

Abstract
Periodontitis is an infectious process characterized by inflammation affecting the supporting structures of the teeth. Porphyromonas gingivalis is a major oral bacterial species implicated in the pathogenesis of periodontitis. Processing of interleukin (IL)-1 family cytokines is regulated by an intracellular innate immune response system, known as the NALP3 [nacht domain-, leucine-rich repeat-, and pyrin domain (PYD)-containing protein 3] inflammasome complex. The aim of the present study was to investigate by quantitative real-time polymerase chain reaction (PCR) the mRNA expression of NALP3, its effector molecule apoptosis associated speck-like protein (ASC), its putative antagonist NLRP2 (NLR family, PYD-containing protein 2), IL-1β and IL-18 (i) in gingival tissues from patients with gingivitis (n = 10), chronic periodontitis (n = 18), generalized aggressive periodontitis (n = 20), as well as in healthy subjects (n = 20), (ii) in vitro in a human monocytic cell line (Mono-Mac-6), in response to P. gingivalis challenge for 6 h. The clinical data indicate that NALP3 and NLRP2, but not ASC, are expressed at significantly higher levels in the three forms of inflammatory periodontal disease compared to health. Furthermore, a positive correlation was revealed between NALP3 and IL-1β or IL-18 expression levels in these tissues. The in vitro data demonstrate that P. gingivalis deregulates the NALP3 inflammasome complex in Mono-Mac-6 cells by enhancing NALP3 and down-regulating NLRP2 and ASC expression. In conclusion, this study reveals a role for the NALP3 inflammasome complex in inflammatory periodontal disease, and provides a mechanistic insight to the host immune responses involved in the pathogenesis of the disease by demonstrating the modulation of this cytokine-signalling pathway by bacterial challenge.
Keywords: ASC, interleukin-1β, NALP3, NLRP2, periodontitis
Introduction
Periodontal disease is defined as any condition affecting the periodontium [1], which is the consortium of tissues surrounding and supporting the tooth, including the gingiva and alveolar bone. The most common are those caused by the accumulation of a bacterial biofilm on the tooth surface, triggering inflammation and subsequent destruction of the periodontium. The inflammatory processes involve activation of the broad axis of innate immunity, specifically by up-regulation of proinflammatory cytokines [2–5]. Proinflammatory cytokines, including members of the interleukin (IL)-1 family such as IL-1β and IL-18, are present in the diseased periodontium, and their unbalanced production appears to mediate periodontal tissue destruction [6–9]. Porphyromonas gingivalis is a Gram-negative bacterial species which constitutes a major component of the pathogenic microbiota implicated in chronic periodontitis [10–13]. This is a form of periodontal disease characterized by the progressive destruction of the alveolar bone, leading eventually to tooth loss.
Recent evidence demonstrates that the downstream processing of IL-1β is regulated by a cytosolic protein complex of the nucleotide-binding oligomerization domain-like receptor (NLR) protein family, namely NALP3 [nacht domain-, leucine-rich repeat-, and pyrin domain (PYD)-containing protein 3]/cryopyrin-inflammasome [14,15]. This is essentially a family of intracellular innate immune sensors that can respond to bacterial challenge, initiating early host responses [16]. The inflammasome cooperates with the Toll-like receptor (TLR) pathways to mediate a rapid response to pathogens [17,18]. NALP3 (also known as PYPAF-1, NLRP3 or cryopyrin) exerts its inflammatory effects through apoptosis-associated speck-like protein (ASC) that functions as an adaptor to downstream pathways [19]. Co-expression of NALP3 and ASC activate caspase-1, which leads in turn to cleavage and activation of IL-1β[20]. Moreover, NLRP2 (PYPAF-2, or pyrin), a protein related to the NALP3 inflammasome, was shown to inhibit NALP3–ASC interactions [21]. Recent studies revealed an essential role for NALP3 inflammasome in mediating IL-1β production in response to several bacterial ligands, including lipopolysaccharide (LPS), peptidoglycan, bacterial and viral RNA [22–26]. Peripheral blood mononuclear cells (PBMCs), particularly monocytes, which are known to be high producers of IL-1β, are reported to express NALP3 mRNA [27,28], and this was highly induced by bacterial LPS [29]. Moreover, NALP3-deficient macrophages do not produce IL-1β in response to bacterial stimulation [23,24,30,31]. Interestingly, monocytes from patients with a mutation within the nucleotide-binding oligomerization domain of the NALP3 gene exhibit spontaneous activation of IL-1β production that can be potentiated further by LPS [32]. Notably, treatment of these patients with an IL-1 receptor antagonist reverses the clinical symptoms, suggesting a cause–effect relationship between IL-1β production and the development of disease [33].
The fundamental involvement of inflammasome complexes in inflammatory responses is been emphasized by the fact that mutations in the NALP3 gene are associated strongly with autoinflammatory conditions, such as rheumatoid arthritis, Muckle–Wells syndrome, Crohn's disease, familial cold autoinflammatory syndrome, but also septic shock [34–38]. Hyperproduction of IL-1β is considered to be a central event in the pathogenesis of autoinflammatory conditions [40].
The potential involvement of NALP3 in the pathogenesis of more common inflammatory disorders prompted us to investigate its role in periodontal diseases [26]. While it is known that the NALP3 inflammasome is expressed in monocytic cells, the regulation of this by periodontal pathogens has yet to be revealed. We hypothesized that the inflammasome complex would be altered in patients with periodontal disease, and that its expression would be regulated in monocytic cells challenged by periodontal pathogens. Therefore, the aim of this study was to investigate the gene expression of NALP3, NLRP2, ASC, IL-18 and IL-1β in clinical samples of gingival tissues from patients with various forms of periodontal disease and healthy subjects. The further aim was to investigate in vitro the regulation of this complex by P. gingivalis in a human monocytic cell line, namely Mono-Mac-6.
