is 2–3 orders of magnitude lower than HCN1 and HCN2 regardless of age or genotype (Chen et al., 2008). It is also worth pointing out that our electrophysiological recordings were performed in cells from young mice (2–3 weeks old), whereas older animals were used for behavioral studies (2–4 months old). However, since developmental changes in HCN expression are essentially complete in other pyramidal cells by postnatal day 14 (P14)–P18 (Surges et al., 2006; Brewster et al., 2007), and because HCN2 channels are insensitive to ketamine and propofol, it is unlikely that any minor residual HCN2 current would influence behavioral responses to these anesthetics in either juveniles or adults.
HCN1 channels and hypnotic effects of other anesthetics
We found a good correlation between effects of two other anesthetic drugs on HCN1 subunits and their hypnotic actions in HCN1 knock-out mice; etomidate had no effect on HCN1 channels and its ability to evoke a LORR was unaltered in HCN1 knock-out mice, whereas propofol selectively inhibits HCN1 subunits (Cacheaux et al., 2005; Chen et al., 2005a; Lyashchenko et al., 2007), and we found a significant decrease in sensitivity of HCN1 knock-out mice to hypnotic effects of propofol. Other molecular targets, such as the GABAA receptor β2 or β3 subunit, likely contribute to residual hypnotic effects of propofol in HCN1 knock-out mice (Rudolph and Antkowiak, 2004). Since bradycardic drugs that block I h do not discriminate among HCN subunits (e.g., ZD-7288), their effects cannot be unambiguously tested in these HCN1 knock-out mice. Likewise, this mouse model was not suitable for testing sensitivity to volatile anesthetics that inhibit both HCN1 and HCN2 subunits (Chen et al., 2005b, 2008), and which activate the upregulated GABAA current in cortical neurons from HCN1 knock-out mice (X. Chen and D. A. Bayliss, unpublished observations). Nevertheless, it is noteworthy that compounds as dissimilar as ketamine and propofol share this common HCN1 molecular target, and that deletion of HCN1 leads to decreased hypnotic sensitivity to both drugs. The mechanism by which ketamine modulates HCN1 channels remains to be determined. However, HCN1 channel inhibition by ketamine shares a number of characteristics with that described for propofol, including HCN1 subunit-selectivity, insensitivity to cAMP and higher apparent affinity for shifts in V½ than for suppression of maximal current (Fig. 1) (Cacheaux et al., 2005; Chen et al., 2005a; Lyashchenko et al., 2007). For propofol, an allosteric inhibitory mechanism has been described that involves the conserved membrane channel core, independent of the N terminus or the C-terminal regulatory domains (Lyashchenko et al., 2007).
Hypnosis and anesthetic modulation of I h in cortical neurons
Our results indicate that effects of ketamine and propofol on excitability and dendritosomatic integration in cortical pyramidal neurons are primarily due to effects on I h, and specifically on HCN1 subunits. Both compounds inhibit only HCN1-containing channels in HEK293 cells and, accordingly, inhibition of I h in cortical neurons by ketamine and propofol was not seen in HCN1 knock-out mice. In addition, their effects on input resistance, sag, resonance and synaptic summation are consistent with inhibition of I h and completely absent in HCN1 knock-out mice. Finally, current inhibition by these drugs has similar characteristics in both HEK293 cells and cortical neurons (i.e., it involves a negative shift in V½ and decreased maximal current amplitude). Although this latter correspondence is reassuring, strict quantitative comparison of anesthetic effects on channel properties obtained in these two preparations should be avoided; it is unlikely that voltage control of dendritically located channels in cortical neurons can match that obtained for HCN channels expressed in HEK293 cells. Nonetheless, these data argue strongly that ketamine and propofol inhibit HCN1-containing channels and dendritic I h in cortical neurons to mediate enhanced dendritosomatic synaptic integration. Interestingly, a recent report noted that ketamine/xylazine anesthesia enhanced calcium spikes and dendritic excitability in layer 5 cortical pyramidal neurons, both in vitro and in vivo, although the ionic mechanism was not determined (Potez and Larkum, 2008); our results suggest that this could be due, at least in part, to ketamine-mediated inhibition of I h.
