

Abstract
The cause of the conformational change of normal cellular prion protein (PrP) into its disease-associated form is unknown. Posttranslational modifications, such as glycosylation, acetylation, S-nitrosylation, and phosphorylation, are known to induce protein conformational changes. Here, we investigated whether phosphorylation could induce the conformational change of PrP because PrP contains several kinase motifs and has been found recently in the cytosol, in which kinases generally reside. Neuronal cyclin-dependent kinase 5 (Cdk5) phosphorylated recombinant PrP23–231 at serine 43 (S43) in an in vitro kinase assay. Cdk5-phosphorylated PrP became proteinase K resistant, formed Congo Red-positive fibrils, and formed aggregates that were immunostained with anti-PrP and anti-phospho-PrPS43 (anti-pPrPS43). pPrPS43 was detected in PrP/Cdk5/p25 cotransfected N2a cells. Roscovitine inhibition of Cdk5 activity or transfection of N2a cells with mutant PrP S43A eliminated the anti-pPrPS43-immunopositive protein. Alkaline phosphatase-sensitive and proteinase K-resistant pPrPS43 immunoreactivity was observed in scrapie-infected but not control-injected mice brains. These results raise the possibility that phosphorylation could represent a physiological mechanism of PrP conversion in vivo.
Introduction
Although it is widely accepted that conversion of normal cellular prion protein (PrP) into a proteinase K (PK) resistant (PKRES or PrPRES) aberrant conformational form is associated with transmissible spongiform encephalopathies, the underlying molecular mechanism of this conversion is not clear. The hypothesis that prion protein in the form of PrPRES or PrP scrapie (PrPSc) is responsible for conversion of normal cellular PrP is well supported in vivo and in vitro. PrPSc seeded conversion is observed by infecting live animals (Prusiner, 1982; Caughey, 1993), specific cell lines (Race et al., 1987), and cell-free mammalian protein systems (Neary et al., 1991; Kocisko et al., 1994; Bessen et al., 1995; Saborio et al., 2001; Deleault et al., 2007).
Conversion is also observed in the absence of PrPSc seed in vivo. Single point and insertional octapeptide repeat mutations of the Prnp gene generate PrPRES (Monari et al., 1994; Tateishi and Kitamoto, 1995; Mastrianni et al., 2001; Piccardo et al., 2001; Grasbon-Frodl et al., 2004). Transgenic mice expressing the Gertsmann–Sträussler–Scheinker-associated PrP P101L mutation or PrP with a nine octapeptide repeat insertion result in a mild PrPRES form of PrP (Hsiao et al., 1990; Chiesa et al., 1998). In the absence of a PrP mutation, endoplasmic reticulum associated degradation pathway-generated cytosolic PrP becomes PKRES in mouse neuroblastoma Neuro2a (N2a) cells; however, this does not occur in human primary neurons or human neuroblastoma cell lines (Ma and Lindquist, 2002; Ma et al., 2002; Roucou et al., 2003).
In vitro, purified human, hamster, or mouse PrP90–231 or PrP23–231 convert under acidic pH, mild denaturant conditions, treatment with 0.1% SDS and sonication, and protein misfolded cyclic amplification (PMCA) assay in the presence of RNA (Swietnicki et al., 1997; Jackson et al., 1999; Swietnicki et al., 2000; Deleault et al., 2003; Bocharova et al., 2006; Lührs et al., 2006; Atarashi et al., 2007; Wang et al., 2007). Methionine oxidation, high-pressure, Al3+ and Zn2+ also promote conversion of PrP and PrP fragments (Torrent et al., 2004; Breydo et al., 2005; Ricchelli et al., 2006). Conversion of PrP appears to first involve the formation of oligomers that evolve into fibrillar structures with time and give PKRES fragments between 8 and 16 kDa (Jackson et al., 1999; Swietnicki et al., 2000; Xiong et al., 2001; Sokolowski et al., 2003; Breydo et al., 2005; Lührs et al., 2006). Transmissibility of disease from PrPSc-free conversions has been observed only in two situations: by the injection of Escherichia coli-purified fibrillar PrP89–231 in brains of transgenic mice overexpressing PrP89–231 (Legname et al., 2004) and by infecting wild-type hamsters with purified mammalian PrP submitted to PMCA in the presence of poly-anions (Deleault et al., 2007).
Several anionic conditions, such as anionic detergents, synthetic poly-anions, RNA, and low pH conditions, favor the conformational change of PrP in vitro (Deleault et al., 2003, 2005, 2007; Supattapone, 2004; Geoghegan et al., 2007). Therefore, here, we considered the hypothesis that phosphorylation of PrP, which would also provide anionic conditions, could affect PrP conformation.
Materials and Methods
Antibodies.
The following commercially available antibodies were used: monoclonal 3F4 anti-PrP109–112 (Kascsak et al., 1987), monoclonal 6H4 anti-PrP144–156 (Prionics), monoclonal phospho-Tyr (pTyr-100) (Cell Signaling Technology), HRP-conjugated goat anti-rabbit or anti-mouse IgG (GE Healthcare), and β-actin (Sigma-Aldrich). The polyclonal R155 anti-PrP36–56 was produced in our laboratory. The human PrP peptide Gly-phospho-Ser-Pro-Gly-Gly-Asn-Arg-Tyr-Pro terminating with an added Cys was synthesized, purified, conjugated to keyhole limpet hemocyanin, and injected into rabbits by Sigma-Genosys. ELISA performed by Sigma-Genosys gave a titer of 1:25,000 for non-phospho-peptide and 1:500,000 for phospho-peptide after the first production bleed. The antiserum anti-phospho-PrpS43 (pPrPS43) was used at a titer of 1:100 for Western blots and 1:250 for immunoprecipitation.
Site-directed mutagenesis of PrP and PrP purification.