Materials and methods
Study population and clinical examination
A total of 58 subjects were included in this study, recruited from the Department of Periodontology, School of Dentistry, Ege University, İzmir. The use of human subjects satisfied the requirements of Ege University Faculty of Medicine Institutional Review Board and was conducted in accordance with the Helsinki Declaration. Written and informed consent was obtained from each subject prior to enrolment in the study. Selection of the patients was made according to the clinical and radiographic criteria proposed by the 1999 International World Workshop for a Classification of Periodontal Disease and Conditions [41]. Complete medical and dental histories were taken from all subjects. None of the subjects had history of systemic disease or cigarette smoking, and had not taken medications such as antibiotics or contraceptives that could affect their periodontal status for at least 3 months prior to the study. Clinical periodontal examination included measurement of probing pocket depth, clinical attachment level at six sites around each tooth with a manual probe, full-mouth papilla bleeding index and plaque index. The healthy group consisted of three females and seven males (aged 16–36 years, mean age 24·2 ± 8·4) with no clinical signs of gingival inflammation or radiographic evidence of alveolar bone loss, mean probing pocket depth 1·75 ± 0·63 mm, plaque index 0·8 ± 0·9 and papilla bleeding index 0·3 ± 0·4. The gingival tissue samples were taken from non-bleeding sites. The gingivitis group included four female and six male patients (aged 22–48 years, mean age 34·2 ± 8·7), which exhibited varying degrees of gingival inflammation, no radiographic evidence of alveolar bone loss, mean probing pocket depth 2·7 ± 0·6 mm, plaque index 2·2 ± 1·3 and papilla bleeding index 2·6 ± 0·84. The generalized aggressive periodontitis (G-AgP) group consisted of 12 females and eight males (aged 21–38 years, mean age 28·3 ± 4·6), with mean probing pocket depth 7·0 ± 1·2 mm, clinical attachment loss 7·4 ± 1·6 mm, plaque index 2·3 ± 0·8 and papilla bleeding index 2·7 ± 1·4. The chronic periodontitis (CP) group included eight females and 10 males (aged 35–61 years, mean age of 46·6 ± 6·2), with mean probing pocket depth 6·6 ± 0·84 mm, clinical attachment loss 7·8 ± 1·7 mm, plaque index 2·3 ± 0·77 and papilla bleeding index 3·1 ± 1·78.
Collection of gingival tissue samples
Gingival tissue samples, including both epithelium and connective tissue, were taken from the approximal sites of single rooted teeth prior to non-surgical periodontal therapy in the instance of periodontally diseased subjects, and during tooth extractions for orthodontic reasons or crown-lengthening procedures in healthy subjects. One tissue sample from each subject was obtained and submerged immediately in a sterile tube containing RNAlater solution (Ambion Inc., Austin, TX, USA), stored at −40°C until further laboratory analysis.
RNA isolation and reverse transcription
Total RNA from gingival tissue biopsies was extracted using the RiboPure RNA Isolation Kit (Ambion Inc.), according to the manufacturer's instructions. The extracted total RNA concentration was quantified using NanoDrop® ND-1000 spectrophotometer (NanoDrop Technologies, Wilmington, DE, USA). One µg of total RNA was incubated with 0·5 µg/ml of oligo dT primer (Promega, Southampton, UK) at 70°C for 5 min and cooled on ice. A master mix was then added to the samples, comprising of 10 mM 2′-deoxynucleosides 5′-triphosphate (dNTPs), 200 units of Moloney murine leukaemia virus (M-MLV) reverse-transcriptase and buffer (Promega). For the reverse-transcription reaction, these samples were incubated at 40°C for 60 min, 70°C for 15 min and then cooled to 4°C. The resulting cDNA was stored at −20°C until further use.
Quantitative real-time PCR (qPCR) analysis
To quantify the mRNA expression levels, qPCR was performed on the prepared cDNA samples. TaqMan® gene expression assays (Applied Biosystems, Warrington, UK) and ROX qPCR mastermix were used for the amplification reactions, and the qPCR analyses were performed in an ABI Prism 7900HT Sequence Detection System (Applied Biosystems). Expressions of the following target genes (Assay ID) were investigated: NALP3 (Hs00918085-m1), ASC (Hs01547324-m1), NLRP2 (Hs01546938-m1), IL-18 (Hs01038787-ml) and IL-1β (Hs00174097-m1). The housekeeping genes used to normalize the target gene Ct values in the clinical samples were glyceraldehyde-3-phosphate-dehydrogenase (GAPDH) (Hs99999905-m1) and 18S rRNA (Hs99999901-s1), while the ones used for the in vitro experiments were ubiquitin C (UBC) (Hs01871556-m1) and GAPDH. The amplification conditions were: 10 min at 95°C, followed by 40 cycles at 95°C for 15 s and 60°C for 1 min. Each qPCR run also included non-template controls, which generated a Ct greater than 40 in all experiments. The expression levels were calculated using the comparative Ct method (2−ΔCt formula) after normalization against the selected housekeeping genes.
Bacterial culture
P. gingivalis wild-type W50 strain was grown on blood agar plates supplemented with 5% horse blood in an anaerobic environment containing 80% nitrogen, 10% hydrogen and 10% CO2 at 37°C, and was then subcultured into 10 ml of media consisting of brain heart infusion broth supplemented with 5 µg/ml of haemin. The following day, the 10 ml culture was inoculated into 90 ml of fresh media. At day 6, the liquid cultures were centrifuged at 10 000 g for 45 min at 4°C. The resulting culture supernatant was collected, aliquoted and stored at −80°C, to be used for further experimentation. Total bacterial protein concentration in 6-day culture supernatants of P. gingivalis strain W50 was determined using the Bio-Rad Protein Assay (Bio-Rad, Hemel Hempstead, Hertfordshire, UK).