This cellular analysis of anesthetic actions in cortical pyramidal neurons suggests at least one site where inhibition of I h could contribute to hypnosis. The combination of membrane hyperpolarization and reduced dendritic shunt associated with inhibition of I h are expected to support coherent oscillatory behavior (Carr et al., 2007); indeed, blocking I h with ZD-7288 prolongs recurrent network activity in the cortex (Wang et al., 2007) and decreases in I h have been observed in a number of epilepsy models in rodents (Shah et al., 2004; Jung et al., 2007; Kole et al., 2007). In animals anesthetized with ketamine (in combination with urethane, xylazine or N2O) (Steriade et al., 1993), a prominent slow EEG oscillation (<1 Hz) is observed that resembles the so-called slow rhythm of deep sleep (Amzica and Steriade, 1998). This slow oscillation is cortically derived, accompanied by membrane hyperpolarization and maintained by balanced excitatory and inhibitory intracortical synaptic inputs (Amzica and Steriade, 1998; Shu et al., 2003). So, we propose that inhibition of I h by ketamine promotes synaptically mediated synchronization of rhythmic cortical activity by contributing to the membrane hyperpolarization required for oscillatory activity while simultaneously decreasing dendritic shunt to enhance synaptic efficacy. In this way, ketamine may modulate endogenous cortical mechanisms that contribute to normal sleep to bring about its own unique state of altered consciousness.
Footnotes
This work was supported by American Heart Association Beginning Grant-in-Aid 0665349U to X.C. and National Institutes of Health–National Institutes of General Medical Sciences Grant GM66181 to D.A.B. We thank Drs. E. M. Talley and P. Q. Barrett for comments on this manuscript, and Drs. F. Viana and P. Álvarez for advice on performance and analysis of ZAP function experiments.
References
- Amzica and Steriade, 1998.Amzica F, Steriade M. Electrophysiological correlates of sleep delta waves. Electroencephalogr Clin Neurophysiol. 1998;107:69–83. doi: 10.1016/s0013-4694(98)00051-0. [DOI] [PubMed] [Google Scholar]
- Andrásfalvy et al., 2008.Andrásfalvy BK, Makara JK, Johnston D, Magee JC. Altered synaptic and non-synaptic properties of CA1 pyramidal neurons in Kv4.2 knockout mice. J Physiol. 2008;586:3881–3892. doi: 10.1113/jphysiol.2008.154336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antkowiak, 2002.Antkowiak B. In vitro networks: cortical mechanisms of anaesthetic action. Br J Anaesth. 2002;89:102–111. doi: 10.1093/bja/aef154. [DOI] [PubMed] [Google Scholar]
- Bazhenov et al., 1998.Bazhenov M, Timofeev I, Steriade M, Sejnowski TJ. Computational models of thalamocortical augmenting responses. J Neurosci. 1998;18:6444–6465. doi: 10.1523/JNEUROSCI.18-16-06444.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewster et al., 2007.Brewster AL, Chen Y, Bender RA, Yeh A, Shigemoto R, Baram TZ. Quantitative analysis and subcellular distribution of mRNA and protein expression of the hyperpolarization-activated cyclic nucleotide-gated channels throughout development in rat hippocampus. Cereb Cortex. 2007;17:702–712. doi: 10.1093/cercor/bhk021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cacheaux et al., 2005.Cacheaux LP, Topf N, Tibbs GR, Schaefer UR, Levi R, Harrison NL, Abbott GW, Goldstein PA. Impairment of hyperpolarization-activated, cyclic nucleotide-gated channel function by the intravenous general anesthetic propofol. J Pharmacol Exp Ther. 2005;315:517–525. doi: 10.1124/jpet.105.091801. [DOI] [PubMed] [Google Scholar]
