

Abstract
CCL19 and CCL21 and their receptor CCR7 are expressed constitutively within lymphoid organs, regulating lymphocyte homing. Recent studies suggest that these chemokines may have inflammatory properties. We hypothesized a role of CCL19/CCL21 in human immunodeficiency virus (HIV) infection by promoting inflammation. We examined the expression of CCL19 and CCL21 in mononuclear cells from peripheral blood mononuclear cells (PBMC) and bone marrow mononuclear cells (BMMC) in HIV-infected patients before and during highly active anti-retroviral therapy (HAART). We also examined the ability of CCL19/CCL21 to promote inflammatory responses in these patients. PBMC from untreated HIV-infected patients (n = 29) released enhanced levels of CCL19 spontaneously compared with cells from controls (n = 20), particularly in those with symptomatic disease (n = 15, P < 0·01 versus controls). During HAART (n = 9), there was a decrease in the spontaneous CCL19 release and an increase in the phytohaemagglutinin-stimulated CCL19 release in both PBMC (P < 0·01) and BMMC (P < 0·05). In patients with enhanced HIV replication there was an increased proportion of inflammatory CD8+CCR7-CD45RA- T cells in peripheral blood [P < 0·01 and P < 0·05 versus controls, untreated (n = 9) and treatment failure (n = 8), respectively]. In vitro, CCL19/CCL21 promoted an inflammatory response in PBMC when accompanied by high viral load, irrespective of HAART. The HIV-tat protein significantly boosted the inflammatory effect of CCL19/CCL21 in PBMC. These findings link a dysregulated CCL19/CCL21/CCR7 system in HIV-infected patients to persistent inflammation and HIV replication, not only in untreated HIV infection, but also in treatment failure during HAART.
Keywords: chemokines, HIV infection, inflammation, lymphocytes
Introduction
Chemokines are known as regulatory molecules in leucocyte maturation, lymphocyte homing and the development of lymphoid tissues [1,2]. Some of these chemokines may be protective in human immunodeficiency virus (HIV)-infected individuals by their ability to block HIV entry into T cells and macrophages through the HIV co-receptors CXCR4 and CCR5 [3,4]. However, HIV infection is characterized by persistent immune activation that contributes to immunodeficiency. Based on their potent inflammatory properties, chemokines could also have harmful effects in HIV-infected individuals [5].
In contrast to inflammatory chemokines, the homeostatic chemokines such as CCL19 and CCL21 and their cognate receptor (i.e. CCR7) are expressed constitutively within secondary lymphoid tissue, regulating lymphocyte and dendritic cell (DC) homing [6,7]. Recent studies suggest that these chemokines also could promote inflammatory responses [8,9]. In fact, CCL19, CCL21 and CCR7 have been implicated in the pathogenesis of various inflammatory and infectious disorders (e.g. atherosclerosis, viral hepatitis and influenza) [10–12].
There are some studies on CCL19/CCL21/CCR7 expression during HIV infection. CCL19 and CCL21 have been reported to contribute to persistent HIV infection in secondary lymphoid tissue by promoting viral replication in activated T cells as well as facilitating infection of resting CD4+ T cells and establishing viral latency [13]. Moreover, Choi et al. have reported altered CCL19 and CCL21 expression in the simian immunodeficiency virus (SIV)/macaque model, potentially promoting alterations in the networks of antigen-presenting cells in lymphoid tissues, contributing ultimately to systemic immunodeficiency [14]. Furthermore, we have recently shown increased serum levels of CCL19 and CCL21 in relation to disease progression in HIV-infected patients, and during highly active anti-retroviral therapy (HAART) a decrease in CCL19/CCL21 levels was restricted to virological responders [15]. However, there are no data on CCL19/CCL21 regulation at the cellular level in HIV-infected patients. Moreover, while the role of CCL19 and CCL21 in lymphocyte homing has been studied extensively, the ability of these chemokines to promote inflammatory responses in HIV-infected individuals is far from clear.
To elucidate further the role of CCL19 and CCL21 in HIV infection we examined the expression of these chemokines at the cellular levels in mononuclear cells from peripheral blood and bone marrow aspirates both before and during HAART. We also examined the ability of CCL19 and CCL21 to promote inflammatory responses in HIV-infected patients in relation to ongoing HIV replication.
Materials and methods
Patients and control subjects
Twenty-nine HIV-infected patients were included in a cross-sectional study where none of the patients received HAART {18 males and 11 females, 30 ± 6 years [mean ± standard error of the mean (s.e.m.)]}. Fourteen HIV-infected patients were classified as asymptomatic [Centers for Disease Control and Prevention (CDC) group A; CD4+ T cell count, 485 ± 55 × 106/l; CD8+ T cell count, 1004 ± 85 × 106/l; HIV RNA level, 110 ± 35 × 103 copies/ml plasma (mean ± s.e.m.)] and 15 as symptomatic [10 in CDC group B and five in CDC group C; CD4+ T cell count, 95 ± 45 × 106/l; CD8+ T cell count, 450 ± 64 × 106/l; HIV RNA level, 755 ± 76 × 103 copies/ml plasma (mean ± s.e.m.)]. For comparison, blood samples were also collected from 20 sex- and age-matched healthy controls. A subgroup of nine patients from the cross-sectional study was also followed during HAART with samples taken before and 4 and 26 weeks after initiation of therapy.