Cell culture
The human myelomonocytic cell line Mono-Mac-6 was obtained from the German Collection of Microorganisms and Cell Cultures (Mascheroder, Braunschweig, Germany). Mono-Mac-6 cells were cultured in RPMI-glutamax, supplemented with 10% fetal bovine serum, 1% non-essential amino acids, 1% sodium pyruvate (all from Gibco BRL Life Technologies, Paisley, UK) and 9 µg/ml bovine insulin (Sigma-Aldrich, Poole, UK). The cells were confirmed to be free of mycoplasma infection prior to experimentation (MycoAlert® Mycoplasma Detection Assay, Lonza, Switzerland). Mono-Mac-6 cells (1 × 106/ml) were used for induction assays in a total volume of 200 µl per well in the absence or presence of P. gingivalis. The doses used were based on results from previous studies [42]. Each experiment was carried out in triplicate cultures.
Statistical analysis
For the clinical data, non-parametrical statistical analysis was employed. Comparisons between the groups were performed using the Kruskal–Wallis test, with Dunn's multiple comparison test. Spearman's rank correlation analysis was used to investigate the possible correlations between NALP3 and IL-1β, or IL-18 expression levels. For the in vitro experimental data, the statistical significances were analysed by a one-way analysis of variance (anova) with a Bonferroni post-hoc test. P < 0·05 values were considered to be statistically significant. All data analysis was performed with GraphPad Prism Software cersion 4·0.
Results
NALP3, ASC, NLRP2, IL-1β and IL-18 mRNA expression analysis in gingival tissues by qPCR
Expressions of NALP3, its effector protein ASC and its putative antagonist NLRP2 were examined in gingival tissues from patients with gingivitis, CP and G-AgP, as well as healthy subjects. All three gene products were readily measurable by qPCR in all tissues. In comparison to healthy tissues, NALP3 expression was significantly (P < 0·05) higher in either of the periodontitis groups or gingivitis groups (Fig. 1a). In particular, NALP3 expression was higher by 7·4-fold in G-AgP, 4·3-fold in CP and 7·7-fold in gingivitis, respectively. Interestingly, when the three disease groups were compared to each other, no significant differences were observed in the expression of these genes. Similarly, NLRP2 expression was increased in all disease groups compared to healthy tissues (Fig. 1a), but this was statistically significant (P < 0·05) only in G-AgP (sixfold) and gingivitis (8·2-fold). In contrast, there were no significant differences in ASC expression between any of the disease groups and health (Fig. 1c). In comparison to healthy tissues, both IL-1β and IL-18 mRNA expressions were significantly (P < 0·05) higher in gingivitis, CP and G-AgP. In the case of IL-1β mRNA expression, this was higher by 7·1-fold in G-AgP, 8·2-fold in CP and 5·8-fold in gingivitis, respectively. When the disease groups were compared to each other, there were no significant differences between them in either IL-1β or IL-18 mRNA expression.
Fig. 1.
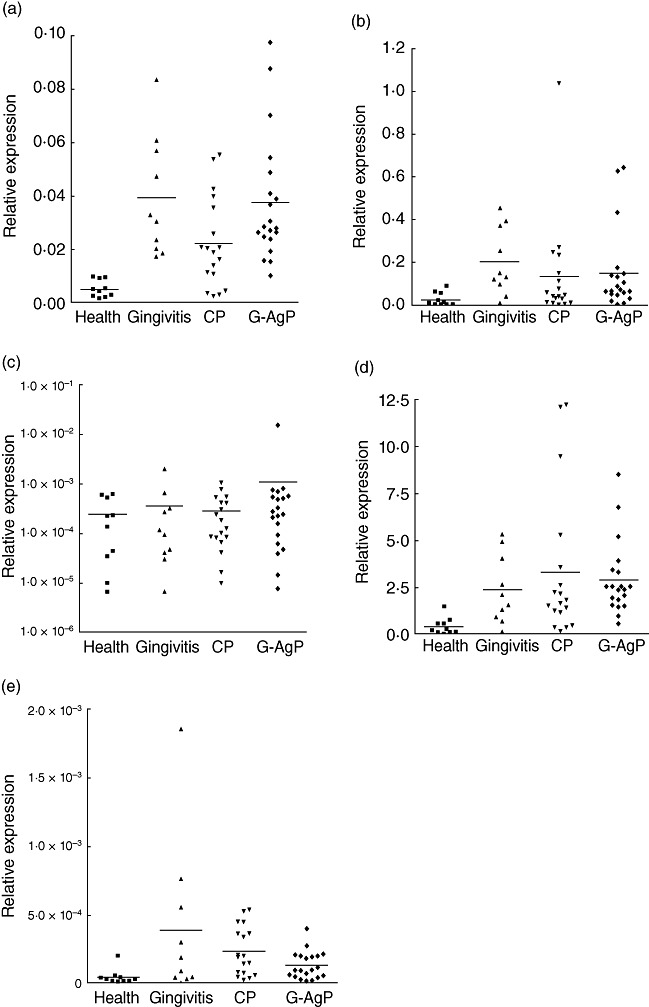
NALP3 [nacht domain-, leucine-rich repeat-, and pyrin domain (PYD)-containing protein 3] inflammasome complex expression levels in periodontal diseases. Distribution of NALP3 (a), NLRP2 (NLR family, PYD-containing protein 2) (b), ASC (c), interleukin (IL)-1β (d) and IL-18 (e) gene expression in gingival tissues of healthy subjects (n = 10) and patients with gingivitis (n = 10), chronic periodontitis (CP) (n = 18) and generalized aggressive periodontitis (G-AgP) (n = 20). The mRNA expression levels were measured by quantitative polymerase chain reaction (qPCR) analysis, normalized against the expression levels of glyceraldehyde-3-phosphate-dehydrogenase (GAPDH) and 18S RNA.
