

Abstract
Eight-hydroxy-2′-deoxyguanosine (8-OHdG) is increased in the brain in late-stage Alzheimer’s disease (LAD) and mild cognitive impairment (MCI). To determine if decreased base-excision repair contributes to these elevations, we measured oxoguanine glycosylase 1 (OGG1) protein and incision activities in nuclear and mitochondrial fractions from frontal (FL), temporal (TL), and parietal (PL) lobes from 8 MCI and 7 LAD patients, and 6 age-matched normal control (NC) subjects. OGG1 activity was significantly (P<0.05) decreased in nuclear specimens of FL, TL, and PL in MCI and LAD and in mitochondria from LAD FL and TL and MCI TL. Nuclear OGG1 protein was significantly decreased in LAD FL and MCI and LAD PL. No differences in mitochondrial OGG1 protein levels were found. Overall, our results suggest that decreased OGG1 activity occurs early in the progression of AD, possibly mediated by 4-hydroxynonenal inactivation and may contribute to elevated 8-OHdG in the brain in MCI and LAD.
Keywords: Oxoguanine glycosylase, Lipid peroxidation, Alzheimer’s disease, Mild cognitive impairment, 4-Hydroxynonenal
Introduction
Increasing evidence supports a role for oxidative damage in the pathogenesis of a variety of neurodegenerative diseases including Alzheimer’s disease (AD). Reactive oxygen species (ROS) may be particularly damaging to brain because of its lipid-rich environment and relatively low antioxidant capacity. ROS may interact with and damage lipids, proteins, RNA, and DNA (reviewed in [1]). Studies show significantly elevated levels of oxidative damage to lipids including formation of aldehydic by-products 4-hydroxynonenal (HNE) and acrolein in vulnerable regions of the brain in late-stage AD (LAD) [2–4] and in mild cognitive impairment (MCI) [5-7], the earliest clinical manifestation of AD. In addition, several studies show significant elevations of markers of oxidation in nuclear and mitochondrial DNA isolated from LAD (reviewed in [8]) and MCI [9] subjects and in RNA from LAD [10–14] and MCI [15] as well as other neurologic disorders including Parkinson’s disease [16] and diffuse Lewy body disease (DLB) [17]. Because mitochondria generate most free radicals, their DNA are particularly susceptible to oxidative damage. Comparison of nuclear and mitochondrial DNA oxidative damage shows that mitochondrial levels of 8-hydroxy-2′-deoxyguanosine (8-OHdG) are significantly higher than those observed in nuclear DNA in MCI [9] and LAD [18,19].
Although several oxidized bases can result from attack of DNA by ROS, the predominant marker is 8-OHdG [20]. 8-OHdG is potentially mutagenic and, if unrepaired, improperly pairs with adenine during replication and induces G:C → A:T transversion mutations [21]. In postmitotic neurons, accumulation of oxidatively modified DNA bases may result in diminished cellular activity and death [22]. Protection against accumulation of 8-OHdG in human cells is mediated in part by the base-excision repair (BER) pathway that is initiated by excision of oxidized guanine by 8-oxoguanine DNA glycosylase (OGG1).
There are few studies of OGG1 and its activity in the progression of AD. Initial studies from our laboratory showed decreased OGG1 activity in nuclear fractions from vulnerable regions of LAD brain compared to age-matched normal control (NC) subjects [23]. Using immunohistochemistry, Mao et al. [24] showed OGG1 mutations that led to significant decreases in OGG1 activity in 4 of 14 LAD subjects analyzed. In contrast, studies of OGG1 in MCI have been limited. Recent studies of Weissman et al. [25] showed no significant change in OGG1 excision activity in MCI IPL compared to NC subjects. No studies of OGG1 excision activities in mitochondrial fractions from MCI or LAD brain have been reported.
To determine if elevations of 8-OHdG in mitochondrial and nuclear DNA from vulnerable regions of MCI and LAD brain are due to altered OGG1 activity, we determined OGG1 protein levels and 8-OHdG incision activities in nuclear and mitochondrial enriched fractions from frontal (FL), parietal (PL), and temporal (TL) lobe and cerebellum (CER) from MCI, LAD, and age-matched NC.
Materials and methods
Subject selection and neuropathologic examination
To isolate sufficient amounts of mitochondria for activity analyses relatively large brain specimens (∼2 g) were obtained from frontal, parietal, and temporal lobes and CER from short postmortem interval (PMI) autopsies of 7 (4 men, 3 women) LAD patients, 8 (2 men, 6 women) MCI patients, and 6 (4 men, 2 women) age-matched NC subjects through the Neuropathology Core of the University of Kentucky Alzheimer’s Disease Center (UK-ADC). These brain regions were chosen for analysis to allow comparison of OGG1 activity and protein to levels of 8-OHG measured in previous studies [9,19]. All LAD, MCI, and NC subjects were followed longitudinally in the Clinical Core of the UK-ADC and had annual neuropsychological testing and physical and neurological examinations. LAD patients demonstrated progressive cognitive decline and met NINDS/ADRDA accepted standard criteria for the clinical diagnosis of probable AD [26]. All NC subjects had neuropsychologic scores in the normal range and showed no evidence of memory decline. Subjects with amnestic MCI were derived from the control group and were normal on enrollment into the longitudinal study and developed MCI during follow-up. The clinical criteria for diagnosis of amnestic MCI are those of Petersen et al. (1999) and include: (a) memory complaints, (b) objective memory impairment for age and education, (c) intact general cognitive function, (d) intact activities of daily living (ADLs), and (e) the subject is not demented. Objective memory test impairment was based on a score of ≤1.5 standard deviations from the mean of controls on the CERAD Word List Learning Task [27] and corroborated in some cases with the Free and Cued Selective Reminding Test.
All subjects had neuropathological examination of multiple sections of neocortical association areas, hippocampus, entorhinal cortex, amygdala, basal ganglia, nucleus basalis of Meynert, midbrain, pons, medulla, and cerebellum using hematoxylin and eosin stain, the modified Bielschowsky stain, and Aβ and α-synuclein immunostains. Braak staging [28] scores were determined using the Gallyas stain on sections of entorhinal cortex, hippocampus, and amygdala and the Bielschowsky stain on neocortical sections. All LAD patients met accepted criteria for the histopathologic diagnosis of AD (high likelihood NIA-Reagan Institute [NIA-RI] criteria) [27,29,30] and typically demonstrated Braak staging scores of VI.