In another substudy, examining CCR7 expression and the effect of CCL19/CCL21, blood samples were collected from HIV-infected patients before initiating HAART (n = 9), HIV-infected patients on successful HAART (HIV RNA < 40 copies/ml, n = 10), HIV-infected patients with virological treatment failure (HIV RNA > 5000 copies/ml, n = 8) and healthy controls (n = 9), although not all experiments could be performed in all individuals.
Informed consent for sampling was obtained from all subjects. All parts of the study were conducted according to the ethical guidelines at our hospital in conformity with the Helsinki declaration, and approved by the local ethical committee.
Bone marrow culture
Heparinized bone marrow samples were obtained by aspiration from the posterior iliac crest, and bone marrow mononuclear cells (BMMC) were prepared by density gradient centrifugation (Lymphoprep; Nycomed Pharma, Oslo, Norway). BMMC were incubated in flat-bottomed 96-well trays (2 × 106/ml; Costar, Cambridge, MA, USA), in medium alone [RPMI-1640 with 2 mM L-glutamine and 25 mM HEPES buffer (Gibco, Paisley, UK) supplemented with 10% fetal calf serum (FCS; Sigma, St Louis, MO, USA)] or stimulated with phytohaemagglutinin (PHA; final dilution 1:100; Murex, Dartford, UK). The concentration of PHA was based on previous dose–response experiments (data not shown).
PBMC culture
PBMC, obtained from heparinized blood by density gradient centrifugation (Lymphoprep; Nycomed Pharma), were incubated in flat-bottomed 96-well trays (2 × 106/ml; Costar) in medium alone [RPMI-1640 with 2 mM L-glutamine and 25 mM HEPES buffer (PAA Laboratories, Pasching, Austria) supplemented with 10% FCS (PAA Laboratories)] or stimulated with CCL19 (100 ng/ml; R&D Systems, Minneapolis, MN, USA), CCL21 (100 ng/ml; R&D Systems), PHA (final dilution 1:100; Murex), recombinant HIV-1 tat protein (HIV-tat; 1 µg/ml; ImmunoDiagnostics, Woburn, MA, USA), recombinant HIV-1 gp120 protein (HIV-gp120; 1 µg/ml; ImmunoDiagnostics) and recombinant HIV-1 p24 protein (HIV-p24; 1 µg/ml; ImmunoDiagnostics).
Separation and stimulation of CD8+ T cell subsets
Separation of CD3+CD8+CCR7+CD45RA+, CD3+CD8+CCR7-CD45RA+, CD3+CD8+CCR7+CD45RA- and CD3+CD8+CCR7-CD45RA- T cells from freshly isolated PBMC were performed by staining with fluorescein isothiocyanate (FITC)-conjugated anti-CD3, peridinin chlorophyll protein (PerCP)-conjugated anti-CD8, allophycocyanin (APC)-conjugated anti-CD45RA (all from Becton Dickinson, San Diego, CA, USA), and phycoerythrin (PE)-conjugated anti-CCR7 (R&D Systems), with subsequent use of a fluorescence activated cell sorter (FACS)Diva cell sorter equipped with the FACSDiva software (Becton Dickinson). The CD8+ T cell subsets were resuspended in RPMI-1640 with 2 mM L-glutamine and 25 mM HEPES buffer (Gibco), supplemented with 10% FCS (Sigma) and incubated in flat-bottomed 96-well plates (106/ml; 0·2 ml/well; Costar). The cells were stimulated with immunomagnetic beads pre-coated with anti-CD3 and anti-CD28 (Dynabeads, CD3/CD28 T Cell Expander, cat. no. 11131D; Dynal, Oslo, Norway) at a cell-to-bead ratio of 1:1. In all in vitro experiments (BMMC, PBMC and CD8+ T cell subsets), cell-free supernatants were harvested at different time-points and stored at –80°C until analysis.
Flow cytometry
Cryopreserved PBMC were thawed as described previously [16,17]. Staining was performed using FITC-conjugated anti-CD3, PerCP-conjugated anti-CD4 and anti-CD8, and APC-conjugated anti-CD45RA (all from Becton-Dickinson) and PE-conjugated anti-CCR7 (R&D Systems). Isotype controls were used as appropriate. Flow cytometry was performed using a FACSCalibur instrument equipped with the CellQuest software (Becton Dickinson).