Because NALP3 is involved in IL-1β and IL-18 activation, its possible correlation with IL-1β and IL-18 mRNA levels in the gingival tissues was investigated (Fig. 2). Spearman's rank correlation analysis indicated that the expression levels of NALP3 were correlated positively with IL-1β and IL-18 (r = 0·62, P < 0·0001 and r = 0·45, P < 0·0004, respectively).
Fig. 2.
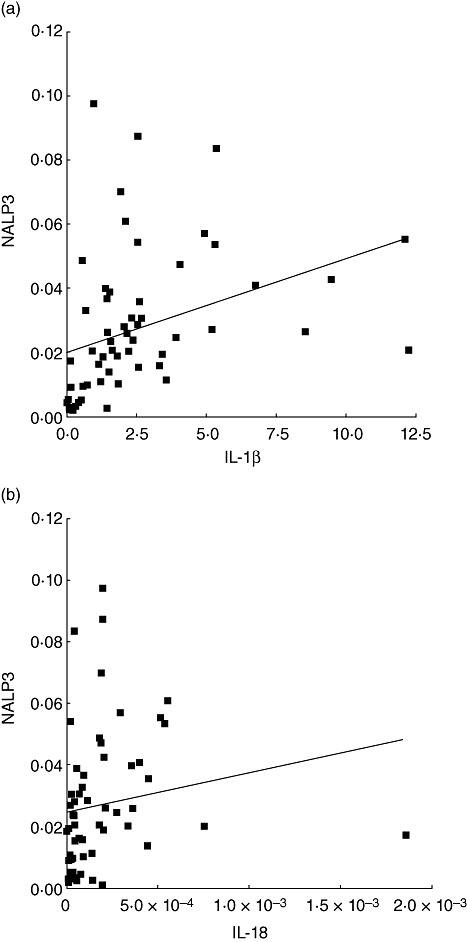
Scatter-plot showing the correlation between the NALP3 [nacht domain-, leucine-rich repeat-, and pyrin domain (PYD)-containing protein 3] and interleukin (IL)-1β (a) or IL-18 (b) mRNA expression in gingival tissues from all subjects (n = 58).
Regulation of NALP3, ASC, NLRP2, IL-1β and IL-18 mRNA expression by P. gingivalis in monocytic cells
The regulation of the inflammasome complex by P. gingivalis was also studied in vitro. Mono-Mac-6 cells were challenged with ascending protein concentrations of P. gingivalis W50 strain culture supernatants for 6 h. The untreated (control) cell cultures expressed low but detectable levels of NALP3 mRNA. Nevertheless, P. gingivalis challenge elicited a dose-dependent increase in NALP3 mRNA expression, which was up to threefold greater to the control (Fig. 3a). In contrast, NLRP2 mRNA expression was highly expressed in the untreated cells, and this was down-regulated in a dose-dependent manner by P. gingivalis challenge (Fig. 3b). Compared to the control, there was a 9·3-fold reduction in NLRP2 expression levels with the highest P. gingivalis protein concentration used (6·4 µg/ml). Furthermore, ASC was highly expressed in the untreated control, but this was down-regulated up to 15·3-fold with increasing P. gingivalis concentrations (Fig. 3c). During this period, the expressions of IL-1β (Fig. 3d) and IL-18 (Fig. 3e) were enhanced in response to P. gingivalis challenge.
Fig. 3.
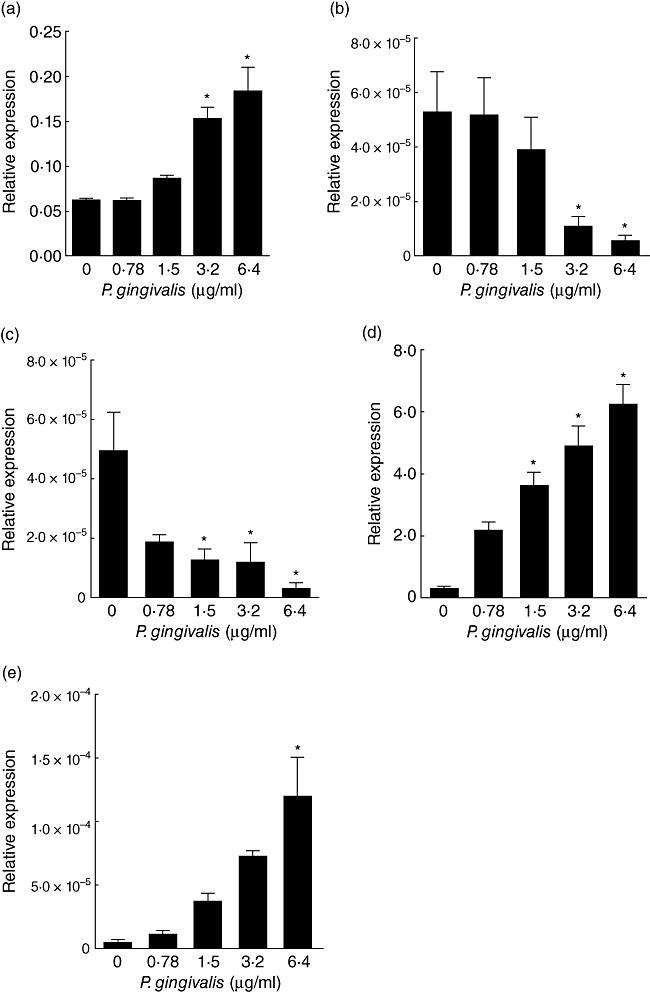
Regulation of NALP3 [nacht domain-, leucine-rich repeat-, and pyrin domain (PYD)-containing protein 3] inflammasome complex expression by Porpyromonas gingivalis. Mono-Mac-6 cell cultures were challenged with ascending protein concentrations of P. gingivalis culture supernatant for 6 h. The mRNA expression levels of NALP-3 (a), NLRP2 (NLR family, PYD-containing protein 2) (b), ASC (c), interleukin (IL)-1β (d) and IL-18 (e) were measured by quantitative polymerase chain reaction (qPCR) analysis and normalized against the expression levels of glyceraldehyde-3-phosphate-dehydrogenase (GAPDH) and ubiquitin C (UBC). Bars represent mean values ± standard error of the mean from three independent experiments. The asterisk indicates the groups that were significantly different to the control group (P < 0·05).