Histopathologic examination of NC subjects generally showed only age-associated changes and Braak staging scores of I to III and met NIA-RI low likelihood criteria for the histopathologic diagnosis of AD. The major distinction between MCI and NC subjects was a significant increase in neuritic plaques in neocortical regions and a significant increase in NFT in entorhinal cortex, hippocampus, and amygdala in MCI compared to NC subjects [31]. Braak staging scores of MCI subjects typically ranged from III to IV.
Preparation of nuclear and mitochondrial enriched fractions
Enriched nuclear and mitochondrial fractions were prepared using the method of Mecocci et al. [32] with modification [19]. Briefly, brain specimens were homogenized on ice using a motor-driven Teflon-coated Dounce homogenizer in MSB-Ca2+ buffer (0.21 M mannitol, 0.07 M sucrose, 0.05 M Tris-HCl, 3 mM CaCl2, pH 7.5). Disodium EDTA (0.01 M) was added to the homogenate, which was centrifuged at 1500 g and 4°C for 20 min. The pellet was resuspended in MSB-Ca2+ buffer and centrifuged again to yield the nuclear fraction. The combined supernatant was centrifuged at 20,000 g and 4°C for 20 min to yield a crude mitochondrial pellet. The pellet was washed with MSB-Ca2+ buffer and resuspended in 2 ml MSB-Ca2+, loaded onto a 1:1 Percoll/ MSB-Ca2+ gradient, and centrifuged at 50,000 g and 4°C for 1 h. Mitochondria were isolated at a density of 1.035 g/ml. The mitochondria were resuspended in MSB-Ca2+ buffer and centrifuged through a second Percoll gradient leading to purified intact mitochondria. The mitochondrial pellet was resuspended in 300 μl 20 mM Hepes-KOH (pH 7.6) containing 1 mM EDTA, 2 mM DTT, 300 μM KCl, 5% glycerol, and 0.05% Triton-X (MSHE buffer). Purity of isolated fractions was verified using Western blot analysis of representative mitochondrial and nuclear specimens and antibodies against lamin A, a nuclear specific marker and porin, a mitochondria specific marker.
8-Oxoguanine glycosylase activity assay
8-Oxoguanine glycosylase activity assays were carried out as previously described [33] using an oligonucleotide duplex consisting of 32P-labeled 5′-GAACGACTGT(oxoG)ACTTGACTGCTACTGAT-3′ and unlabeled complementary strand (5′-ATCAGTAGCAGTCAAGTCACAGTCGTTC-3′) (Midland Certified Reagent Company). For OGG1 reactions, a 20 μl reaction mixture containing 40 mM Hepes-KOH (pH 7.6), 5 mM EDTA, 2 mM DTT, 75 mM KCl, 10% glycerol and 88.7 fmol 32P-labeled duplex oligonucleotide was mixed with 20 μg nuclear or mitochondrial protein and incubated at 37°C for 1 h. Reactions were quenched by adding an equal volume of loading buffer containing 90% formamide, 0.002% bromophenol blue, and 0.002% xylene green and heating at 95°C for 5 min. The reaction mixture was cooled to room temperature and separated on a 20% polyacrylamide gel containing 7 M urea prerun at 18 W for 30 min. The samples were separated by electrophoresis at 16 W for 90 min using 1X Tris borate EDTA (TBE) (pH 8.0) as a running buffer. Blanks for the assay consisted of reaction mixture and 20 μg protein specimens that were heat-inactivated by boiling at 95°C for 5 min before initiating the activity assay. Samples were analyzed with NC and MCI or NC and LAD samples on the same gel to allow comparison. Radiolabeled bands were visualized using a Fuji FLA-2000 phosphorimager after exposure of the imaging plate for 24 h. Band intensities of the cleaved product and parent oligonucleotide bands in each lane were quantified using Image-Quant software (Molecular Dynamics). Results of the assay are reported as the mean±SEM amount of radioactivity in the incised product band relative to the total radioactivity in the lane (incised product + intact duplex oligonucleotide).
Single-strand conformational polymorphism analysis
To determine possible mutations in the OGG1 gene, PCR-based single-strand conformational polymorphism was used as previously described [24] with primers against all seven exons and flanking splice sites of the OGG1 gene.
Western blot analyses for OGG1
Specimens (25 μg) of nuclear or mitochondrial fractions were subjected to SDS-PAGE and transferred to nitrocellulose. Gels were loaded with NC and MCI or NC and LAD specimens by brain region to allow comparison. The Western blots were blocked for 2 h in 5% dry milk/Tris buffered saline containing 0.1% Tween 20 (TTBS) followed by incubation overnight in a 1:100 dilution of rabbit anti-human OGG1 (Santa Cruz Biotechnology) in 5% dry milk/TTBS. Following 3 washes in TTBS, the blots were incubated with a 1:1000 dilution of horseradish peroxidase-labeled goat anti-rabbit IgG (Vector). Following 3 washes in TTBS the bands corresponding to OGG1 (40 kDa) bands were visualized using enhanced chemiluminescence (ECL) (Invitrogen) per the manufacturer’s instructions. Gels were stripped and reprobed for lamin B (Abcam), a nuclear specific marker (nuclear specimens) or porin (Oncogene) (mitochondrial specimens) as loading controls. Gels were scanned and band intensities were quantified using Scion Image Analysis (NIH). To allow comparison of data from multiple gels, results are reported as mean±SEM % control (for each gel).
Immunoprecipitation of HNE-positive OGG1
To determine if OGG1 in specimens with diminished OGG1 activity was modified by HNE, an aldehydic by-product of lipid peroxidation, 3 LAD, 3 MCI, and 3 NC nuclear specimens and 3 NC, 3 MCI, and 3 LAD mitochondrial fractions were subjected to immunoprecipitation using a polyclonal antibody against OGG1 (Santa Cruz Biotechnology) using standard methods. Briefly, rabbit anti-OGG1 antibody was covalently crosslinked to protein A/protein G Sepharose beads by incubation at room temperature for 2 h. The beads were centrifuged at 1000 g for 3 min and washed 3 to 5 times with 500 μl PBS (pH 7.2). The beads were then added to 200 μg protein samples and incubated overnight on a rotation plate at 4°C. The beads were isolated and rinsed 3 times with PBS. The beads were suspended in 30 μl gel loading buffer and immunoprecipitated proteins eluted by heating at 100°C in gel loading buffer. The eluted proteins were then separated using a 10 to 20% gradient gel and subjected to Western blot analysis using anti-HNE antibody (Alpha Diagnostics). The immunoprecipitation blank consisted of all reagents without protein. Results are expressed as mean±SEM % control OGG1 isolated by immunopositive for HNE.