Cytokine measurements
Concentrations of CCL19, CCL21 tumour necrosis factor (TNF)-α, and interferon (IFN)-γ in supernatants from PBMC and BMMC (CCL19 and CCL21) were measured by enzyme immunoassays (R&D Systems). Cytokine levels in CD8+ T cell subset supernatants were measured by multiplex suspension array technology using the BioPlex (Bio-Rad, Hercules, CA, USA). TNF-α and IFN-γ multiplexable beads were purchased from R&D Systems. The quantification was accomplished by using the BioPlex Manager Software (Bio-Rad).
HIV RNA quantification
HIV RNA levels were measured by quantitative reverse polymerase chain reaction (Amplicor HIV Monitor; Roche Diagnostic Systems, Branchburg, NJ, USA; detection limit 40 copies/ml plasma).
Statistical analysis
In this study we were looking for rather large differences (> 50% changes), and based on previous studies from our group on inflammatory markers in HIV infection a sample size as in the present study would be adequate. For comparisons of two groups of individuals, the Mann–Whitney U-test was used. When more than two groups were compared, the Kruskal–Wallis test was used. If a significant difference was found, the Mann–Whitney U-test was used to determine the differences between each pair of groups. The Wilcoxon signed-rank test was used for comparison of paired data. Coefficients of correlation were calculated by Spearman's rank test. Probabilities are two-sided and considered to be significant when P < 0·05.
Results
CCL19 and CCL21 levels in PBMC
We first examined the release of CCL19 and CCL21 in un-stimulated PBMC from 29 treatment-naive HIV-infected patients and 20 healthy controls. As shown in Fig. 1, PBMC from untreated HIV-infected patients released significantly enhanced levels of CCL19 spontaneously compared with cells from healthy controls, correlated significantly with CD4+ T cell counts (r = −0·45, P < 0·05) and viral load (r = 0·57, P < 0·05) in these patients. In contrast, CCL21 was not detected in PBMC supernatants from either patients or controls.
Fig. 1.
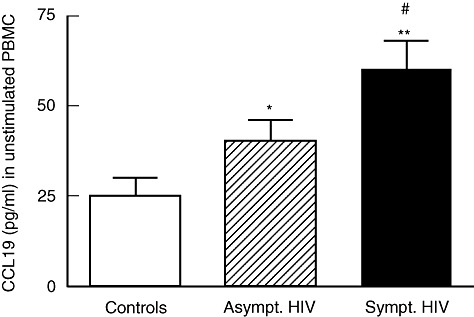
The spontaneous release of CCL19 in peripheral blood mononuclear cells (PBMC). The spontaneous release of CCL19 in PBMC was assessed by enzyme immunoassay after culturing for 20 h in 20 healthy controls and in 29 treatment-naive human immunodeficiency virus (HIV)-infected patients; 14 with asymptomatic (CDC group A) and 15 with symptomatic (10 in CDC group B and five in CDC group C) HIV infection. *P < 0·05 and **P < 0·001 versus controls. #P < 0·05 versus asymptomatic HIV infection. Data are mean ± standard error of the mean.
CCL19 and CCL21 in PBMC and BMMC during HAART
Nine of the HIV-infected patients were followed during HAART with blood and bone marrow samples taken before and 4 and 26 weeks after initiating therapy. At baseline, the spontaneous release of CCL19 was increased and the PHA-stimulated release of CCL19 was decreased in HIV-infected patients compared with healthy controls in both PBMC and BMMC (Fig. 2). During HAART, this pattern was at least partly reversed. Thus, along with the decrease in viral load and increase in CD4+ T cell counts (data not shown), HAART induced a decrease in the spontaneous CCL19 release and an increase in the PHA-stimulated CCL19 release in both PBMC and BMMC (Fig. 2). Similar to PBMC, CCL21 was not detected in BMMC in either patients or controls.
Fig. 2.
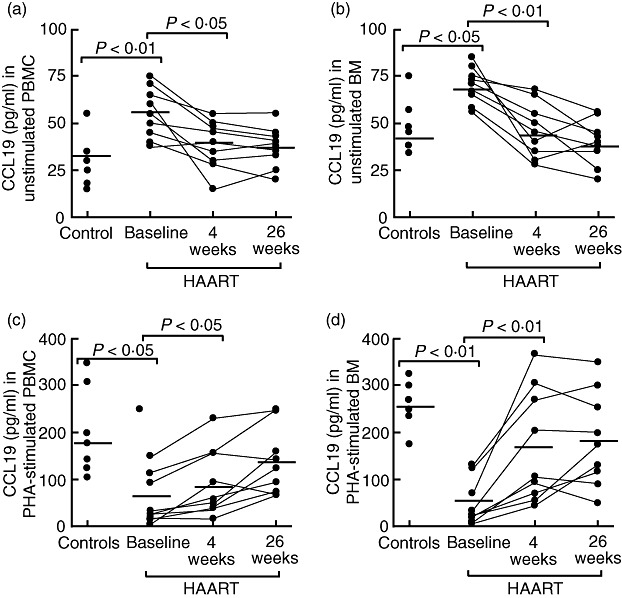
The release of CCL19 from mononuclear cells during highly active anti-retroviral therapy (HAART). The panels show the spontaneous (a, b) and phytohaemagglutinin (PHA)-stimulated (c, d) release of CCL19 in mononuclear cells from peripheral blood mononuclear cells (PBMC, left panels) and bone marrow (BM) aspirates (right panels) after culturing for 20 h in six healthy controls and in nine human immunodeficiency virus (HIV)-infected patients before (baseline) and at different time-points (weeks) after initiating HAART. The bold horizontal line indicates the mean of the observations (PHA final dilution 1:100).