Discussion
The NALP3 inflammasome complex is predicted to be an essential part of the innate immunity system [43]. The present study is the first to investigate the involvement of this complex in periodontal diseases, demonstrating significantly increased levels of NALP3 and NLRP2, but not ASC, mRNA expression in gingival tissues affected by periodontal disease compared to healthy ones. Nevertheless, the impact of age differences between groups as a confounder should not be excluded, especially as this factor may alter immune responses, including the expression of this system. As expected, both IL-1β and IL-18 mRNA expression were also enhanced in periodontal disease compared to health, consistent with previous findings [8,9,44]. Therefore, we considered further a potential relationship between these two cytokines of the IL-1 family and NALP3, and hypothesized a putative correlation at the tissue gene expression level. Indeed, a positive correlation was revealed denoting a functional relationship of NALP3 with its downstream processing targets, IL-1β and IL-18, in periodontal diseases. It is of interest that, while IL-1 cytokines and NALP3 inflammasome gene expressions were significantly higher in gingivitis, CP and G-AgP compared to health, there were no differences among these three disease groups. This indicates that enhancement of the inflammasome complex expression is distinguishable in the presence of inflammatory periodontal disease. However, it cannot differentiate between periodontitis and gingivitis, and therefore may not account for the alveolar bone destruction, which occurs in the case of periodontitis but not gingivitis. We have demonstrated previously that the receptor activator of nuclear factor kappa B (NF-κB) ligand (RANKL)–osteoprotegerin (OPG) system may be a more suitable indicator of bone destruction, rather than an inflammatory determinant of periodontal disease [45,46].
These clinical data indicate high NALP3 inflammasome expression levels in diseased gingival tissues. The inflamed gingival periodontal tissues are characterized by an influx of inflammatory cells, including neutrophils and monocytes/macrophages [47]. These cells have been shown to express the NALP3 inflammasome complex, sensing and responding to bacterial challenge [29,48]. We employed the Mono-Mac-6 cell line and selected P. gingivalis, a major periodontal pathogen, as bacterial challenge to investigate regulation of the NALP3 inflammasome complex. NALP3 expression was low in untreated cells, but this was elevated significantly upon P. gingivalis challenge, along with the expression of IL-1β and IL-18. In agreement with these findings, we have shown previously that P. gingivalis triggers the release of IL-1β and IL-18, the end-point targets of inflammasome activation, in Mono-Mac-6 cells [42]. Whether NALP3 is involved directly in the induction of these cytokines in the present experimental system needs to be addressed by interventional studies.
The present study also shows that Mono-Mac-6 cells express ASC, an important adapter molecule in NALP3-mediated cellular responses, constitutively [29,49,50]. Interestingly, P. gingivalis challenge down-regulated ASC mRNA expression dramatically, providing evidence of the functionality of this molecule in the regulation of the NALP3 in human monocytes [51]. These findings are in agreement with a recent report showing a decrease of ASC mRNA expression in differentiated THP-1 after 6 h of challenge with live P. gingivalis[52]. In contrast, another study has demonstrated that Escherichia coli LPS enhances ASC mRNA expression in monocytic cells [53]. This discrepancy could reflect differences in the cell lines used, or simply the different nature of bacterial stimulation. In fact, P. gingivalis has the capacity to evade immune responses, and thus contribute to the establishment of chronic inflammation [54]. Down-regulation of ASC may be a host response manipulated potentially by P. gingivalis in favour of its survival in the infected periodontal tissues, as also discussed by other researchers [52].
Mono-Mac-6 cells expressed NLRP2 mRNA constitutively, and its levels were reduced after P. gingivalis challenge. NLRP2 was shown to compete with caspase-1 for binding to ASC [21], and has also been proposed as an antagonist of NALP3–ASC interactions [55], thereby exerting a modulatory effect on IL-1β production. Thus, NLRP2 might interfere with NALP3-mediated cytokine production at several points in the signalling cascade [56]. The present demonstration of NLRP2 down-regulation by P. gingivalis may denote that this molecule is part of a negative regulatory loop of the NALP3 inflammasome complex.
In conclusion, the present study demonstrated enhanced expression of the NALP3 inflammasome complex in inflammatory periodontal diseases, occurring both in gingivitis and periodontitis. This cytokine-signalling pathway may therefore be crucial in the regulatory control of inflammatory responses in periodontal diseases. The findings are potentiated further by a mechanistic insight revealing cross-talk between P. gingivalis and the NALP3 inflammasome complex in human monocytic cells. P. gingivalis is likely to affect this complex by regulating its individual components differentially. The up-regulation of NALP3 and down-regulation of its inhibitor NLRP2 denote a positive regulatory effect on IL-1 cytokines. Nevertheless, as ASC promotes the processing of latent into mature IL-1β[18,30], down-regulation by P. gingivalis suggests that it may attempt to dampen down the production of this cytokine. It is therefore tempting to postulate a pathway by which P. gingivalis may manipulate the host inflammatory responses, in order to survive and prevail within the infected periodontal tissues.