Inhibition of 8-oxoguaine glycosylase activity by HNE
To determine if HNE significantly impairs OGG1 activity, NC nuclear specimens were aliquoted into duplicate sets; one set was incubated for 1 h at 37°C with 5 μM HNE (Cayman Chemical), and one with vehicle alone (control). Following incubation, OGG1 incision activity was assayed as described above.
Statistical analyses
OGG1 protein and incision activities were compared using analysis of variance (ANOVA) with Scheffe’s post hoc test for individual differences and the commercially available ABSTAT software (AndersonBell). Correlation coefficients between OGG1 activities or protein and levels of 8-OHdG measured using gas chromatography/mass spectrometry with selective-ion monitoring (GC/MS-SIM) [9,19] were calculated using ABSTAT. Braak staging scores were compared using the Mann-Whitney U test and are reported as median scores.
Results
Subject demographic data are shown in Table 1. No significant differences for age or PMI between any of the subject groups were found. Braak staging scores were significantly higher in MCI (median = III) and LAD subjects (median = VI) compared to age-matched NC subjects (median= I).
Table 1.
Subject demographic data
| Mean±SEM age (years) |
Sex men/ women |
Mean±SEM PMI (h) |
Median Braak staging Score |
|
|---|---|---|---|---|
| NC | 82.8±3.3 | 4M/2W | 3.0±0.4 | I |
| MCI | 89.6±1.6 | 2M/6W | 3.9±0.9 | III* |
| LAD | 85.0±1.9 | 4M/3W | 3.3±0.2 | VI* |
P<0.05.
Nuclear 8-oxoguanine glycosylase activity
Consistent with our previous studies [9,19], nuclear and mitochondrial fractions isolated using the protocol described above were free from cross-contamination (Fig. 1). Fig. 2 shows a representative image of OGG1-mediated incision of the 32P-labeled duplex oligonucleotide by nuclear specimens from NC and MCI patients and by a representative NC specimen heated at 100°C for 5 min before activity assays (blank). Incision of the oligonucleotide at 8-OHdG by OGG1 was measured by quantifying the intensity of the bands representing the incision product and the intact oligonucleotide for each sample. Results of the assay were calculated as the percentage of total radiolabled oligonucleotide present in the smaller incised band relative to the total present in each lane (cleaved + uncleaved). Incision activity was significantly decreased (P<0.05) in nuclear fractions prepared from MCI FL (20.0±0.8% total radiolabeled oligonucleotide) compared to NC specimens (32.9±1.0% total), MCI TL (15.3±0.9% total) compared to NC TL (24.6±1.1% total), and in MCI PL (14.1±1.1% total) compared to NC PL (27.5±2.5% total). Nuclear OGG1 activities were also significantly (P<0.05) decreased in FL (14.5±0.6% total), PL (16.3±0.7% total), and TL (17.8±0.8% total) of LAD subjects compared to NC subjects. Comparison of OGG1 activities between MCI and LAD subjects showed that activities were similarly diminished in MCI and LAD for all brain regions analyzed, suggesting that impairment of OGG1 activity may be an early event in the pathogenesis of AD. Nuclear OGG1 incision activities in CER were not significantly different for any of the subject groups (Fig. 3A). Comparison of OGG1 activity by brain region showed that activities were significantly lower in CER of NC and MCI subjects compared to FP, IP, and TP specimens. In LAD, CER OGG1 activities were significantly lower than FL specimens only. Because of the small number of subjects in each group, data from all subjects were pooled for calculation of correlations between OGG1 activity and levels of 8-OHdG measured using GC/MS-SIM [9,19]. Supplementary Tables 1 to 4 show levels of OGG1 incision activity and protein and corresponding levels of 8-OHdG (number of modified bases/106 bases) for each brain region studied. Correlation coefficients between OGG1 incision activities and levels of 8-OHdG measured using GC/MS-SIM [9,19] showed a significant negative correlation (R2=−0.73; P<0.05) between nuclear OGG1 activity and 8-OHdG levels in PL (Supplementary Table 1) and a significant negative correlation (R2=-0.79, P=0.01) in nuclear fractions of TL (Supplementary Table 2). OGG1 incision activity and 8-OHdG levels in nuclear fractions from FL (Supplementary Table 3) or CER (Supplementary Table 4) revealed no significant relationships.
Fig. 1.

Western blot analysis of representative nuclear (lanes 1–3) and mitochondrial (lanes 4–6) fractions probed for lamin A, a nuclear marker, and porin, a mitochondrial marker. There was no cross-contamination between fractions.
Fig. 2.
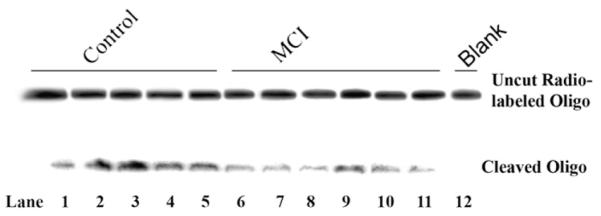
Representative image showing OGG1 incision activity of nuclear specimens from normal control (NC; lanes 1–5) and MCI (lanes 6–11) subjects. OGG1 activity was indicated by incision of the duplex oligonucleotide (top band) to form a smaller fragment (lower band). Lane 12 shows that there was no activity in a representative protein specimen heated at 100°C before initiation of the assay (blank).
Fig. 3.

(A) OGG1-mediated incision activities of nuclear specimens from NC, MCI, and LAD subjects expressed as mean±SEM % activity in the incised band relative to the total radioactivity in the lane. OGG1 activity in frontal lobe (FL), parietal lobe (PL), and temporal lobe (TL) of MCI and LAD subjects was significantly decreased (P<0.05) compared to NC subjects. (B) OGG1-mediated incision of the radiolabeled oligonucleotide by mitochondrial specimens from NC, MCI, and LAD subjects (mean±SEM % total radioactivity/lane). BER activity in frontal lobe (FL) of LAD subjects and temporal lobe (TL) from MCI and LAD subjects was significantly decreased (P<0.05) compared to NC subjects.