PrP S43A was generated by QuikChange site-directed mutagenesis (Jodoin et al., 2007) with the forward primer 5′-CCGGGGCAGGGCGCACCTGGAGG-CAACC-3′ and the reverse primer 5′-GGTTGCCTCCAGGTGCGCCC-TGCCCCGG-3′, from pBKSII–PrP23–231 cDNA. The S43A mutation was confirmed by BglI digestion. PrP and PrP S43A were subcloned into the BamHI and XhoI sites of the pET-23b(+) vector (EMD Chemicals) after PCR amplification with the forward primer 5′-ACGCGGATCCCAA-GAAGCGCCCGAAGCCT-3′ and the reverse primer 5′-GCCGCT-CGAGGCTCGATCCTCTCTGGTA-3′. The expression of C-terminally His-tagged PrP was induced in pET-23b(+)PrP or PrP S43A-transformed E. coli BL21(DE3)pLysS (Stratagene) with isopropyl-β-d-thiogalactopyranoside and purified as described previously (Gilch et al., 2003). In addition, PrP S43A was introduced into pCep4β–PrP full length (Bounhar et al., 2001) by QuikChange site-directed mutagenesis.
Kinase assay.
One microliter of cyclin-dependent kinase 5 (Cdk5) kinase extracted from bovine brain (Paudel et al., 1993), 1.5 U of recombinant glutathione S-transferase (GST)–Cdk5 with 2 U of GST–p25 (Calbiochem), or 500 U of Casein kinase II (CKII) (Biomol Research Laboratories) were added to 0.45 μg/μl PrP (a generous gift from Dr. Witold Surewicz, Case Western Reserve University, Cleveland, OH) in kinase assay buffer containing 110.5 mm HEPES, pH 7.2, 0.15 mm EDTA, 0.15 mm EGTA, 0.07 mm okadaic acid, 11.1 mm sodium fluoride, 11.1 mm MgCl2, 1 μCi of [γ-32P]ATP (2 mCi/ml; PerkinElmer Life and Analytical Sciences), 2 mm ATP, and EDTA-free protease inhibitor cocktail (Roche Applied Science). The Cdk5 inhibitor olomoucine (Biomol Research Laboratories) was added at a concentration of 400 μm. The kinase reaction mix was incubated at 30°C for 4 h, separated on 15% SDS-PAGE gels, and visualized by overnight exposure for autoradiography or by Western blotting with the monoclonal 3F4 antibody or the anti-pPrPS43 antiserum. Immunoreactivity was detected with HRP-conjugated anti-mouse or anti-rabbit secondary antibodies and ImmobilonWestern chemiluminescent HRP substrate reagents (Millipore).
PK treatment of phosphorylated PrP.
Various concentrations of PK (BioShop) in 50 mm Tris-HCl, pH 7.5, ranging from 0 to 50 μg/ml were mixed with 2.3 μg of [γ-32P]phosphorylated or nonphosphorylated PrP in kinase reaction buffer containing freshly added 0.1 mm okadaic acid. The reaction mix was incubated at 4°C for 1 h or at 37°C for 1–4 h. The PK-treated PrP was analyzed by autoradiography and Western blot analyses as described above.
Effect of pPrP on nonphosphorylated PrP aggregation.
Two microliters (0.9 μg of total PrP) of Cdk5-pPrP kinase assay or kinase assay without Cdk5 were added to 5.85 μg of PrP in a volume of 15 μl and incubated at 37°C for 0, 24, 48, and 96 h. After 96 h, 2 μl corresponding to 0.12 μg or 0.012 μg of the original kinase assays were added to 5.85 μg of fresh PrP and incubated for 24 h at 37°C (cycle 1) for serial propagation assays. The reaction was repeated for six cycles, transferring 2 μl to fresh nonphosphorylated PrP at the end of each cycle. At the end of each cycle, 2 μl aliquots were also removed and added to 10 μl containing a final concentration of 0 (−PK) or 10 μg/ml PK (+PK) and digested at 37°C for 1 h before submitting to a 3F4 Western blot.
Transmission electron microscopy.
Kinase reactions were dialyzed against 5 mm Tris-HCl, pH 7.4, using the Slide-A-Lyzer Mini Dialysis unit (Pierce). Approximately 0.5 μl of the dialyzed kinase reaction mix was deposited onto Formvar (Camemco Supplies)-coated copper grids for 2 min, adsorbed with Whatman 1M filter paper, dried for 2 min, and stained with 4% uranyl acetate. Analyses of the samples were done with an FFI TECHNAI 12 120V transmission electron microscope at the Facility for Electron Microscopy at McGill University. Controls consisted of nonphosphorylated PrP lacking Cdk5 in the reaction mix and Cdk5 in the absence of PrP in the kinase reaction mix. Aging was done by incubating phosphorylated or nonphosphorylated dialyzed PrP at 37°C for 16 d. Samples were frozen at −80°C until analysis by transmission electron microscopy.
Immunoelectron microscopy.
Dialyzed PrP or pPrP were placed on the grids as described above and blocked with a solution of 2% bovine serum albumin, 2% casein, and 0.5% ovalbumin (BCO) for 5 min. Anti-PrP antibodies were applied for 1 h at room temperature at a dilution of 1:10 in BCO for 3F4 and anti-pPrPS43 and 1:20 for 6H4. After washes in Dulbecco's PBS (DPBS) and another 5 min blocking step in BCO, samples were incubated for 30 min with 1:20 anti-rabbit or anti-mouse IgG antibodies conjugated with 10 nm gold particles (Sigma). After washing with DPBS, samples were stained with 4% uranyl acetate for 30 s to 1 min. Controls included samples with no primary antibodies and immunostaining of amyloid β peptide Aβ1–42 fibrils generated as described previously (Zhang et al., 2002). Coimmunostaining of PrP with 6H4 antibody and anti-pPrPS43 antiserum was detected with anti-mouse IgG conjugated to 5 nm gold (Sigma) and goat anti-Affinitypure donkey anti-rabbit (heavy and light chains) conjugated to 18 nm gold particles (Jackson ImmunoResearch).
Congo Red staining.