Acknowledgments
This study was supported by the authors' institutions. The authors wish to thank Drs A. Berdeli and K. Eker (Faculty of Medicine, Ege University) for providing the core facilities for processing of the gingival tissue samples.
Disclosures
The authors declare no conflicts of interest.
References
- 1.Armitage GC. Periodontal diagnoses and classification of periodontal diseases. Periodontol 2000. 2004;34:9–21. doi: 10.1046/j.0906-6713.2002.003421.x. [DOI] [PubMed] [Google Scholar]
- 2.Gemmell E, Marshall RI, Seymour GJ. Cytokines and prostaglandins in immune homeostasis and tissue destruction in periodontal disease. Periodontol 2000. 1997;14:112–43. doi: 10.1111/j.1600-0757.1997.tb00194.x. [DOI] [PubMed] [Google Scholar]
- 3.Oido-Mori M, Rezzonico R, Wang PL, et al. Porphyromonas gingivalis gingipain-R enhances interleukin-8 but decreases gamma interferon-inducible protein 10 production by human gingival fibroblasts in response to T-cell contact. Infect Immun. 2001;69:4493–501. doi: 10.1128/IAI.69.7.4493-4501.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Taylor JJ, Preshaw PM, Donaldson PT. Cytokine gene polymorphism and immunoregulation in periodontal disease. Periodontol 2000. 2004;35:158–82. doi: 10.1111/j.0906-6713.2004.003561.x. [DOI] [PubMed] [Google Scholar]
- 5.Landi L, Amar S, Polins AS, Van Dyke TE. Host mechanisms in the pathogenesis of periodontal disease. Curr Opin Periodontol. 1997;4:3–10. [PubMed] [Google Scholar]
- 6.Okada H, Murakami S. Cytokine expression in periodontal health and disease. Crit Rev Oral Biol Med. 1998;9:248–66. doi: 10.1177/10454411980090030101. [DOI] [PubMed] [Google Scholar]
- 7.Bascones A, Noronha S, Gomez M, Mota P, Gonzalez Moles MA, Dorrego MV. Tissue destruction in periodontitis: bacteria or cytokines fault? Quintessence Int. 2005;36:299–306. [PubMed] [Google Scholar]
- 8.Johnson RB, Serio FG. Interleukin-18 concentrations and the pathogenesis of periodontal disease. J Periodontol. 2005;76:785–90. doi: 10.1902/jop.2005.76.5.785. [DOI] [PubMed] [Google Scholar]
- 9.Orozco A, Gemmell E, Bickel M, Seymour GJ. Interleukin-1beta, interleukin-12 and interleukin-18 levels in gingival fluid and serum of patients with gingivitis and periodontitis. Oral Microbiol Immunol. 2006;21:256–60. doi: 10.1111/j.1399-302X.2006.00292.x. [DOI] [PubMed] [Google Scholar]
- 10.Holt SC, Kesavalu L, Walker S, Genco CA. Virulence factors of Porphyromonas gingivalis. Periodontol 2000. 1999;20:168–238. doi: 10.1111/j.1600-0757.1999.tb00162.x. [DOI] [PubMed] [Google Scholar]
- 11.Socransky SS, Haffajee AD, Cugini MA, Smith C, Kent RL., Jr Microbial complexes in subgingival plaque. J Clin Periodontol. 1998;25:134–44. doi: 10.1111/j.1600-051x.1998.tb02419.x. [DOI] [PubMed] [Google Scholar]
- 12.Kesavalu L, Sathishkumar S, Bakthavatchalu V, et al. Rat model of polymicrobial infection, immunity, and alveolar bone resorption in periodontal disease. Infect Immun. 2007;75:1704–12. doi: 10.1128/IAI.00733-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Slots J, Ting M. Actinobacillus actinomycetemcomitans and Porphyromonas gingivalis in human periodontal disease: occurrence and treatment. Periodontol 2000. 1999;20:82–121. doi: 10.1111/j.1600-0757.1999.tb00159.x. [DOI] [PubMed] [Google Scholar]
- 14.Petrilli V, Dostert C, Muruve DA, Tschopp J. The inflammasome: a danger sensing complex triggering innate immunity. Curr Opin Immunol. 2007;19:615–22. doi: 10.1016/j.coi.2007.09.002. [DOI] [PubMed] [Google Scholar]
- 15.Freche B, Reig N, van der Goot FG. The role of the inflammasome in cellular responses to toxins and bacterial effectors. Semin Immunopathol. 2007;29:249–60. doi: 10.1007/s00281-007-0085-0. [DOI] [PubMed] [Google Scholar]
- 16.Mariathasan S, Monack DM. Inflammasome adaptors and sensors: intracellular regulators of infection and inflammation. Nat Rev Immunol. 2007;7:31–40. doi: 10.1038/nri1997. [DOI] [PubMed] [Google Scholar]
- 17.Ting JP, Davis BK. CATERPILLER: a novel gene family important in immunity, cell death, and diseases. Annu Rev Immunol. 2005;23:387–414. doi: 10.1146/annurev.immunol.23.021704.115616. [DOI] [PubMed] [Google Scholar]
- 18.Dowds TA, Masumoto J, Zhu L, Inohara N, Nunez G. Cryopyrin-induced interleukin 1beta secretion in monocytic cells: enhanced activity of disease-associated mutants and requirement for ASC. J Biol Chem. 2004;279:21924–8. doi: 10.1074/jbc.M401178200. [DOI] [PubMed] [Google Scholar]
- 19.Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell. 2002;10:417–26. doi: 10.1016/s1097-2765(02)00599-3. [DOI] [PubMed] [Google Scholar]