Mitochondrial 8-oxoguanine glycosylase activity
Fig. 3B shows mean±SEM OGG1 incision activities (% total radiolabeled oligo) in mitochondrial fractions from FL, PL, and TL and CER of NC, MCI, and LAD subjects. In general, mitochondrial activities were lower than those observed in nuclear fractions (∼50% nuclear activity in PL and TL, ∼90% nuclear levels in FL) in neocortical areas but were elevated in CER (114% nuclear activities). Levels of OGG1 activity were significantly (P<0.05) decreased in mitochondrial fractions from LAD FL (19.2±0.8% total radiolabel) compared to NC FL (26.5±2.0). OGG-mediated incision was also significantly decreased in MCI TL (7.7±0.8% total radiolabel) and LAD TL (8.2±0.6% total) compared to NC specimens (11.3±1.0% total). Comparison of mitochondrial OGG1 activities by brain region showed a statistically significant increase in FL specimens compared to TL, PL, and CER of all three subject groups. Consistent with observations from nuclear specimens, mitochondrial OGG1 incision activities were generally similar between MCI and LAD subjects, suggesting that changes observed in LAD may begin in earlier stages of the disease. Supplementary Tables 1 to 4 show OGG1 incision activities and corresponding 8-OHdG levels measured using GC/MS-SIM and show a significant negative correlation ( R2=-0.73; P=0.03) for PL specimens only (Supplementary Table 1).
OGG1 protein levels in MCI and LAD
To determine if the decreased OGG1 activities we observed were due to a loss of OGG1 protein, 25 μg specimens of the samples used for activity assays were subjected to Western blot analysis and probed for OGG1. Western blots of OGG1 in nuclear specimens are shown from NC and MCI (Fig. 4A—1) or NC and LAD subjects (Fig. 4A—2) and the corresponding blots probed for lamin A as a nuclear loading control. Fig. 4B shows representative Western blots of mitochondrial fractions from NC and MCI (Fig. 4B—1) or NC and LAD (Fig. 4B—2) subjects probed for OGG1 followed by probing for porin (voltage-dependent anion channel) as a mitochondrial loading control and shows a single band at ∼ 40 kDa (mature form). Because of gel to gel variations in staining intensity Western results were normalized to control staining on each individual gel. Fig. 5A shows mean±SEM % control immunostaining for OGG1 and a significant (P<0.05) decrease of OGG1 protein in nuclear fractions in FL of LAD subjects (61.5±6.9% control) compared to age-matched NC (100±11.7%). In contrast, nuclear OGG1 was significantly elevated in TL of MCI patients (140.4±11.4% control). Temporal lobe nuclear OGG1 protein levels were also elevated (124.1±23.2% control), although the difference was not significant. Surprisingly, OGG1 protein levels from nuclear fractions of MCI CER were significantly elevated (177.2±16.4% control) compared to age-matched NC. Although protein levels are depleted in some areas of MCI and LAD brain, the decreases were not as profound as observed for losses of OGG1 incision activities, suggesting that other contributions to the diminished activity we observed. Mitochondrial OGG1 protein levels were not significantly different for any brain region studied (Fig. 5) Supplementary Tables 1 to 4 show levels of OGG1 measured using Western blot analysis and the corresponding levels of 8-OHdG measured using GC/MS-SIM for each brain region studied. Calculation of correlation coefficients between OGG1 protein levels and 8-OHdG levels showed no significant relationship between the two (Supplementary Tables 1 to 4).
Fig. 4.

(A) Representative Western blots of nuclear specimens from NC (4A—1; lanes 1–5) and MCI (4A—1; lanes 6–11) and NC (4A—2; lanes 1–5) and LAD (4A—2; lanes 6–10) subjects probed for OGG1 and lamin A (nuclear loading control). (B) Representative Western blots of mitochondrial specimens from NC (4B—1; lanes 1–5) and MCI (4B—1; lanes 6–11) and NC (4B—2; lanes 1–5) and LAD (4B—2; lanes 6–11) subjects probed for OGG1 and porin (mitochondrial loading control).
Fig. 5.

(A) Nuclear OGG1 protein levels (mean±SEM % control) in MCI, LAD, and NC subjects. OGG1 was significantly decreased in frontal lobe (FL) of LAD subjects. In contrast, OGG1 was significantly increased in MCI temporal lobe (TL) and CER. (B) Mitochondrial OGG1 protein levels (mean±SEM % control) in MCI, LAD, and NC subjects. Mitochondrial OGG1 protein was not significantly different in any of the subjects or brain regions studied.
OGG1 single-strand conformational polymorphism analyses
Previous studies showed ∼30% of LAD subjects demonstrated mutations in OGG1 that resulted in decreased excision activity [24]. Analysis of subjects in the current study using primers for all 7 exons and flanking sites of OGG1 showed 1 LAD patient and 2 MCI patients associated with a base substitution in exon 5 (data not shown). The base substitution observed in the LAD patient resulted in an amino acid change (I274 to V274), whereas the substitutions observed in the 2 MCI patients did not. However, none of the mutations observed were associated with diminished OGG1 activity.
OGG1 and HNE
The decreased OGG1 incision activity in excess of the decrease in protein, particularly for mitochondrial preparations, suggested that it might be functionally inactivated. To determine if OGG1 protein was modified and perhaps inactivated by a neurotoxic by-product of lipid peroxidation (HNE), nuclear specimens from 3 NC, 3 MCI, and 3 LAD subjects and mitochondrial specimens from 3 NC, 3 MCI, and 3 LAD subjects were subjected to immunoprecipitation using the anti-OGG1 antibody. The immunoprecipitated proteins were then probed using a polyclonal anti-HNE antibody. OGG1 immunoprecipitated from MCI mitochondrial preparations had statistically significantly higher levels of HNE immunostaining quantified by Scion Image Analysis (146.4±8.2% control) compared to NC subjects (100±13.2%) (Fig. 6A). LAD mitochondrial specimens also showed elevated HNE immunoreactivity, although the differences were not statistically significant. In contrast to mitochondrial specimens, OGG1 immunoprecipitated from nuclear preparations did not show statistically significant levels of HNE immunoreactivity (Fig. 6B). To determine if HNE can directly impair OGG1-mediated incision, NC nuclear specimens with high OGG1 activity were preincubated 1 h with 5 μM HNE before initiating the OGG1 activity assay. A concentration of 5 μM was chosen because it falls within the range of concentrations observed in LAD brain [4]. Preincubation with HNE led to a significant (P<0.05) decrease in 8-oxoguanine glycosylase activity (56.8±4.3% control) compared to control specimens (100±14.5% control). Together these data suggest that modification of OGG1 by HNE may impair activity of the enzyme and account for the more pronounced loss of incision activity compared to changes in OGG1 protein levels that we observe.
Fig. 6.