Three microliters of 10 μm dialyzed PrP, Cdk5-phosphorylated PrP (pPrP) PK-digested Cdk5-phosphorylated PrP, fibrillar Aβ1–42 and Aβ42–1 (Zhang et al., 2002), and 0.05 μl of bovine brain purified Cdk5 were applied on glass slides and dried overnight at 4°C. Dried samples were stained for 2 h with Congo Red solution (4 mm Congo Red, 50 mm NaCl, and 80% EtOH) filtered on a 0.5 μm membrane. Samples were washed four times with 90% ethanol and dried, and pictures were taken under polarized light microscopy.
Purification of phospho-proteins.
Mouse N2a cells (American Type Culture Collection) were cultured in MEM containing 10% fetal bovine serum (HyClone) and transfected with pCep4β–PrP, pCep4β–PrP/pcDNA3.1–Cdk5/pcDNA3.1–p25, or pCep4β–PrP S43A/pcDNA3.1–Cdk5/pcDNA3.1–p25 (Bounhar et al., 2001; Li et al., 2007) using Lipofectamine2000 reagent (Invitrogen). Cells were maintained in culture in the presence of 100 nm okadaic acid (BioShop). For the roscovitine treatments, 10 μm roscovitine (Biomol Research Laboratories) was added 24 h after the transfection, and cells were maintained in culture for an additional 24 h. Proteins were collected 48 h after the transfection and phospho-proteins purified with the PhosphoPurification kit according to the instructions of the manufacturer (Qiagen).
Immunohistochemistry on mice brains.
C57BL/6 mice were intracerebrally inoculated with 20 μl of a 1% brain homogenate from mice infected with the 22A strain of scrapie (TSE Resource Centre). The animals were killed at onset of clinical symptoms, and the whole brains were fixed in 10% Formalin and then processed and embedded in paraffin wax. Brains of age-matched, mock-infected mice were collected as controls. Four micrometer sections were deparaffinized, rehydrated, autoclaved in sodium citrate antigen retrieval buffer (10 mm sodium citrate and 0.05% Tween 20, pH 6.0) at 121°C 30 min, washed with TBS-T (0.1% Triton X-100, 20 mm Tris, and 150 mm NaCl, pH 7.5), and blocked with Power Block Universal blocking reagent (Inter Medico). The anti-pPrPS43 antiserum (1:200) was incubated overnight at 4°C, followed by washing in TBS-T and incubating with UltraVision One alkaline phosphatase polymer according to the UltraVision ONE Detection System protocol (Thermo Fisher Scientific). The tissue sections were counterstained with hematoxylin. When indicated, before the first antibody incubation, the sections were treated at 37°C for 1 h with 150 U/ml alkaline phosphatase (Fermentas) or 15 min with a 1:50 dilution of ready-to-use proteinase K (Dako). For the adsorption of the anti-pPrPS43 antiserum, diluted antiserum (1:200) was incubated overnight at 4°C with 20 μg/ml pPrPS43 peptide, and centrifuged, and the supernatant was used as adsorbed antiserum.
Results
Cdk5 phosphorylation of PrP23–231
Several kinase motifs are highly conserved in PrP (Table 1). We focused on Cdk5 because it is an abundant neuronal kinase involved in neurodegeneration and neurons are the cell type most affected in prion diseases. Cdk5 phosphorylates serine–proline (SP) motifs. Prion protein has two SP phosphorylation motifs: one in the N terminus part of the mature protein at amino acid Ser43/Pro44 and one in the glycosyl phosphatidylinositol-anchor signal peptide at S237/P238 (Table 1). In vitro phosphorylation of PrP23–231 with Cdk5 showed intense phosphorylation of the full-length and a fragment of PrP23–231, similar to Cdk5 phosphorylation of the known Cdk5 substrate, Tau protein (Fig. 1A,B). The Cdk5 inhibitor olomoucine essentially inhibited PrP phosphorylation (Fig. 1C). To determine whether Cdk5 phosphorylates PrP at S43, we mutated S43 to A43 in PrP23–231. The PrP S43A was labeled poorly compared with the wild-type protein (Fig. 1D).
Table 1.
Conservation of phosphorylation motifs in PrP amongst various species
| Putative motif | Phospho AA | Mammals | Chicken | Turtle | Doppel |
|---|---|---|---|---|---|
| PGQSPGGN | S43 | All | RQ | SN | |
| IHFGSDYED | S143 | Only in human, bovine, and bison | S | R | A |
| FGSDYEDRY | Y145 | W in rat, mouse, and hamster | D | E | |
| YEDRYYREN | Y149 | All | WW | WW | YY |
| PMDEYSNQN | Y169 | All | R | A | |
| HTVTTTTKG | T191 | All | P | P | F |
| TVTTTTKGE | T192 | All | A | N | Q |
| VTTTTKGEN | T193 | All | A | E | K |
| YQRGSSMVL | S230 | All except rabbit | S | S | A |
| QRGSSMVLF | S231 | All except rabbit | G | G | G |
| VLFSSPPVI* | S237 | All | AD | DP | HQ |
Mammalian species examined are human, guar, macaque, kudu, bison, rhesus monkey, baboon, mink, rabbit, camel, rat, nilgai, sheep, mouse, hamster, cattle, and cat. Human Doppel was also included. The phosphorylation sites were identified with NetPhos 2.0 Server in EXPASY and had significant scores, except the last SP motif (*), which has low predictability for phosphorylation.
Figure 1.

Cdk5 phosphorylation of human PrP23–231 at S43. A, Autoradiogram of Cdk5 in vitro kinase assay on PrP23–231 or Tau protein. B, Coomassie blue stain of PrP and Tau protein. C, Autoradiogram of in vitro Cdk5 kinase assay on PrP in the absence or presence of olomoucine (Olo.). D, Autoradiogram and Coomassie blue stain of in vitro Cdk5 kinase assay on PrP and PrP S43A. E, Western blot with anti-pPrPS43 antiserum and anti-PrP109–112 3F4 antibody of 100 ng of nonphosphorylated PrP or 100 ng of Cdk5 pPrP. F, Western blot with 3F4 antibody of PrP and pPrP immunoprecipitated (IP) with anti-pPrPS43 or anti-PrP36–56 R155 antisera. G, Western blot with 3F4 and anti-pPrPS43 of Cdk5 kinase assay on PrP or PrP S43A.