Expression of CCR7 in HIV infection
To characterize further the CCL19/CCL21 system in HIV infection, we examined the expression of their corresponding receptor, CCR7, by flow cytometry in PBMC from HIV-infected patients before initiating HAART (n = 9), HIV-infected patients on successful HAART (HIV RNA < 40 copies/ml, n = 9), HIV-infected patients with virological treatment failure (HIV RNA > 5000 copies/ml, n = 8) and healthy controls (n = 9). While there were no differences in the expression CCR7 in monocytes between HIV-infected patients and controls, the percentage of CCR7 expressing T cells was decreased significantly in HIV-infected patients with high viral load, irrespective of ongoing HAART (data not shown). Further subanalyses showed that this decrease in the percentage of CCR7+ T cells was seen primarily in the CD8+ T cell subset, reflecting a decreased proportion of CCR7 expressing CD8+CD45RA+ T cells (Fig. 3a–d), with no changes in the proportion of CCR7+ CD4+ T cells (data not shown). Consequently, HIV-infected patients with high viral load, irrespective of ongoing HAART, showed an increased proportion of CD8+CCR7-CD45RA- T cells, thought to represent effector memory cells (Fig. 3D).
Fig. 3.
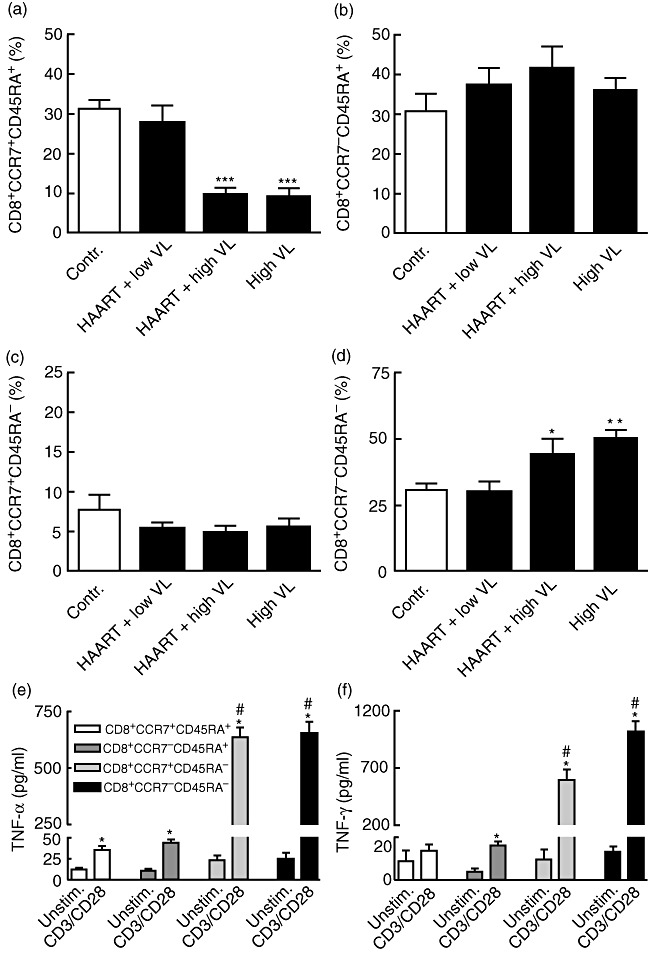
Expression of CCR7 in CD8+ T cells from human immunodeficiency virus (HIV)-infected patients. The proportion of different CD8+ T cell subpopulations according to the expression of CCR7 as assessed by flow cytometry in HIV-infected patients with high viral load (VL) before initiating highly active anti-retroviral therapy (HAART) (HIV RNA > 5000 copies/ml plasma, high VL, n = 9), HIV-infected patients with virological treatment failure (HIV RNA > 5000 copies/ml plasma, HAART + high VL, n = 8), HIV-infected patients on successful HAART (HIV RNA < 40 copies/ml plasma, HAART+ low VL n = 9), and healthy controls (contr. n = 9). *P < 0·05, **P < 0·01, and ***P < 0·001 versus controls and those on successful HAART. The lower panels show tumour necrosis factor (TNF)-α (e) and interferon (IFN)-γ (f) levels in different CD8+ T cell subpopulations after culturing for 24 h with or without anti-CD3/anti-CD28 activation (see Methods). The T cell subpopulations were obtained from four healthy controls, and the data are based in two parallel experiments from these individuals. *P < 0·05 versus unstimulated cells of the same CD8+ T cell subset. #P < 0·05 versus CD8+CCR7+CD45RA+ T cells. Data are mean ± standard error of the mean (panels a–f).