- Carr et al., 2007.Carr DB, Andrews GD, Glen WB, Lavin A. α2-Noradrenergic receptors activation enhances excitability and synaptic integration in rat prefrontal cortex pyramidal neurons via inhibition of HCN currents. J Physiol. 2007;584:437–450. doi: 10.1113/jphysiol.2007.141671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen et al., 2005a.Chen X, Shu S, Bayliss DA. Suppression of Ih contributes to propofol-induced inhibition of mouse cortical pyramidal neurons. J Neurophysiol. 2005a;94:3872–3883. doi: 10.1152/jn.00389.2005. [DOI] [PubMed] [Google Scholar]
- Chen et al., 2005b.Chen X, Sirois JE, Lei Q, Talley EM, Lynch C, 3rd, Bayliss DA. HCN subunit-specific and cAMP-modulated effects of anesthetics on neuronal pacemaker currents. J Neurosci. 2005b;25:5803–5814. doi: 10.1523/JNEUROSCI.1153-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen et al., 2008.Chen X, Shu S, Kennedy DP, Willcox SC, Bayliss DA. Subunit-specific effects of isoflurane on neuronal Ih in HCN1 knockout mice. J Neurophysiol. 2008 doi: 10.1152/jn.01352.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen et al., 1973.Cohen ML, Chan SL, Way WL, Trevor AJ. Distribution in the brain and metabolism of ketamine in the rat after intravenous administration. Anesthesiology. 1973;39:370–376. doi: 10.1097/00000542-197310000-00003. [DOI] [PubMed] [Google Scholar]
- Daniell, 1990.Daniell LC. The noncompetitive N-methyl-d-aspartate antagonists, MK-801, phencyclidine and ketamine, increase the potency of general anesthetics. Pharmacol Biochem Behav. 1990;36:111–115. doi: 10.1016/0091-3057(90)90134-4. [DOI] [PubMed] [Google Scholar]
- Franks and Lieb, 1994.Franks NP, Lieb WR. Molecular and cellular mechanisms of general anaesthesia. Nature. 1994;367:607–614. doi: 10.1038/367607a0. [DOI] [PubMed] [Google Scholar]
- Garfield and Bukusoglu, 1996.Garfield JM, Bukusoglu C. Propofol and ethanol produce additive hypnotic and anesthetic effects in the mouse. Anesth Analg. 1996;83:156–161. doi: 10.1097/00000539-199607000-00027. [DOI] [PubMed] [Google Scholar]
- Hevers et al., 2008.Hevers W, Hadley SH, Lüddens H, Amin J. Ketamine, but not phencyclidine, selectively modulates cerebellar GABAA receptors containing α6 and δ subunits. J Neurosci. 2008;28:5383–5393. doi: 10.1523/JNEUROSCI.5443-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill and Tononi, 2005.Hill S, Tononi G. Modeling sleep and wakefulness in the thalamocortical system. J Neurophysiol. 2005;93:1671–1698. doi: 10.1152/jn.00915.2004. [DOI] [PubMed] [Google Scholar]
- Hutcheon and Yarom, 2000.Hutcheon B, Yarom Y. Resonance, oscillation and the intrinsic frequency preferences of neurons. Trends Neurosci. 2000;23:216–222. doi: 10.1016/s0166-2236(00)01547-2. [DOI] [PubMed] [Google Scholar]
- Idvall et al., 1979.Idvall J, Ahlgren I, Aronsen KR, Stenberg P. Ketamine infusions: pharmacokinetics and clinical effects. Br J Anaesth. 1979;51:1167–1173. doi: 10.1093/bja/51.12.1167. [DOI] [PubMed] [Google Scholar]
- Irifune et al., 2007.Irifune M, Katayama S, Takarada T, Shimizu Y, Endo C, Takata T, Morita K, Dohi T, Sato T, Kawahara M. MK-801 enhances gabaculine-induced loss of the righting reflex in mice, but not immobility. Can J Anaesth. 2007;54:998–1005. doi: 10.1007/BF03016634. [DOI] [PubMed] [Google Scholar]
- Jung et al., 2007.Jung S, Jones TD, Lugo JN, Jr, Sheerin AH, Miller JW, D'Ambrosio R, Anderson AE, Poolos NP. Progressive dendritic HCN channelopathy during epileptogenesis in the rat pilocarpine model of epilepsy. J Neurosci. 2007;27:13012–13021. doi: 10.1523/JNEUROSCI.3605-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelland et al., 1993.Kelland MD, Soltis RP, Boldry RC, Walters JR. Behavioral and electrophysiological comparison of ketamine with dizocilpine in the rat. Physiol Behav. 1993;54:547–554. doi: 10.1016/0031-9384(93)90248-e. [DOI] [PubMed] [Google Scholar]