We then generated an antiserum against pPrP at S43 (anti-pPrPS43). The anti-pPrPS43 recognized the Cdk5-phosphorylated PrP but not the nonphosphorylated PrP (Fig. 1E). Anti-pPrPS43 immunoprecipitated Cdk5-phosphorylated PrP but not the nonphosphorylated PrP, indicating that this antiserum recognized native pPrP (Fig. 1F). In contrast, R155 (anti-PrP36–56) immunoprecipitated both pPrP and PrP. The anti-pPrPS43 did not recognize Cdk5-phosphorylated mutant PrP S43A, thus confirming that S43 is the site of phosphorylation (Fig. 1G). Together, these results indicate that S43 is a major Cdk5 phosphorylation site in PrP.
Cdk5-phosphorylated PrP converts to a PKRES form
To determine whether PrP phosphorylation induces PKRES, we submitted the 32P-phosphorylated PrP to increasing amounts of PK. A 32P-pPrP peptide of ∼10 kDa robustly resisted a 1 h treatment of 10 μg/ml PK at 4°C or 37°C (Fig. 2A) and 4 h of 50 μg/ml PK digestion at 37°C (Fig. 2B). These results indicate that the pPrP has either undergone a conformational change or developed aggregates that resist PK digestion.
Figure 2.
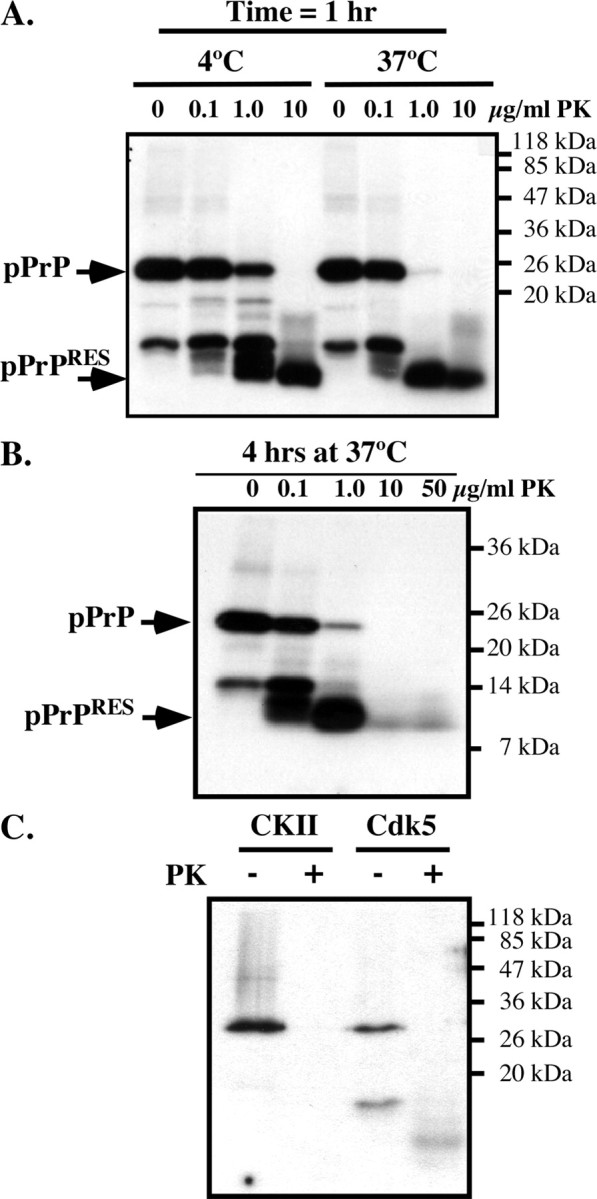
Cdk5-phosphorylated, but not CKII-phosphorylated, PrP is resistant to PK. A, Autoradiogram of Cdk5-phosphorylated PrP incubated with 0–10 μg/ml PK for 1 h at 4°C or 37°C. B, Autoradiogram of Cdk5-phosphorylated PrP incubated with 0–50 μg/ml PK for 4 h at 37°C. C, Autoradiogram of Cdk5- or CKII-phosphorylated PrP incubated with 0 or10 μg/ml PK for 1 h at 37°C.
To assess whether phosphorylation at other amino acid residues can also induce PKRES of PrP, PrP was phosphorylated with CKII, known to phosphorylate bovine PrP at Ser154 (equivalent to S143 in humans) (Negro et al., 2000). However, CKII-phosphorylated human PrP did not become PKRES (Fig. 2C). These results indicate that Cdk5-phosphorylated PrP specifically becomes PKRES.
Cdk5-phosphorylated PrP induces the aggregation of nonphosphorylated PrP
To determine whether non-32P-labeled PrP in the reaction mixture was also resistant to PK, the Cdk5-phosphorylated PrP was immunoblotted (Fig. 3A). The nonphosphorylated PrP was completely degraded by PK, but the Cdk5-phosphorylated PrP was not. The 3F4 antibody detected PKRES 25 kDa full-length, the 16 and 18 kDa copurified PrP fragments, and faintly the 10 kDa radiolabeled PrPRES fragment. Together, these results show that, first, the PKRES radiolabeled 10 kDa fragment contains the phosphorylated S43, and, second, that nonphosphorylated 25 kDa full-length PrP can become PKRES in the kinase assay. The 16 and 18 kDa nonradiolabeled PKRES fragments either represent pPrP after the proteolytic cleavage of the N terminus of PrP containing the pS43 epitope or nonphosphorylated PKRES PrP fragments. Seeding PrP from the Cdk5 kinase assay generated a small amount of PKRES nonphosphorylated full-length PrP and PrP fragments after 24 h of incubation, whereas seeding PrP from the kinase assay lacking Cdk5 did not (Fig. 3B). Furthermore, a longer exposure demonstrated an increase of the 10 kDa PrP fragment at 24 and 48 h. At 48 and 96 h of incubation, aggregated PrP representing oligomers of varying sizes were also generated as evidenced by the smear at the top of the Western blot (Fig. 3B).