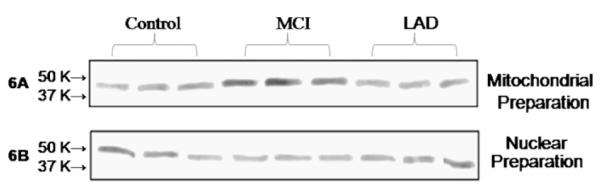
Representative mitochondrial specimens from 3 NC (lanes 1–3), 3 MCI (lanes 4–6), and 3 LAD (lanes 7–9) subjects were subjected to immunoprecipitation (IP) using anti-OGG1 and the isolated proteins probed for HNE, a by-product of lipid peroxidation (A). Nuclear specimens from 3 NC (lanes 1–2), 3 MCI (lanes 3–5), and 3 LAD subjects (lanes 6–8) were analyzed in the same way. Results of the assay show significantly more HNE-positive OGG1 in MCI mitochondrial specimens compared to NC specimens. HNE levels were not significantly different in nuclear immunoprecipitations.
Discussion
Previous studies from our laboratory and others show increased levels of markers of nuclear and mitochondrial DNA oxidation, particularly 8-OHdG in LAD [18,19,34-37] and MCI [9]. Because elevated levels of 8-OHdG may be due to increased oxidative stress, diminished repair capacity, or a combination of the two, we carried out studies to determine if nuclear and mitochondrial OGG1 activities were impaired in the progression of AD. Our results show a significant, disease-dependent decrease in OGG1 incision activity in nuclear fractions from neocortical regions of MCI and LAD brain compared to age-matched NC. These data are in contrast to those of Weissman et al. [25] who showed no significant alterations in nuclear OGG1 activity in a single brain region (inferior parietal lobule) of MCI subjects, although there was a trend toward decreased excision activity with disease progression. We found no significant differences in OGG1 activity in CER of MCI or LAD subjects compared to NC subjects. Of particular interest our data show that nuclear and generally mitochondrial OGG1 activities are significantly decreased in MCI neocortex to levels that are comparable to those in LAD, suggesting that diminished repair capacities occur early in the disease process and may play a meaningful role in the accumulation of DNA oxidative damage in AD. Because diminished OGG1 activities could be due to neuron loss in disease progression, protein levels were measured using a monoclonal antibody against OGG1. Our data show that OGG1 protein levels were not diminished to the same degree as the loss of excision activity, suggesting that the protein may be functionally inactivated. Our data further indicate that a neurotoxic by-product of lipid peroxidation (HNE) present in LAD and MCI brain [4,5] inactivates OGG1 activity and that OGG1 in mitochondrial specimens from MCI subjects shows elevated HNE immunostaining. These data are consistent with our previous studies that showed significant elevations of free HNE in MCI and LAD brain compared to levels in NC subjects [4,5]. Together these data suggest that aldehydic modification of OGG1 may contribute to diminished OGG1 activities in MCI and LAD.
To our knowledge, this is the first study to evaluate OGG1 activities in mitochondrial fractions from MCI and LAD brain. In contrast to nuclear OGG1, results of our study show that mitochondrial OGG1 protein levels are not significantly altered in MCI or LAD neocortex or CER compared to age-matched NC. Measures of OGG1 incision activity in mitochondrial fractions showed significant decreases in only FL specimens of LAD subjects and in TL specimens from MCI and LAD subjects. The observation of minimal alteration of OGG1 activity in mitochondria is a bit surprising in light of the pronounced elevation of 8-OHdG in mitochondrial DNA in MCI and LAD neocortex and suggests a compensatory increase in OGG1 in disease progression or perhaps potential deficiencies in other DNA repair pathways. During oxidative phosphorylation ∼2% of total oxygen consumed by cells is converted into ROS in mitochondria [20] leaving their DNA particularly susceptible to oxidative damage. Previous studies showed significantly elevated 8-OHdG in mitochondrial DNA compared to nuclear DNA from the same specimens in MCI [9] and LAD brain [18]. Although we found no significant differences in mitochondrial OGG1 activities in PL, we observed a significant negative correlation between OGG1 activity and levels of 8-OHdG.
Because of its high electron density guanine is paricularly vulnerable to oxidative modificaiton by hydroxyl radicals [38] leading to formation of 8-OHdG as the predominant marker of oxidative damage to DNA in MCI and LAD brain (reviewed in [8]). Free radical attack of C8 of guanine leads to formation of 8-hydroxyguanine radicals that result in production of 8-OHdG or following ring opening, 2,6-diamino—4-hydroxy—5-formamidopyrimidine (fapyguanine) [38,39]. To limit the potential impact of these oxidative modifications on binding of transcription factors, neurons employ BER in which the first step is excision of 8-OHdG by OGG1 or uridine by uracil DNA glycosylase [40]. OGG1 functions in BER by releasing the modified base and creating an abasic site [41]. Following removal of the modified base, the gap is filled by DNA polymerase and ligated by DNA ligase [6]. Mammalian cells code for OGG1, a functional homologue of E. coli MutM that recognizes and excises 8-OHdG residues paired with C in DNA [42] and is present in mitochondria and nuclei. Nuclear OGG1 (OGG1-α) and the mitochondrial isoform (OGG1-β) are both coded by the same gene and have an identical sequence of 315 amino acids from the N-terminus[43-46]. Differences in the 2 proteins are in their C-termini where OGG1-α has a nuclear localization sequence coded by exon 7, whereas OGG1-β has a mitochondrial targeting sequence coded by exon 8 (reviewed in [47]). Following translocation to mitochondria, the targeting sequence is cleaved by the mitochondrial processing peptidase in the matrix [48,49].
Our initial study of nuclear OGG1 activity in LAD brain showed significant loss of activity in vulnerable regions (hippocampus, superior and middle temporal gyrus) of the LAD brain compared with age-matched NC [23]. However, we did not measure OGG1 protein levels or mitochondrial protein or activity nor were we able to correlate OGG1 activities and 8-OHdG levels. Studies of nuclear OGG1 in IPL and CER from MCI and LAD subjects showed no significant alterations of OGG1 activity in IPL, although there was a trend toward decreased activity with disease progression [25]. Immunohistochemical studies of OGG1-β in LAD showed that the protein is decreased in orbitofrontal gyrus and entorhinal cortex in AD compared to NC subjects [50]. Additionally, immunoreactivity of OGG1-β was associated with NFT-bearing neurons and dystrophic neurites in AD [50]. Mao et al. showed that 4 of 14 LAD subjects studied exhibited mutations in OGG1 that led to diminished excision activity [24].