- 20.Mariathasan S. ASC, Ipaf and Cryopyrin/Nalp3: bona fide intracellular adapters of the caspase-1 inflammasome. Microbes Infect. 2007;9:664–71. doi: 10.1016/j.micinf.2007.01.017. [DOI] [PubMed] [Google Scholar]
- 21.Chae JJ, Komarow HD, Cheng J, et al. Targeted disruption of pyrin, the FMF protein, causes heightened sensitivity to endotoxin and a defect in macrophage apoptosis. Mol Cell. 2003;11:591–604. doi: 10.1016/s1097-2765(03)00056-x. [DOI] [PubMed] [Google Scholar]
- 22.Kanneganti TD, Ozoren N, Body-Malapel M, et al. Bacterial RNA and small antiviral compounds activate caspase-1 through cryopyrin/Nalp3. Nature. 2006;440:233–6. doi: 10.1038/nature04517. [DOI] [PubMed] [Google Scholar]
- 23.Mariathasan S, Weiss DS, Newton K, et al. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature. 2006;440:228–32. doi: 10.1038/nature04515. [DOI] [PubMed] [Google Scholar]
- 24.Kanneganti TD, Body-Malapel M, Amer A, et al. Critical role for Cryopyrin/Nalp3 in activation of caspase-1 in response to viral infection and double-stranded RNA. J Biol Chem. 2006;281:36560–8. doi: 10.1074/jbc.M607594200. [DOI] [PubMed] [Google Scholar]
- 25.Martinon F, Agostini L, Meylan E, Tschopp J. Identification of bacterial muramyl dipeptide as activator of the NALP3/cryopyrin inflammasome. Curr Biol. 2004;14:1929–34. doi: 10.1016/j.cub.2004.10.027. [DOI] [PubMed] [Google Scholar]
- 26.Becker CE, O'Neill LA. Inflammasomes in inflammatory disorders: the role of TLRs and their interactions with NLRs. Semin Immunopathol. 2007;29:239–48. doi: 10.1007/s00281-007-0081-4. [DOI] [PubMed] [Google Scholar]
- 27.Anderson JP, Mueller JL, Rosengren S, et al. Structural, expression, and evolutionary analysis of mouse CIAS1. Gene. 2004;338:25–34. doi: 10.1016/j.gene.2004.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kummer JA, Broekhuizen R, Everett H, et al. Inflammasome components NALP 1 and 3 show distinct but separate expression profiles in human tissues suggesting a site-specific role in the inflammatory response. J Histochem Cytochem. 2007;55:443–52. doi: 10.1369/jhc.6A7101.2006. [DOI] [PubMed] [Google Scholar]
- 29.Sutterwala FS, Ogura Y, Szczepanik M, et al. Critical role for NALP3/CIAS1/Cryopyrin in innate and adaptive immunity through its regulation of caspase-1. Immunity. 2006;24:317–27. doi: 10.1016/j.immuni.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 30.Srinivasula SM, Poyet JL, Razmara M, Datta P, Zhang Z, Alnemri ES. The PYRIN-CARD protein ASC is an activating adaptor for caspase-1. J Biol Chem. 2002;277:21119–22. doi: 10.1074/jbc.C200179200. [DOI] [PubMed] [Google Scholar]
- 31.Mariathasan S, Newton K, Monack DM, et al. Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature. 2004;430:213–18. doi: 10.1038/nature02664. [DOI] [PubMed] [Google Scholar]
- 32.Gattorno M, Tassi S, Carta S, et al. Pattern of interleukin-1beta secretion in response to lipopolysaccharide and ATP before and after interleukin-1 blockade in patients with CIAS1 mutations. Arthritis Rheum. 2007;56:3138–48. doi: 10.1002/art.22842. [DOI] [PubMed] [Google Scholar]
- 33.Hawkins PN, Lachmann HJ, McDermott MF. Interleukin-1-receptor antagonist in the Muckle–Wells syndrome. N Engl J Med. 2003;348:2583–4. doi: 10.1056/NEJM200306193482523. [DOI] [PubMed] [Google Scholar]
- 34.Fahy RJ, Exline MC, Gavrilin MA, et al. Inflammasome mRNA expression in human monocytes during early septic shock. Am J Respir Crit Care Med. 2008;177:983–8. doi: 10.1164/rccm.200703-418OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hoffman HM, Mueller JL, Broide DH, Wanderer AA, Kolodner RD. Mutation of a new gene encoding a putative pyrin-like protein causes familial cold autoinflammatory syndrome and Muckle–Wells syndrome. Nat Genet. 2001;29:301–5. doi: 10.1038/ng756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hull KM, Shoham N, Chae JJ, Aksentijevich I, Kastner DL. The expanding spectrum of systemic autoinflammatory disorders and their rheumatic manifestations. Curr Opin Rheumatol. 2003;15:61–9. doi: 10.1097/00002281-200301000-00011. [DOI] [PubMed] [Google Scholar]
- 37.Agostini L, Martinon F, Burns K, McDermott MF, Hawkins PN, Tschopp J. NALP3 forms an IL-1beta-processing inflammasome with increased activity in Muckle–Wells autoinflammatory disorder. Immunity. 2004;20:319–25. doi: 10.1016/s1074-7613(04)00046-9. [DOI] [PubMed] [Google Scholar]
- 38.Saitoh T, Fujita N, Jang MH, et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature. 2008;456:264–8. doi: 10.1038/nature07383. [DOI] [PubMed] [Google Scholar]