Figure 3.

Cdk5-phosphorylated PrP induces the aggregation of nonphosphorylated PrP in vitro. A, Autoradiogram and Western blot analysis with 3F4 antibody of nonphosphorylated or Cdk5-phosphorylated PrP treated with 10 μg/ml PK for 1 h at 37°C. The arrow indicates the 10 kDa PrPRES fragment detected on the autoradiogram or Western blot. B, PrP Western blot of nonphosphorylated PrP seeded with kinase assays performed with (pPrP) or without Cdk5 and incubated for the indicated time without (−PK) or with (+PK) PK treatment. The bottom shows a longer exposure of another pPrP seeded experiment revealing the 10 kDa PrP fragment in +PK. C, PrP Western blot of nonphosphorylated PrP seeded with a 2 μl aliquot of the 96 h time point (0 cycle) in B and incubated 24 h (cycle 1). Subsequent cycles represent samples in which 2 μl at the end of the incubation period was added into fresh nonphosphorylated PrP and incubated 24 h. The bottom represents an original seed of 0.2 μl of the 96 h time point in B.
To assess whether pPrP can convert nonphosphorylated PrP into a PKRES protein in a serial propagation assay, 2 or 0.2 μl of the 96 h reaction mix in Figure 3B was incubated 24 h with fresh nonphosphorylated PrP before testing for PKRES, and this was repeated six times (cycles), each time seeding fresh nonphosphorylated PrP with 2 μl of the incubation mix at the end of the 24 h incubation period. Increasing amounts of PKRES full-length PrP and PrP fragments were obtained from the Cdk5-containing kinase assay seeded PrP mix after three cycles but not from the non-Cdk5-containing kinase assay (Fig. 3C). However, no additional amplification of the 10 kDa PKRES PrP fragment was obtained. Seeding with 0.2 μl of the Cdk5 kinase assay also resulted in PKRES PrP after six cycles. These results indicate that pPrP enhances nonphosphorylated PrP aggregation but not conversion.
Cdk5-phosphorylated PrP forms aggregates and fibrils
To evaluate the structural state of PrP after phosphorylation, we analyzed PrP and pPrP by transmission electron microscopy. Globular aggregates were detected in the pPrP and much less abundantly in the nonphosphorylated PrP reaction mixture (Fig. 4). The Cdk5 alone did not contain these structures (data not shown). The globular structures had various appearances: some were monomeric, whereas others were aggregated. Immunodecoration with the anti-PrP 6H4 and 3F4 antibodies (3F4 not shown) and anti-pPrPS43 antiserum confirmed that PrP and pPrP were present in these globular structures (Fig. 4A,B). Both epitopes colocalized but did not overlap in these globular aggregates (Fig. 4C). No immunoreactivity was observed in the absence of primary antibodies (data not shown). These two antibodies did not recognize Aβ1–42 fibrils, thus indicating specificity (Fig. 4D). The 6H4 antibody recognized nonphosphorylated PrP (Fig. 4E), but anti-pPrPS43 did not (Fig. 4F).
Figure 4.
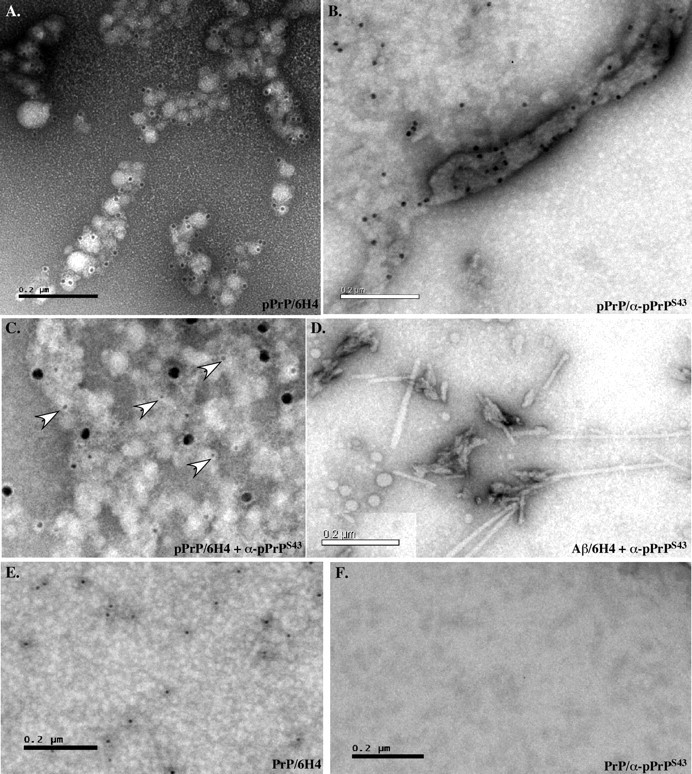
Cdk5-phosphorylated PrP forms aggregates. A–C, Immunostaining of pPrP with 6H4 (10 nm gold particle size) (A), anti-pPrPS43 (10 nm) (B), or both anti-pPrPS43 (18 nm) and 6H4 (5 nm) (C) antibodies. D, Control of fibrillar Aβ1–42 immunostaining with anti-pPrPS43 (10 nm). E, F, Immunostaining of nonphosphorylated PrP with 6H4 (10 nm) (E) and anti-pPrPS43 (10 nm) (F) antibodies.
In addition, large fibrils were detected in transmission electron micrographs of pPrP (Fig. 5A). These increased in abundance and diversity with an incubation of the Cdk5-phosphorylated PrP at 37°C for 16 d (Fig. 5B), a method commonly used to enhance fibrillization of peptides. The globular aggregates also seem to become more compacted with time (Fig. 5C). In contrast, nonphosphorylated PrP remained amorphous with time of incubation (Fig. 5D).
Figure 5.