In studies of OGG1 in aging, incision activity was decreased in peripheral blood lymphocytes [51] and fibroblasts [52,53] as a function of age. In studies of aging mice, OGG1 activity was significantly lower in aged mice in multiple brain regions [6]. In contrast, studies of OGG1 in mitochondrial fractions prepared from aged human fibroblasts and liver of aged mice showed significantly higher levels of OGG1-β activity [54]. Although these data appear contradictory in light of elevated levels of 8-OHdG in aged mice, Szczesny et al. [54] showed that a large fraction of mitochondrial OGG1-β in aged animals is stuck to the mitochondrial membrane in the precursor form and is not translocated to and processed in the matrix that is essential for functional activity. Although we did not assess the specific mitochondrial distribution in our specimens, this may explain our observation of minimal loss of mitochondrial OGG1 activity in the presence of significantly elevated levels of 8-OHdG in MCI and LAD brain.
Overall, our findings suggest that OGG1 activity is significantly decreased in nuclear and mitochondrial fractions from neocortex of subjects with MCI and LAD compared to NC and that the decreases in MCI are generally as low as those in LAD, suggesting that diminished excision of 8-OHdG in nuclear DNA occurs early and may contribute to the pathogenesis of neuron degeneration in AD. Our data also suggest that loss of activity is in excess of decreased protein levels and may be due to HNE-mediated impairment of enzyme activity.
Supplementary Material
Acknowledgments
Supported by NIH Grants 5-P01-AG05119 and 5-P30-AG028383, and by a grant from the Abercrombie Foundation. The authors thank Ms. Paula Thomason for technical and editorial assistance, and Ms. Sonya Anderson for subject demographic data.
Abbreviations:
- AD
Alzheimer’s disease
- BER
base-excision repair
- CER
cerebellum
- DTT
dithiothreitol
- FL
frontal lobe
- HNE
4-hydroxynonenal
- LAD
late-stage Alzheimer’s disease
- MCI
mild cognitive impairment
- NC
normal control
- NFT
neurofibrillary tangle
- PL
parietal lobe
- PMI
postmortem interval
- OGG1
oxoguanine glycosylase 1
- 8-OHdG
eight-hydroxy-2′-deoxyguanosine
- PBS
phosphate-buffered saline
- ROS
reactive oxygen species
- SDS-PAGE
sodium dodecyl sulfate-polyacrylamide gel electrophoresis
- TL
temporal lobe
Footnotes
Appendix A. Supplementary data Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.freeradbiomed.2008.06.003.
References
- [1].Moreira PI, Honda K, Liu Q, Santos MS, Oliveira CR, Aliev G, Nunomura A, Zhu X, Smith MA, Perry G. Oxidative stress: the old enemy in Alzheimer’s disease pathophysiology. Curr. Alzheimer Res. 2005;2:403–408. doi: 10.2174/156720505774330537. [DOI] [PubMed] [Google Scholar]
- [2].Lovell MA, Ehmann WD, Mattson MP, Markesbery WR. Elevated 4-hydroxynonenal in ventricular fluid in Alzheimer’s disease. Neurobiol. Aging. 1997;18:457–461. doi: 10.1016/s0197-4580(97)00108-5. [DOI] [PubMed] [Google Scholar]
- [3].Lovell MA, Xie C, Markesbery WR. Acrolein is increased in Alzheimer’s disease brain and is toxic to primary hippocampal cultures. Neurobiol. Aging. 2001;22:187–194. doi: 10.1016/s0197-4580(00)00235-9. [DOI] [PubMed] [Google Scholar]
- [4].Markesbery WR, Lovell MA. Four-hydroxynonenal, a product of lipid peroxidation, is increased in the brain in Alzheimer’s disease. Neurobiol. Aging. 1998;19:33–36. doi: 10.1016/s0197-4580(98)00009-8. [DOI] [PubMed] [Google Scholar]
- [5].Williams TI, Lynn BC, Markesbery WR, Lovell MA. Increased levels of 4-hydroxynonenal and acrolein, neurotoxic markers of lipid peroxidation, in the brain in mild cognitive impairment and early Alzheimer’s disease. Neurobiol. Aging. 2006;27:1094–1099. doi: 10.1016/j.neurobiolaging.2005.06.004. [DOI] [PubMed] [Google Scholar]
- [6].Imam SZ, Karahalil B, Hogue BA, Souza-Pinto NC, Bohr VA. Mitochondrial and nuclear DNA-repair capacity of various brain regions in mouse is altered in an age-dependent manner. Neurobiol. Aging. 2006;27:1129–1136. doi: 10.1016/j.neurobiolaging.2005.06.002. [DOI] [PubMed] [Google Scholar]
- [7].Keller JN, Schmitt FA, Scheff SW, Ding Q, Chen Q, Butterfield DA, Markesbery WR. Evidence of increased oxidative damage in subjects with mild cognitive impairment. Neurology. 2005;64:1152–1156. doi: 10.1212/01.WNL.0000156156.13641.BA. [DOI] [PubMed] [Google Scholar]
- [8].Markesbery WR, Lovell MA. DNA oxidation in Alzheimer’s disease. Antioxid. Redox Signal. 2006;8:2039–2045. doi: 10.1089/ars.2006.8.2039. [DOI] [PubMed] [Google Scholar]
- [9].Wang J, Markesbery WR, Lovell MA. Increased oxidative damage in nuclear and mitochondrial DNA in mild cognitive impairment. J. Neurochem. 2006;96:825–832. doi: 10.1111/j.1471-4159.2005.03615.x. [DOI] [PubMed] [Google Scholar]
- [10].Ding Q, Markesbery WR, Cecarini V, Keller JN. Decreased RNA, and increased RNA oxidation, in ribosomes from early Alzheimer’s disease. Neurochem. Res. 2006;31:705–710. doi: 10.1007/s11064-006-9071-5. [DOI] [PubMed] [Google Scholar]
- [11].Ding Q, Markesbery WR, Chen Q, Li F, Keller JN. Ribosome dysfunction is an early event in Alzheimer’s disease. J. Neurosci. 2005;25:9171–9175. doi: 10.1523/JNEUROSCI.3040-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Nunomura A, Perry G, Aliev G, Hirai K, Takeda A, Balraj EK, Jones PK, Ghanbari H, Wataya T, Shimohama S, Chiba S, Atwood CS, Petersen RB, Smith MA. Oxidative damage is the earliest event in Alzheimer disease. J. Neuropathol. Exp. Neurol. 2001;60:759–767. doi: 10.1093/jnen/60.8.759. [DOI] [PubMed] [Google Scholar]