Cytokine profile in CD8+ T cell subsets
HIV-infected patients with high viral load were characterized by an increased proportion of CD8+CCR7-CD45RA- T cells on the cost of a decrease in CD8+CCR7+CD45RA+ T cells. In an attempt to elucidate any immunological consequences of this pattern, we examined the cytokine profile in these two CD8+ T cell subsets. Unfortunately, we were not able to isolate these subsets (cell sorter) from HIV-infected patients, and accordingly, this experiment was performed only in healthy individuals (n = 4). Anti-CD3/anti-CD28-activated CD8+CCR7-CD45RA- T cells released large amounts of TNF-α and IFN-γ compared with CD8+CCR7+CD45RA+ T cells, suggesting that the subpopulation that dominates within the CD8+ T cell subset in HIV-infected individuals with high viral load represents an inflammatory phenotype (Fig. 3e and f). The other CD8+CD45RA- subset (CD8+CCR7+CD45RA- T cells) also released large amounts of TNF-α and IFN-γ upon activation compared with CD8+CD45RA+ cells (Fig. 3e and f). However, based on the lack of data from HIV-infected patients, these results should be interpreted with caution.
Effect of CCL19 and CCL21 on cytokine responses in PBMC
In addition to CCR7- and CCR7+ CD45RA- T cells, CCR7 expressing monocytes may have an inflammatory potential upon activation [18]. To elucidate further the consequences of a dysregulated CCL19/CCL21/CCR7 system in HIV-infected individuals, with and without accompanying high viral load, we examined the CCL19/CCL21-mediated cytokine response in PBMC from HIV-infected patients with low viral load (n = 10, HIV RNA < 40 copies/ml, all on HAART), HIV-infected patients with high viral load (n = 14, > 5000 HIV RNA copies/ml; seven on HAART with drug resistance and seven without anti-retroviral therapy), and in PBMC from healthy controls (n = 8). CCL19, and to a lesser degree CCL21, promoted a marked increase in TNF-α and IFN-γ release in PBMC from those with high viral load, irrespective of ongoing HAART, with no (CCL21) or only modest (CCL19) responses in those on successful HAART (Fig. 4a and b). While PBMC from healthy controls showed no release of TNF-α and IFN-γ when stimulated with CCL19 and CCL21 alone (Fig. 4a and b), co-stimulation with HIV-tat protein, but not with HIV-gp120 and HIV-p24 protein (data not shown), boosted the CCL19- and CCL21-mediated cytokine response in these cells (Fig. 4c and d).
Fig. 4.
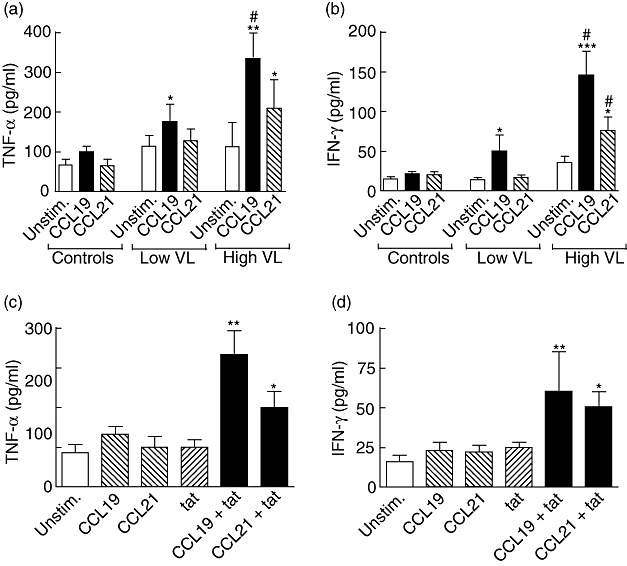
Effect of CCL19 and CCL21 on cytokine responses in peripheral blood mononuclear cells (PBMC). The CCL19/CCL21 (100 ng/ml for both) mediated release of tumour necrosis factor (TNF)-α (a) and interferon (IFN)-γ (b) in PBMC from human immunodeficiency virus (HIV)-infected patients with low viral load (VL) [n = 10, HIV RNA < 40 copies/ml plasma, all on highly active anti-retroviral therapy (HAART)], HIV-infected patients with high VL (n = 14, > 5000 HIV RNA copies/ml plasma; seven on HAART with drug resistance and seven without antiretroviral therapy), and in PBMC from healthy controls (n = 8) (culture time 72 h). *P < 0·05, **P < 0·01, and ***P < 0·001 versus unstimulated (unstim.) cells. #P < 0·05 versus those with low VL and healthy controls. The lower panels show the effects of CCL19 (100 ng/ml), CCL21 (100 ng/ml), HIV-1 tat protein (tat; 1 µg/ml), or a combination thereof on the release of TNF-α (c) and IFN-γ (d) in PBMC from five of the healthy controls after culturing for 72 h. *P < 0·05 and **P < 0·01 versus unstimulated cells. Data are mean ± standard error of the mean (panels a–d).