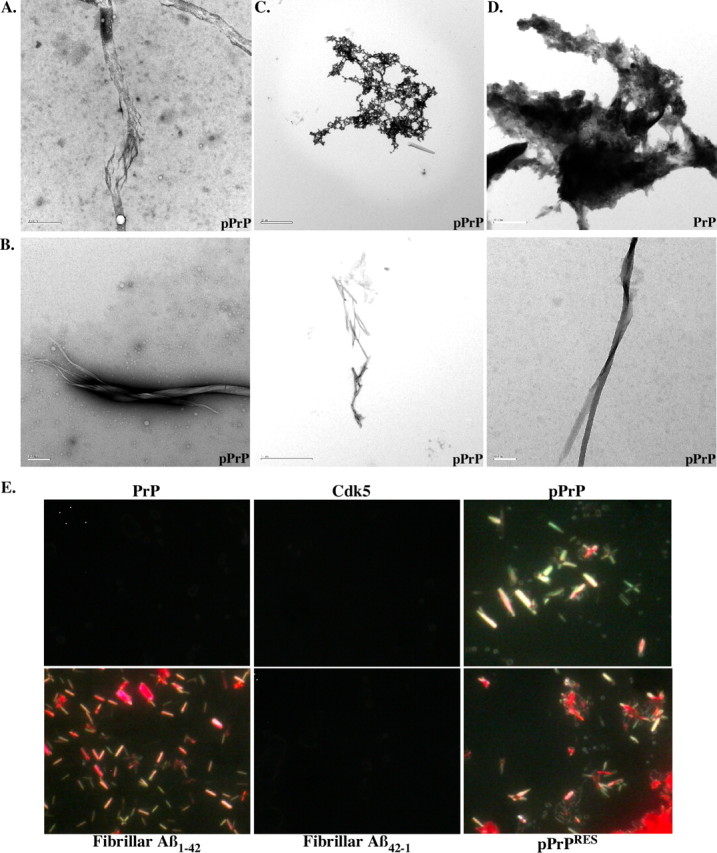
Cdk5-phosphorylated PrP forms fibrils. A, Electron micrograph of dialyzed Cdk5-phosphorylated PrP showing fibrillar-like structures. Scale bar, 0.5 μm. B, Fibrils detected in pPrP after a 16 d incubation at 37°C. Scale bar, 0.2 μm for left and right; 2 μm for middle. C, Compact structure of pPrP after a 16 d incubation at 37°C. Scale bar, 2 μm. D, Amorphous appearance of nonphosphorylated PrP after a 16 d incubation at 37°C. Scale bar, 0.5 μm. E, Congo Red staining of dialyzed PrP, Cdk5 alone, Cdk5-pPrP, PrPRES, fibrillar Aβ1–42, and Aβ42–1. Pictures were taken under polarized light microscopy.
To determine whether pPrP had taken an amyloid conformation, we conducted Congo Red staining (Fig. 5E). Whereas neither the PrP nor the Cdk5 preparation stained with Congo Red, Cdk5-pPrP displayed fibril-like structures with the expected apple green birefringence appearance under polarized light. These structures resembled those observed with Aβ1–42. The reverse control peptide Aβ42–1 was negative, as expected. Treatment of the proteins with PK did not remove these Congo Red-positive structures in Cdk5-phosphorylated PrP. Together, these results show that phosphorylation of PrP induces a conformational change in PrP.
Phosphorylated PrP is detected in PrP/Cdk5/p25 cotransfected N2a cells
To examine whether PrP can be phosphorylated in live cells, we transfected mouse neuroblastoma N2a cells with wild-type human PrP, Cdk5, and p25 cDNAs and isolated phospho-proteins from total cellular protein extracts. PrP was expressed at high levels, and most was recuperated in the flow-through of the phospho-column (Fig. 6A). Four 3F4-positive 25–30 kDa proteins were recovered in fractions 2–4, with most of the protein eluting in fraction 3 (F3). Immunoblotting with an anti-pTyr antiserum confirmed that F3 contained most of the phospho-proteins. The anti-pPrPS43 antiserum recognized one of the four 3F4-positive proteins in F3. To determine whether Cdk5 phosphorylated any of these pPrP, we treated the transfected cells with the Cdk5 inhibitor roscovitine. Three 3F4-positive PrP proteins were observed in the phospho fraction (Fig. 6B). Furthermore, transfection of N2a cells with full-length PrP S43A rather than wild-type PrP or wild-type PrP in the absence of Cdk5/p25 also yielded only three proteins (Fig. 6C,D). As expected, no 3F4 immunoreactivity was detected in the phospho-proteins purified from untransfected N2a cells (Fig. 6E). These results indicate that PrP is phosphorylated at S43 by a roscovitine-sensitive kinase. In addition, the results show additional Cdk5-independent PrP phosphorylation.
Figure 6.

Purification of phosphorylated PrP from PrP/Cdk5/p25 cotransfected N2a cells protein extracts. A–D, Western blot analyses with 3F4, pTyr, anti-pPrPS43, and β-actin on phospho-column fractionated proteins from PrP/Cdk5/p25 cotransfected N2a cells (A), roscovitine-treated PrP/Cdk5/p25 cotransfected N2a cells (B), PrP S43A/Cdk5/p25 cotransfected N2a cells (C), PrP-transfected N2a (D), or untransfected N2a cells (E).
Alkaline phosphatase-sensitive and proteinase K-resistant pPrPS43 immunoreactivity in scrapie-infected mice brains
To assess whether pPrPS43 could be part of the pathogenic process in scrapie infections, we immunostained coronal sections of mock-infected or 22A scrapie-infected mouse brains with the anti-pPrPS43 antiserum (Fig. 7). No immunoreactivity was detected in the mock-infected brains. However, widespread immunostaining was detected in the 22A-infected brains, especially in the medulla and thalamus region, which are the regions normally affected by the 22A strain. In contrast, no immunoreactivity was detected in the hypothalamus, which shows relatively little prion-associated pathology in this model. The pPrPS43 immunoreactivity was mostly located in diffuse deposits of PrPSc and in the cytoplasm of neurons (Fig. 8). Occasionally strong staining was also detected in the nucleus of some cells (Fig. 8). The anti-pPrPS43 immunoreactivity was eliminated with a pretreatment of the tissue sections with alkaline phosphatase, thus confirming detection of the phospho-epitope by the antiserum (Fig. 7). In contrast, the immunoreactivity to pPrPS43 was preserved in PK-treated tissue sections, indicating that pPrP is part of the pathogenic PrP. Immunoreactivity was completely absorbed with pPrPS43peptide and was not detected in the absence of primary antiserum. These results indicate that PKRES pPrPS43 is also present in infectious PrP diseases.