- 39.Rosengren S, Hoffman HM, Bugbee W, Boyle DL. Expression and regulation of cryopyrin and related proteins in rheumatoid arthritis synovium. Ann Rheum Dis. 2005;64:708–14. doi: 10.1136/ard.2004.025577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Martinon F, Tschopp J. Inflammatory caspases: linking an intracellular innate immune system to autoinflammatory diseases. Cell. 2004;117:561–74. doi: 10.1016/j.cell.2004.05.004. [DOI] [PubMed] [Google Scholar]
- 41.Armitage GC. Development of a classification system for periodontal diseases and conditions. Ann Periodontol. 1999;4:1–6. doi: 10.1902/annals.1999.4.1.1. [DOI] [PubMed] [Google Scholar]
- 42.Hamedi M, Belibasakis GN, Cruchley AT, Rangarajan M, Curtis MA, Bostanci N. Porphyromonas gingivalis culture supernatants differentially regulate interleukin-1beta and interleukin-18 in human monocytic cells. Cytokine. 2009;45:99–104. doi: 10.1016/j.cyto.2008.11.005. [DOI] [PubMed] [Google Scholar]
- 43.Franchi L, Warner N, Viani K, Nunez G. Function of Nod-like receptors in microbial recognition and host defense. Immunol Rev. 2009;227:106–28. doi: 10.1111/j.1600-065X.2008.00734.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Duarte PM, de Oliveira MC, Tambeli CH, et al. Overexpression of interleukin-1beta and interleukin-6 may play an important role in periodontal breakdown in type 2 diabetic patients. J Periodont Res. 2007;42:377–81. doi: 10.1111/j.1600-0765.2006.00961.x. [DOI] [PubMed] [Google Scholar]
- 45.Bostanci N, Ilgenli T, Emingil G, et al. Gingival crevicular fluid levels of RANKL and OPG in periodontal diseases: implications of their relative ratio. J Clin Periodontol. 2007;34:370–6. doi: 10.1111/j.1600-051X.2007.01061.x. [DOI] [PubMed] [Google Scholar]
- 46.Bostanci N, Ilgenli T, Emingil G, et al. Differential expression of receptor activator of nuclear factor-kappaB ligand and osteoprotegerin mRNA in periodontal diseases. J Periodont Res. 2007;42:287–93. doi: 10.1111/j.1600-0765.2006.00946.x. [DOI] [PubMed] [Google Scholar]
- 47.Zappa U, Reinking-Zappa M, Graf H, Espeland M. Cell populations and episodic periodontal attachment loss in humans. J Clin Periodontol. 1991;18:508–15. doi: 10.1111/j.1600-051x.1991.tb00082.x. [DOI] [PubMed] [Google Scholar]
- 48.Franchi L, Park JH, Shaw MH, et al. D-like receptors in innate immunity, infection and disease. Cell Microbiol. 2008;10:1–8. doi: 10.1111/j.1462-5822.2007.01059.x. [DOI] [PubMed] [Google Scholar]
- 49.Ozoren N, Masumoto J, Franchi L, et al. Distinct roles of TLR2 and the adaptor ASC in IL-1beta/IL-18 secretion in response to Listeria monocytogenes. J Immunol. 2006;176:4337–42. doi: 10.4049/jimmunol.176.7.4337. [DOI] [PubMed] [Google Scholar]
- 50.Yamamoto M, Yaginuma K, Tsutsui H, et al. ASC is essential for LPS-induced activation of procaspase-1 independently of TLR-associated signal adaptor molecules. Genes Cells. 2004;9:1055–67. doi: 10.1111/j.1365-2443.2004.00789.x. [DOI] [PubMed] [Google Scholar]
- 51.Gumucio DL, Diaz A, Schaner P, et al. the role of pyrin domain-containing proteins in inflammation and apoptosis. Clin Exp Rheumatol. 2002;4(20 Suppl. 26):S45–53. [PubMed] [Google Scholar]
- 52.Taxman DJ, Zhang J, Champagne C, et al. Cutting edge: ASC mediates the induction of multiple cytokines by Porphyromonas gingivalis via caspase-1-dependent and -independent pathways. J Immunol. 2006;177:4252–6. doi: 10.4049/jimmunol.177.7.4252. [DOI] [PubMed] [Google Scholar]
- 53.Stehlik C, Lee SH, Dorfleutner A, Stassinopoulos A, Sagara J, Reed JC. Apoptosis-associated speck-like protein containing a caspase recruitment domain is a regulator of procaspase-1 activation. J Immunol. 2003;171:6154–63. doi: 10.4049/jimmunol.171.11.6154. [DOI] [PubMed] [Google Scholar]
- 54.Darveau RP, Belton CM, Reife RA, Lamont RJ. Local chemokine paralysis, a novel pathogenic mechanism for Porphyromonas gingivalis. Infect Immun. 1998;66:1660–5. doi: 10.1128/iai.66.4.1660-1665.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dowds TA, Masumoto J, Chen FF, Ogura Y, Inohara N, Nunez G. Regulation of cryopyrin/Pypaf1 signaling by pyrin, the familial Mediterranean fever gene product. Biochem Biophys Res Commun. 2003;302:575–80. doi: 10.1016/s0006-291x(03)00221-3. [DOI] [PubMed] [Google Scholar]
- 56.Bruey JM, Bruey-Sedano N, Newman R, Chandler S, Stehlik C, Reed JC. PAN1/NALP2/PYPAF2, an inducible inflammatory mediator that regulates NF-kappaB and caspase-1 activation in macrophages. J Biol Chem. 2004;279:51897–907. doi: 10.1074/jbc.M406741200. [DOI] [PubMed] [Google Scholar]