Discussion
We have previously reported enhanced serum levels of CCL19 and CCL21 in HIV-infected patients in relation to disease severity in untreated HIV infection [15]. In the present study we extend these findings by showing that HAART-naive HIV-infected patients are characterized by increased spontaneous and decreased stimulated release of CCL19 in mononuclear cells from bone marrow aspirates and peripheral blood, and these changes were reversed at least partly by HAART. In those with enhanced HIV replication, changes in CCL19 levels were accompanied by a decreased proportion of CCR7+ T cells in peripheral blood, irrespective of ongoing HAART, reflecting primarily an increased percentage of CD8+CCR7-CD45RA- T cells, representing an inflammatory T cell phenotype. Our in vitro experiments showed that CCL19 and CCL21 promoted an inflammatory response in PBMC when accompanied by high viral load and, again, irrespective of ongoing HAART. These findings link a dysregulated CCL19/CCL21/CCR7 system in HIV-infected patients to persistent inflammation and HIV replication, not only in untreated HIV infection but also in those with treatment failure during HAART.
Previous studies on CCL19/CCL21/CCR7 in HIV infection have focused upon their role in lymphocyte/DC interactions in secondary lymphoid organs. Desai et al. reported impaired CCR7 up-regulation on plasmacytoid DC in HIV-infected infants and adolescents, contributing to immunological or virological failure [19]. In the SIV/macaque model, disruption of homeostatic chemokine expression was found to promote alterations in the networks of antigen-presenting cells in lymphoid tissues [14]. CCL19 and CCL21 have been identified as key regulators of homeostatic T cell trafficking to secondary lymphoid organs, and recent studies suggest that these chemokines also could promote inflammatory responses in T cells and macrophages [8,9,18]. Here, we show that CCL21 and particularly CCL19 induced a marked increase in TNF-α and IFN-γ release in PBMC from HIV-infected patients with high viral load, irrespective of ongoing HAART. The fact that HIV-infected patients with high viral load had a decreased proportion of CCR7 positive T cells may apparently seem in conflict with this finding. However, these CCL19/CCL21-mediated responses could potentially reflect interaction with pre-activated CD8+CCR7+CD45RA- T cells, demonstrated in the present study to be a major cellular source of TNF-α and IFN-γ within the CD8+ T cell population. The predominance of the inflammatory CD8+CCR7-CD45RA- T cell subset in HIV-infected patients with high viral load might contribute further to enhanced inflammation in these patients.
In the present study we show that while untreated HIV-infected patients with immunodeficiency were characterized by enhanced spontaneous release of CCL19 in mononuclear cells (MNC) from peripheral blood and bone marrow aspirates, MNC from these patients had an attenuated CCL19 release upon PHA activation. Of note, this pattern seemed, at least partly, to be reversed during HAART. While a persistent and enhanced spontaneous release of CCL19 may be unfavourable, contributing to persistent inflammation and tissue damage, the ability to mount a balanced peak response upon cell activation might be beneficial, contributing to functional networks of antigen-presenting cells and T cells in lymphoid and peripheral tissues. We believe that this pattern of enhanced spontaneous and decreased stimulated response in MNC is part of the immune dysregulation characterizing HIV infection. Indeed, we have reported previously a similar pattern for TNF-α release in PBMC from HIV-infected patients [20].
While PBMC and BMMC released significant amounts of CCL19, these cells did not release any detectable amounts of CCL21 in either HIV-infected patients or controls. We have seen a similar pattern in other populations, such as patients with atherosclerosis, and we have not detected any amount of CCL21 transcripts in PBMC from either HIV-infected patients or controls (P. Aukrust and J. K. Damås, unpublished data). We have reported previously markedly enhanced serum levels of CCL21 in HIV infection [15], and our findings in the current study suggest that this does not reflect increased release from MNC. CCL19 and CCL21 are produced primarily by stromal cells and lymphatics in secondary lymphoid tissue, but ectopic production by endothelial cells and tissue macrophages has been observed in non-lymphoid organs during inflammatory disorders [3,10,11]. Some of these cellular sources could, potentially, contribute to the increased CCL21 levels in HIV infection.