- Kohrs and Durieux, 1998.Kohrs R, Durieux ME. Ketamine: teaching an old drug new tricks. Anesth Analg. 1998;87:1186–1193. doi: 10.1097/00000539-199811000-00039. [DOI] [PubMed] [Google Scholar]
- Kole et al., 2007.Kole MH, Bräuer AU, Stuart GJ. Inherited cortical HCN1 channel loss amplifies dendritic calcium electrogenesis and burst firing in a rat absence epilepsy model. J Physiol. 2007;578:507–525. doi: 10.1113/jphysiol.2006.122028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyashchenko et al., 2007.Lyashchenko AK, Redd KJ, Yang J, Tibbs GR. Propofol inhibits HCN1 pacemaker channels by selective association with the closed states of the membrane embedded channel core. J Physiol. 2007;583:37–56. doi: 10.1113/jphysiol.2007.136465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magee, 2000.Magee JC. Dendritic integration of excitatory synaptic input. Nat Rev Neurosci. 2000;1:181–190. doi: 10.1038/35044552. [DOI] [PubMed] [Google Scholar]
- Nolan et al., 2003.Nolan MF, Malleret G, Lee KH, Gibbs E, Dudman JT, Santoro B, Yin D, Thompson RF, Siegelbaum SA, Kandel ER, Morozov A. The hyperpolarization-activated HCN1 channel is important for motor learning and neuronal integration by cerebellar Purkinje cells. Cell. 2003;115:551–564. doi: 10.1016/s0092-8674(03)00884-5. [DOI] [PubMed] [Google Scholar]
- Nolan et al., 2004.Nolan MF, Malleret G, Dudman JT, Buhl DL, Santoro B, Gibbs E, Vronskaya S, Buzsáki G, Siegelbaum SA, Kandel ER, Morozov A. A behavioral role for dendritic integration: HCN1 channels constrain spatial memory and plasticity at inputs to distal dendrites of CA1 pyramidal neurons. Cell. 2004;119:719–732. doi: 10.1016/j.cell.2004.11.020. [DOI] [PubMed] [Google Scholar]
- Nolan et al., 2007.Nolan MF, Dudman JT, Dodson PD, Santoro B. HCN1 channels control resting and active integrative properties of stellate cells from layer II of the entorhinal cortex. J Neurosci. 2007;27:12440–12451. doi: 10.1523/JNEUROSCI.2358-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orser et al., 1997.Orser BA, Pennefather PS, MacDonald JF. Multiple mechanisms of ketamine blockade of N-methyl-d-aspartate receptors. Anesthesiology. 1997;86:903–917. doi: 10.1097/00000542-199704000-00021. [DOI] [PubMed] [Google Scholar]
- Petrenko et al., 2004.Petrenko AB, Yamakura T, Fujiwara N, Askalany AR, Baba H, Sakimura K. Reduced sensitivity to ketamine and pentobarbital in mice lacking the N-methyl-d-aspartate receptor GluRε1 subunit. Anesth Analg. 2004;99:1136–1140. doi: 10.1213/01.ANE.0000131729.54986.30. [DOI] [PubMed] [Google Scholar]
- Petrenko et al., 2006.Petrenko AB, Yamakura T, Askalany AR, Kohno T, Sakimura K, Baba H. Effects of ketamine on acute somatic nociception in wild-type and N-methyl-d-aspartate (NMDA) receptor ε1 subunit knockout mice. Neuropharmacology. 2006;50:741–747. doi: 10.1016/j.neuropharm.2005.11.019. [DOI] [PubMed] [Google Scholar]
- Potez and Larkum, 2008.Potez S, Larkum ME. Effect of common anesthetics on dendritic properties in layer 5 neocortical pyramidal neurons. J Neurophysiol. 2008;99:1394–1407. doi: 10.1152/jn.01126.2007. [DOI] [PubMed] [Google Scholar]
- Rudolph and Antkowiak, 2004.Rudolph U, Antkowiak B. Molecular and neuronal substrates for general anaesthetics. Nat Rev Neurosci. 2004;5:709–720. doi: 10.1038/nrn1496. [DOI] [PubMed] [Google Scholar]