- [13].Nunomura A, Perry G, Hirai K, Aliev G, Takeda A, Chiba S, Smith MA. Neuronal RNA oxidation in Alzheimer’s disease and Down’s syndrome. Ann. N. Y. Acad. Sci. 1999;893:362–364. doi: 10.1111/j.1749-6632.1999.tb07855.x. [DOI] [PubMed] [Google Scholar]
- [14].Shan X, Lin CL. Quantification of oxidized RNAs in Alzheimer’s disease. Neurobiol. Aging. 2006;27:657–662. doi: 10.1016/j.neurobiolaging.2005.03.022. [DOI] [PubMed] [Google Scholar]
- [15].Lovell MA, Markesbery WR. Oxidatively modified RNA in mild cognitiveimpairment. Neurobiol. Dis. 2008;29:169–175. doi: 10.1016/j.nbd.2007.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Zhang J, Perry G, Smith MA, Robertson D, Olson SJ, Graham DG, Montine TJ. Parkinson’s disease is associated with oxidative damage to cytoplasmic DNA and RNA in substantia nigra neurons. Am. J. Pathol. 1999;154:1423–1429. doi: 10.1016/S0002-9440(10)65396-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Nunomura A, Chiba S, Kosaka K, Takeda A, Castellani RJ, Smith MA, Perry G. Neuronal RNA oxidation is a prominent feature of dementia with Lewy bodies. Neuroreport. 2002;13:2035–2039. doi: 10.1097/00001756-200211150-00009. [DOI] [PubMed] [Google Scholar]
- [18].Mecocci P, MacGarvey U, Beal MF. Oxidative damage to mitochondrial DNA is increased in Alzheimer’s disease. Ann. Neurol. 1994;36:747–751. doi: 10.1002/ana.410360510. [DOI] [PubMed] [Google Scholar]
- [19].Wang J, Xiong S, Xie C, Markesbery WR, Lovell MA. Increased oxidative damage in nuclear and mitochondrial DNA in Alzheimer’s disease. J. Neurochem. 2005;93:953–962. doi: 10.1111/j.1471-4159.2005.03053.x. [DOI] [PubMed] [Google Scholar]
- [20].Halliwell B, Dizdaroglu M. The measurement of oxidative damage to DNA by HPLC and GC/MS techniques. Free Radic. Res. Commun. 1992;16:75–87. doi: 10.3109/10715769209049161. [DOI] [PubMed] [Google Scholar]
- [21].Shibutani S, Takeshita M, Grollman AP. Insertion of specific bases during DNA synthesis past the oxidation-damaged base 8-oxodG. Nature. 1991;349:431–434. doi: 10.1038/349431a0. [DOI] [PubMed] [Google Scholar]
- [22].Barnett YA, King CM. An investigation of antioxidant status, DNA repair capacity and mutation as a function of age in humans. Mutat. Res. 1995;338:115–128. doi: 10.1016/0921-8734(95)00017-z. [DOI] [PubMed] [Google Scholar]
- [23].Lovell MA, Xie C, Markesbery WR. Decreased base excision repair and increased helicase activity in Alzheimer’s disease brain. Brain Res. 2000;855:116–123. doi: 10.1016/s0006-8993(99)02335-5. [DOI] [PubMed] [Google Scholar]
- [24].Mao G, Pan X, Zhu BB, Zhang Y, Yuan F, Huang J, Lovell MA, Lee MP, Markesbery WR, Li GM, Gu L. Identification and characterization of OGG1 mutations in patients with Alzheimer’s disease. Nucleic Acids Res. 2007;35:2759–2766. doi: 10.1093/nar/gkm189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Weissman L, Jo DG, Sorensen MM, de Souza-Pinto NC, Markesbery WR, Mattson MP, Bohr VA. Defective DNA base excision repair in brain from individuals with Alzheimer’s disease and amnestic mild cognitive impairment. Nucleic Acids Res. 2007;35:5545–5555. doi: 10.1093/nar/gkm605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology. 1984;34:939–944. doi: 10.1212/wnl.34.7.939. [DOI] [PubMed] [Google Scholar]
- [27].Morris JC, Heyman A, Mohs RC, Hughes JP, van Belle G, Fillenbaum G, Mellits ED, Clark C. The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD). Part I. Clinical and neuropsychological assessment of Alzheimer’s disease. Neurology. 1989;39:1159–1165. doi: 10.1212/wnl.39.9.1159. [DOI] [PubMed] [Google Scholar]
- [28].Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. (Berl) 1991;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- [29].Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ, Brownlee LM, Vogel FS, Hughes JP, van Belle G, Berg L. The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology. 1991;41:479–486. doi: 10.1212/wnl.41.4.479. [DOI] [PubMed] [Google Scholar]
- [30].National National institute on aging and Reagan institute working group on diagnostic criteria for the neuropathological assessment of Alzheimer’s disease. Neurobiol. Aging. 1997;18(S4):S1–S2. [PubMed] [Google Scholar]
- [31].Markesbery WR, Schmitt FA, Kryscio RJ, Davis DG, Smith CD, Wekstein DR. Neuropathologic substrate of mild cognitive impairment. Arch Neurol. 2006;63:38–46. doi: 10.1001/archneur.63.1.38. [DOI] [PubMed] [Google Scholar]
- [32].Mecocci P, MacGarvey U, Kaufman AE, Koontz D, Shoffner JM, Wallace DC, Beal MF. Oxidative damage to mitochondrial DNA shows marked age-dependent increases in human brain. Ann. Neurol. 1993;34:609–616. doi: 10.1002/ana.410340416. [DOI] [PubMed] [Google Scholar]
- [33].Croteau DL, Bohr VA. Repair of oxidative damage to nuclear and mitochondrial DNA in mammalian cells. J. Biol. Chem. 1997;272:25409–25412. doi: 10.1074/jbc.272.41.25409. [DOI] [PubMed] [Google Scholar]
- [34].Birincioglu M, Jaruga P, Chowdhury G, Rodriguez H, Dizdaroglu M, Gates KS. DNA base damage by the antitumor agent 3-amino-1,2,4-benzotriazine 1,4-dioxide (tirapazamine) J. Am. Chem. Soc. 2003;125:11607–11615. doi: 10.1021/ja0352146. [DOI] [PubMed] [Google Scholar]