Signalling via CCR7 has been reported previously to enhance HIV replication in activated T cells [21]. Pretreatment of resting CD4+ T cells with CCL19 or CCL21 allows for efficient HIV integration, contributing potentially to persistent HIV latency during HAART [13]. Previously we have reported persistently raised serum levels of CCL19 and CCL21 in those with virological treatment failure during HAART [15]. Our findings in the present study may suggest further a link between HIV replication and these chemokines by showing that HIV may boost the inflammatory response to CCL19 and CCL21. Thus, we showed that HIV-tat protein enhanced the release of TNF-α and IFN-γ in CCL19 and CCL21 activated PBMC from healthy controls. Several studies have showed that HIV tat protein may be of particular importance for certain HIV-related responses such as inflammation and neurotoxicity [22,23]. The high unbound tat levels that have been detected in sera of HIV patients underscore the in vivo relevance of these findings [24]. Moreover, while CCL19 and CCL21 induced no or only a modest release of inflammatory cytokines in PBMC from control individuals and HIV-infected patients on successful HAART, these chemokines enhanced markedly the inflammatory potential of PBMC in untreated HIV-infected patients and in those with treatment failure during HAART. However, it is unlikely that this enhancement of inflammatory response in relation to HIV replication is restricted to tat-related mechanisms but, rather, reflects the effects of full HIV replication of the intact virus. Nevertheless, our findings may suggest a pathogenic loop between HIV and CCL19/CCL21, contributing to persistent HIV replication and inflammation in HIV-infected patients irrespectively on ongoing HAART.
Although relatively few patients were examined, our findings suggest that a dysregulated CCL19/CCL21/CCR7 system could play a pathogenic role in HIV infection by contributing to an inappropriate inflammation, being part of pathogenic loop, involving inflammatory interactions between HIV, CCL19/CCL21 and their receptor, CCR7. This pathogenic loop seems to be operating not only in untreated HIV-infected patients, but also in those with virological treatment failure during HAART, contributing to persistent inflammation and HIV replication in these individuals.
Acknowledgments
This work was supported by grants from the Research Council of Norway, Medinnova Foundation and Rikshospitalet University Hospital.
Disclosure
There are no conflicts of interest.
References
- 1.Moser B, Wolf M, Walz A, et al. Chemokines: multiple levels of leukocyte migration control. Trends Immunol. 2004;25:75–84. doi: 10.1016/j.it.2003.12.005. [DOI] [PubMed] [Google Scholar]
- 2.Allen SJ, Crown SE, Handel TM. Chemokine: receptor structure, interactions, and antagonism. Annu Rev Immunol. 2007;25:787–820. doi: 10.1146/annurev.immunol.24.021605.090529. [DOI] [PubMed] [Google Scholar]
- 3.DeVico AL, Gallo RC. Control of HIV-1 infection by soluble factors of the immune response. Nat Rev Microbiol. 2004;2:401–13. doi: 10.1038/nrmicro878. [DOI] [PubMed] [Google Scholar]
- 4.Berger EA, Murphy PM, Farber JM. Chemokine receptors as HIV-1 coreceptors: roles in viral entry, tropism, and disease. Annu Rev Immunol. 1999;17:657–700. doi: 10.1146/annurev.immunol.17.1.657. [DOI] [PubMed] [Google Scholar]
- 5.Boasso A, Shearer GM. Chronic innate immune activation as a cause of HIV-1 immunopathogenesis. Clin Immunol. 2008;126:235–42. doi: 10.1016/j.clim.2007.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Förster R, Schubel A, Breitfeld D, et al. CCR7 coordinates the primary immune response by establishing functional microenvironments in secondary lymphoid organs. Cell. 1991;99:23–33. doi: 10.1016/s0092-8674(00)80059-8. [DOI] [PubMed] [Google Scholar]
- 7.Müller G, Hopken UE, Lipp M. The impact of CCR7 and CXCR5 on lymphoid organ development and systemic immunity. Immunol Rev. 2003;195:117–35. doi: 10.1034/j.1600-065x.2003.00073.x. [DOI] [PubMed] [Google Scholar]
- 8.Marsland BJ, Bättig P, Bauer M, et al. CCL19 and CCL21 induce a potent proinflammatory differentiation program in licensed dendritic cells. Immunity. 2005;22:493–505. doi: 10.1016/j.immuni.2005.02.010. [DOI] [PubMed] [Google Scholar]