Figure 7.
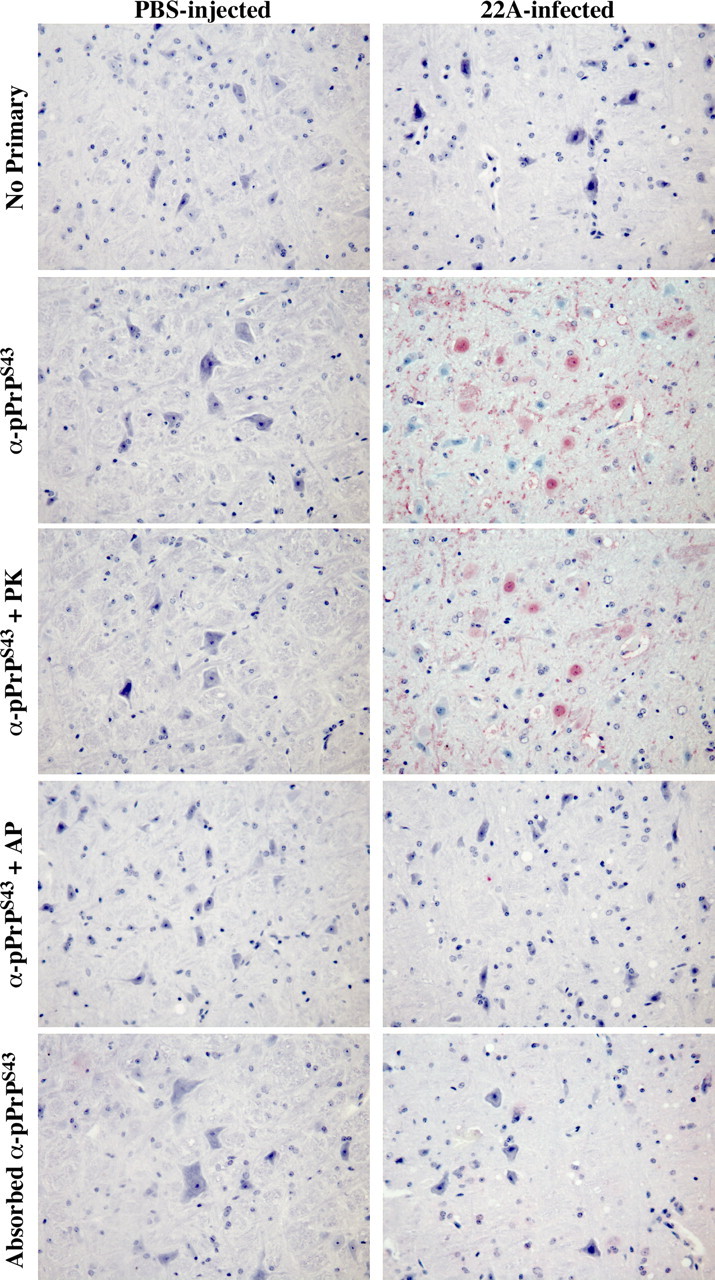
Immunohistological staining of control PBS- and 22A scrapie-infected mice brains. Micrographs of control PBS-injected or 22A-infected mice brain tissue sections of the medulla untreated (no primary, anti-pPrPS43, adsorbed anti-pPrPS43), pretreated with PK (anti-pPrPS43 + PK), or pretreated with alkaline phosphatase (anti-pPrPS43 + AP) with anti-pPrPS43, no primary antiserum, or adsorbed anti-pPrPS43.
Figure 8.

Cytosolic and nuclear pPrPS43 staining of 22A scrapie-infected mice brains. Higher magnification of micrographs of 22A-infected mice brain tissue sections from the thalamus or the medulla immunostained with anti-PrPS43.
Discussion
Here, we show that neuronal Cdk5 phosphorylates PrP at amino acid residue S43 and that this phosphorylation results in the conversion of PrP. That phosphorylated PrP is converted into a conformationally different form is evidenced by (1) PKRES of a 10 kDa phosphorylated PrP fragment, (2) the transformation of the phosphorylated PrP into amyloid structures that project an apple green birefringence under polarized light, and (3) the formation in time of fibrils detected by transmission electron microscopy. The conversion of PrP seems specific to Cdk5 phosphorylation at S43 because phosphorylation by CKII at S143 does not generate phosphorylated PKRES fragments of PrP.
The converted pPrP does not appear to have the ability to convert nonphosphorylated PrP into a 10 kDa PKRES PrP fragment in vitro. Although a small amount of PKRES 10 kDa PrP fragment is detected after 24 and 48 h of incubation of nonphosphorylated PrP seeded with sixfold less of the pPrP mix, it does not further amplify in time. Furthermore, consecutive cycles of dilutions and incubations did not amplify the 10 kDa fragment. However, as discussed below, the pPrP has the ability to enhance nonphosphorylated PrP aggregation. The lack of amplification of converted PrP in these experiments is consistent with the inability to convert bacterially purified PrP with scrapie prions in vitro (Deleault et al., 2005). Nevertheless, the conversion of PrP by Cdk5 phosphorylation provides a physiological molecular mechanism that could explain conversion in vivo. Alkaline phosphatase-sensitive and proteinase K-resistant pPrPS43 immunoreactivity is observed in the expected strain-specific pattern of PrPSc deposition in 22A scrapie-infected mice brains, indicating that phosphorylation of PrP occurs in pathological conditions. Phosphorylation is a well known mechanism of enzymatic activation through the induction of protein conformational change. Phosphorylation either changes the tertiary structure of globular proteins or has a direct effect on the dihedral backbone of the protein (Tholey et al., 1999). Because the prion protein N terminus is highly flexible (Donne et al., 1997; Zahn et al., 2000), it is possible that the phosphorylation at S43 provides a higher-ordered structure or allows electrostatic interactions with other amino acids intramolecularly or intermolecularly to generate PK resistance.