- [35].Gabbita SP, Lovell MA, Markesbery WR. Increased nuclear DNA oxidation in the brain in Alzheimer’s disease. J. Neurochem. 1998;71:2034–2040. doi: 10.1046/j.1471-4159.1998.71052034.x. [DOI] [PubMed] [Google Scholar]
- [36].Lyras L, Cairns NJ, Jenner A, Jenner P, Halliwell B. An assessment of oxidative damage to proteins, lipids, and DNA in brain from patients with Alzheimer’s disease. J. Neurochem. 1997;68:2061–2069. doi: 10.1046/j.1471-4159.1997.68052061.x. [DOI] [PubMed] [Google Scholar]
- [37].Williams T. Islam, Lovell MA, Lynn BC. Analysis of derivatized biogenic aldehydes by HPLC tandem mass spectrometry. Anal. Chem. 2005;77:3383–3389. doi: 10.1021/ac048265+. [DOI] [PubMed] [Google Scholar]
- [38].Dizdaroglu M, Jaruga P, Birincioglu M, Rodriguez H. Free radical-induced damage to DNA: mechanisms and measurement. Free Radic. Biol. Med. 2002;32:1102–1115. doi: 10.1016/s0891-5849(02)00826-2. [DOI] [PubMed] [Google Scholar]
- [39].Breen AP, Murphy JA. Reactions of oxyl radicals with DNA. Free Radic. Biol. Med. 1995;18:1033–1077. doi: 10.1016/0891-5849(94)00209-3. [DOI] [PubMed] [Google Scholar]
- [40].Perez-Campo R, Lopez-Torres M, Cadenas S, Rojas C, Barja G. The rate of free radical production as a determinant of the rate of aging: evidence from the comparative approach. J. Comp. Physiol. B. 1998;168:149–158. doi: 10.1007/s003600050131. [DOI] [PubMed] [Google Scholar]
- [41].Souza-Pinto NC, Croteau DL, Hudson EK, Hansford RG, Bohr VA. Age-associated increase in 8-oxo-deoxyguanosine glycosylase/AP lyase activity in rat mitochondria. Nucleic Acids Res. 1999;27:1935–1942. doi: 10.1093/nar/27.8.1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Tchou J, Kasai H, Shibutani S, Chung MH, Laval J, Grollman AP, Nishimura S. 8-Oxoguanine (8-hydroxyguanine) DNA glycosylase and its substrate specificity. Proc. Natl. Acad. Sci. USA. 1991;88:4690–4694. doi: 10.1073/pnas.88.11.4690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Aburatani H, Hippo Y, Ishida T, Takashima R, Matsuba C, Kodama T, Takao M, Yasui A, Yamamoto K, Asano M. Cloning and characterization of mammalian 8-hydroxyguanine-specific DNA glycosylase/apurinic, apyrimidinic lyase, a functional mutM homologue. Cancer Res. 1997;57:2151–2156. [PubMed] [Google Scholar]
- [44].Nishioka K, Ohtsubo T, Oda H, Fujiwara T, Kang D, Sugimachi K, Nakabeppu Y. Expression and differential intracellular localization of two major forms of human 8-oxoguanine DNA glycosylase encoded by alternatively spliced OGG1 mRNAs. Mol. Biol. Cell. 1999;10:1637–1652. doi: 10.1091/mbc.10.5.1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Roldan-Arjona T, Wei YF, Carter KC, Klungland A, Anselmino C, Wang RP, Augustus M, Lindahl T. Molecular cloning and functional expression of a human cDNA encoding the antimutator enzyme 8-hydroxyguanine-DNA glycosylase. Proc. Natl. Acad. Sci. USA. 1997;94:8016–8020. doi: 10.1073/pnas.94.15.8016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Rosenquist TA, Zharkov DO, Grollman AP. Cloning and characterization of a mammalian 8-oxoguanine DNA glycosylase. Proc. Natl. Acad. Sci. USA. 1997;94:7429–7434. doi: 10.1073/pnas.94.14.7429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Boiteux S, Radicella JP. The human OGG1 gene: structure, functions, and its implication in the process of carcinogenesis. Arch. Biochem. Biophys. 2000;377:1–8. doi: 10.1006/abbi.2000.1773. [DOI] [PubMed] [Google Scholar]
- [48].Gakh O, Cavadini P, Isaya G. Mitochondrial processing peptidases. Biochim. Biophys. Acta. 2002;1592:63–77. doi: 10.1016/s0167-4889(02)00265-3. [DOI] [PubMed] [Google Scholar]
- [49].Pfanner N. Protein sorting: recognizing mitochondrial presequences. Curr. Biol. 2000;10:R412–415. doi: 10.1016/s0960-9822(00)00507-8. [DOI] [PubMed] [Google Scholar]
- [50].Iida T, Furuta A, Nishioka K, Nakabeppu Y, Iwaki T. Expression of 8-oxoguanine DNA glycosylase is reduced and associated with neurofibrillary tangles in Alzheimer’s disease brain. Acta Neuropathol. (Berl.) 2002;103:20–25. doi: 10.1007/s004010100418. [DOI] [PubMed] [Google Scholar]
- [51].Chen SK, Hsieh WA, Tsai MH, Chen CC, Hong AI, Wei YH, Chang WP. Age-associated decrease of oxidative repair enzymes, human 8-oxoguanine DNA glycosylases (hOgg1), in human aging. J. Radiat. Res. (Tokyo) 2003;44:31–35. doi: 10.1269/jrr.44.31. [DOI] [PubMed] [Google Scholar]
- [52].Goukassian D, Gad F, Yaar M, Eller MS, Nehal US, Gilchrest BA. Mechanisms and implications of the age-associated decrease in DNA repaircapacity. FASEB J. 2000;14:1325–1334. doi: 10.1096/fj.14.10.1325. [DOI] [PubMed] [Google Scholar]
- [53].Kaneko T, Tahara S, Taguchi T, Kondo H. Accumulation of oxidative DNA damage, 8-oxo-2′-deoxyguanosine, and change of repair systems during in vitro cellular aging of cultured human skin fibroblasts. Mutat. Res. 2001;487:19–30. doi: 10.1016/s0921-8777(01)00100-8. [DOI] [PubMed] [Google Scholar]
- [54].Szczesny B, Hazra TK, Papaconstantinou J, Mitra S, Boldogh I. Age-dependent deficiency in import of mitochondrial DNA glycosylases required for repair of oxidatively damaged bases. Proc. Natl. Acad. Sci. USA. 2003;100:10670–10675. doi: 10.1073/pnas.1932854100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.