- 9.Flanagan K, Moroziewicz D, Kwak H, et al. The lymphoid chemokine CCL21 costimulates naive T cell expansion and Th1 polarization of non-regulatory CD4+ T cells. Cell Immunol. 2004;231:75–84. doi: 10.1016/j.cellimm.2004.12.006. [DOI] [PubMed] [Google Scholar]
- 10.Bonacchi A, Petrai I, Defranco RM, et al. The chemokine CCL21 modulates lymphocyte recruitment and fibrosis in chronic hepatitis C. Gastroenterology. 2003;125:1060–76. doi: 10.1016/s0016-5085(03)01194-6. [DOI] [PubMed] [Google Scholar]
- 11.Rangel-Moreno J, Moyron-Quiroz JE, Hartson L, et al. Pulmonary expression of CXC chemokine ligand 13, CC chemokine ligand 19, and CC chemokine ligand 21 is essential for local immunity to influenza. Proc Natl Acad Sci USA. 2007;104:10577–82. doi: 10.1073/pnas.0700591104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Damås JK, Smith C, Øie E, et al. Enhanced expression of the homeostatic chemokines CCL19 and CCL21 in clinical and experimental atherosclerosis: possible pathogenic role in plaque destabilization. Arterioscler Thromb Vasc Biol. 2007;27:614–20. doi: 10.1161/01.ATV.0000255581.38523.7c. [DOI] [PubMed] [Google Scholar]
- 13.Saleh S, Solomon A, Wightman F, et al. CCR7 ligands CCL19 and CCL21 increase permissiveness of resting memory CD4+ T-cells to HIV-1 infection: a novel model of HIV-1 latency. Blood. 2007;110:4161–4. doi: 10.1182/blood-2007-06-097907. [DOI] [PubMed] [Google Scholar]
- 14.Choi YK, Fallert BA, Murphey-Corb MA, et al. Simian immunodeficiency virus dramatically alters expression of homeostatic chemokines and dendritic cell markers during infection in vivo. Blood. 2003;101:1684–91. doi: 10.1182/blood-2002-08-2653. [DOI] [PubMed] [Google Scholar]
- 15.Damås JK, Landrø L, Fevang B, et al. Enhanced levels of the CCR7 ligands CCL19 and CCL21 in HIV infection: correlation to viral load, disease progression and response to HAART. AIDS. 2009;23:135–8. doi: 10.1097/QAD.0b013e32831cf595. [DOI] [PubMed] [Google Scholar]
- 16.Hansen JB, Halvorsen DS, Haldorsen BC, et al. Retention of phagocytic functions in cryopreserved human monocytes. J Leukoc Biol. 1995;57:235–41. doi: 10.1002/jlb.57.2.235. [DOI] [PubMed] [Google Scholar]
- 17.Holm AM, Sivertsen EA, Tunheim SH, et al. Gene expression analysis of peripheral T cells in a subgroup of common variable immunodeficiency shows predominance of CCR7(-) effector-memory T cells. Clin Exp Immunol. 2004;138:278–89. doi: 10.1111/j.1365-2249.2004.02630.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Byrnes HD, Kaminski H, Mirza A, et al. Macrophage inflammatory protein-3 beta enhances IL-10 production by activated human peripheral blood monocytes and T cells. J Immunol. 1999;163:4715–20. [PubMed] [Google Scholar]
- 19.Desai S, Chaparro A, Liu H, et al. Impaired CCR7 expression on plasmacytoid dendritic cells of HIV-infected children and adolescents with immunologic and virologic failure. J Acquir Immune Defic Syndr. 2007;45:501–7. doi: 10.1097/QAI.0b013e3180654811. [DOI] [PubMed] [Google Scholar]
- 20.Aukrust P, Müller F, Lien E, et al. Tumor necrosis factor (TNF) system levels in human immunodeficiency virus-infected patients during highly active antiretroviral therapy: persistent TNF activation is associated with virologic and immunologic treatment failure. J Infect Dis. 1999;179:74–82. doi: 10.1086/314572. [DOI] [PubMed] [Google Scholar]
- 21.Nagira M, Sato A, Miki S, et al. Enhanced HIV-1 replication by chemokines constitutively expressed in secondary lymphoid tissues. Virology. 1999;264:422–6. doi: 10.1006/viro.1999.0011. [DOI] [PubMed] [Google Scholar]
- 22.Kalantari P, Harandi OF, Hankey PA, Henderson AJ. HIV-1 Tat mediates degradation of RON receptor tyrosine kinase, a regulator of inflammation. J Immunol. 2008;181:1548–55. doi: 10.4049/jimmunol.181.2.1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Norman JP, Perry SW, Reynolds HM, et al. HIV-1 Tat activates neuronal ryanodine receptors with rapid induction of the unfolded protein response and mitochondrial hyperpolarization. PLoS ONE. 2008;3:e3731. doi: 10.1371/journal.pone.0003731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xiao H, Neuveut C, Tiffany HL, et al. Selective CXCR4 antagonism by Tat: implications for in vivo expansion of coreceptor use by HIV-1. Proc Natl Acad Sci USA. 2000;97:11466–71. doi: 10.1073/pnas.97.21.11466. [DOI] [PMC free article] [PubMed] [Google Scholar]