- Santoro et al., 2000.Santoro B, Chen S, Luthi A, Pavlidis P, Shumyatsky GP, Tibbs GR, Siegelbaum SA. Molecular and functional heterogeneity of hyperpolarization-activated pacemaker channels in the mouse CNS. J Neurosci. 2000;20:5264–5275. doi: 10.1523/JNEUROSCI.20-14-05264.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato et al., 2005.Sato Y, Kobayashi E, Murayama T, Mishina M, Seo N. Effect of N-methyl-d-aspartate receptor epsilon1 subunit gene disruption of the action of general anesthetic drugs in mice. Anesthesiology. 2005;102:557–561. doi: 10.1097/00000542-200503000-00013. [DOI] [PubMed] [Google Scholar]
- Schnoebel et al., 2005.Schnoebel R, Wolff M, Peters SC, Bräu ME, Scholz A, Hempelmann G, Olschewski H, Olschewski A. Ketamine impairs excitability in superficial dorsal horn neurones by blocking sodium and voltage-gated potassium currents. Br J Pharmacol. 2005;146:826–833. doi: 10.1038/sj.bjp.0706385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah et al., 2004.Shah MM, Anderson AE, Leung V, Lin X, Johnston D. Seizure-induced plasticity of h channels in entorhinal cortical layer III pyramidal neurons. Neuron. 2004;44:495–508. doi: 10.1016/j.neuron.2004.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu et al., 2003.Shu Y, Hasenstaub A, McCormick DA. Turning on and off recurrent balanced cortical activity. Nature. 2003;423:288–293. doi: 10.1038/nature01616. [DOI] [PubMed] [Google Scholar]
- Sinner and Graf, 2008.Sinner B, Graf BM. Ketamine. Handb Exp Pharmacol. 2008;182:313–333. doi: 10.1007/978-3-540-74806-9_15. [DOI] [PubMed] [Google Scholar]
- Stabernack et al., 2003.Stabernack C, Sonner JM, Laster M, Zhang Y, Xing Y, Sharma M, Eger EI., 2nd Spinal N-methyl-d-aspartate receptors may contribute to the immobilizing action of isoflurane. Anesth Analg. 2003;96:102–107. doi: 10.1097/00000539-200301000-00022. table of contents. [DOI] [PubMed] [Google Scholar]
- Steriade et al., 1993.Steriade M, Nuñez A, Amzica F. A novel slow (<1 Hz) oscillation of neocortical neurons in vivo: depolarizing and hyperpolarizing components. J Neurosci. 1993;13:3252–3265. doi: 10.1523/JNEUROSCI.13-08-03252.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surges et al., 2006.Surges R, Brewster AL, Bender RA, Beck H, Feuerstein TJ, Baram TZ. Regulated expression of HCN channels and cAMP levels shape the properties of the h current in developing rat hippocampus. Eur J Neurosci. 2006;24:94–104. doi: 10.1111/j.1460-9568.2006.04880.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang et al., 2007.Wang M, Ramos BP, Paspalas CD, Shu Y, Simen A, Duque A, Vijayraghavan S, Brennan A, Dudley A, Nou E, Mazer JA, McCormick DA, Arnsten AF. Alpha2A-adrenoceptors strengthen working memory networks by inhibiting cAMP-HCN channel signaling in prefrontal cortex. Cell. 2007;129:397–410. doi: 10.1016/j.cell.2007.03.015. [DOI] [PubMed] [Google Scholar]
- White et al., 1980.White PF, Ham J, Way WL, Trevor AJ. Pharmacology of ketamine isomers in surgical patients. Anesthesiology. 1980;52:231–239. doi: 10.1097/00000542-198003000-00008. [DOI] [PubMed] [Google Scholar]
- Wolff and Winstock, 2006.Wolff K, Winstock AR. Ketamine: from medicine to misuse. CNS Drugs. 2006;20:199–218. doi: 10.2165/00023210-200620030-00003. [DOI] [PubMed] [Google Scholar]
- Yamakura et al., 2000.Yamakura T, Sakimura K, Shimoji K. The stereoselective effects of ketamine isomers on heteromeric N-methyl-d-aspartate receptor channels. Anesth Analg. 2000;91:225–229. doi: 10.1097/00000539-200007000-00042. [DOI] [PubMed] [Google Scholar]
- Zarate et al., 2006.Zarate CA, Jr, Singh JB, Carlson PJ, Brutsche NE, Ameli R, Luckenbaugh DA, Charney DS, Manji HK. A randomized trial of an N-methyl-d-aspartate antagonist in treatment-resistant major depression. Arch Gen Psychiatry. 2006;63:856–864. doi: 10.1001/archpsyc.63.8.856. [DOI] [PubMed] [Google Scholar]