Although we could not observe an amplification of the 10 kDa pPrP fragment in nonphosphorylated PrP, the phosphorylation of PrP induced PKRES of nonphosphorylated PrP in vitro. These PKRES forms of PrP are likely produced by aggregation because of the coexistence, but not overlap, of the pPrPS43 and 6H4 epitopes in PrP protein aggregates detected by electron microscopy. Furthermore, pPrP promotes PKRES of full-length and 16 or 18 kDa fragments of PrP. Because the size of the nonphosphorylated PrP does not shift with PK treatment, these results infer that pPrP induces nonphosphorylated PrP aggregation rather than conversion. This assumption is further supported by a time-dependent increase in high-molecular-weight PrP aggregates and full-length PrP when recombinant nonphosphorylated PrP is seeded with sixfold less pPrP reaction mix and incubated at 37°C. Furthermore, serial propagation of pPrP, originally diluted 50- or 500-fold from the kinase assay, into recombinant nonphosphorylated PrP, resulted in the production of PKRES PrP after three and six cycles of incubation, respectively. Therefore, pPrP enhances aggregation rather than converts nonphosphorylated PrP. However, we cannot exclude the possibility that, over a very long period of time, the pPrP may actually convert nonphosphorylated PrP.
In transmissible prion diseases, PrP conversion is thought to occur either by template-directed conversion of PrP or by seeded nucleation (for review, see Aguzzi and Polymenidou, 2004). In template-directed conversion, the converted PrP molecule recruits and changes the conformation of normal PrP. In seeded nucleation, the conformationally abnormal protein recruits additional abnormal proteins to form a scaffold of abnormal proteins. However, the mechanism involved in the initial conversion of PrP is unknown. Phosphorylation may represent a physiological event that can originally convert PrP. The negative charges of the phosphate group may act in a manner similar to other anionic molecules, such as RNA, anionic lipid bicelles, low pH conditions, and synthetic poly-anions that have been shown to induce PrP conversion in vitro (Deleault et al., 2003, 2005, 2007; Supattapone, 2004; Geoghegan et al., 2007).
We cannot affirm that the pPrPS43 epitope in vivo is the result of only Cdk5 phosphorylation because other kinases, such as mitogen-activated protein kinase and GSK3β, may also phosphorylate this residue. However, given that Cdk5 is known as a neuronal kinase associated with several pathological events in neurodegenerative diseases, including translocation from the nuclei to the cytosol (Zhang et al., 2008), our results suggest neuronal specificity to this modification of PrP. In addition, PrP is phosphorylated at sites other than S43 and independently from Cdk5 in N2a cells. The three additional pPrP forms observed in N2a cells could represent alternative phosphorylation by other kinases or phosphorylated PrP that is differentially glycosylated or posttranslationally modified. Others have reported in vitro phosphorylation of bovine PrP with protein kinase C, CKII, and two tyrosine kinases, Lyn and c-Fgr (Negro et al., 2000). However, the effect of phosphorylation on PrP conformation was not reported. We have shown here that CKII phosphorylation of PrP does not induce PKRES. Additional investigations into the role of phosphorylation by the various kinases in either the normal PrP function or in the pathological PrP disease mechanism are warranted by these initial findings.
Phosphorylation-dependent conversion of non-enzyme proteins may be a general mechanism associated with neurodegenerative diseases. Phosphorylation is associated with conversion of α-synuclein and Tau protein in Parkinson disease and Alzheimer disease, respectively (Okochi et al., 2000; Fujiwara et al., 2002). Phosphorylated α-synuclein becomes PKRES and misfolded in disease (Neumann et al., 2002), and phosphorylated Tau resists calpain- and thrombin-mediated degradation (Litersky and Johnson, 1992; Arai et al., 2005). However, there is no direct evidence that phosphorylation induces a conformational change of these proteins. Our results with recombinant prion protein show that Cdk5-dependent phosphorylation is directly responsible for the conversion of prion protein. These results raise the possibility that other cytosolic proteins undergo a phosphorylation-dependent conformational change in disease.
In summary, we showed in this study an entirely physiological condition that could explain PrP conversion in prion diseases. This work has two important implications. First, the phosphorylated PrP epitope at S43 may be an excellent candidate for diagnostic purposes. Second, if phosphorylation of PrP at S43 is involved in the pathophysiology of disease, inhibitors of SP-directed kinases could be used as a therapeutic intervention against prion diseases.
Footnotes
This work was supported by the National Institutes of Health Grant 1RO1 NS40431, Canadian Institutes for Health Research Grant MOP-49594, and Fonds de Recherche en Santé du Québec (A.C.L.) and the Public Health Agency of Canada, the Canadian Biotechnology Strategy Fund: Genomics Initiative for Government Laboratories and Public Health Service and Prionet Canada (S.A.B.). We thank Dr. Witold Surewicz (Case Western Reserve University, Cleveland, OH) for providing recombinant PrP and Dr. Hajatollah Vali (Department of Anatomy and Cell Biology, McGill University, Montréal, Québec, Canada) for helping with the electron microscopy work.
References
- Aguzzi and Polymenidou, 2004.Aguzzi A, Polymenidou M. Mammalian prion biology: one century of evolving concepts. Cell. 2004;116:313–327. doi: 10.1016/s0092-8674(03)01031-6. [DOI] [PubMed] [Google Scholar]
- Arai et al., 2005.Arai T, Guo JP, McGeer PL. Proteolysis of non-phosphorylated and phosphorylated tau by thrombin. J Biol Chem. 2005;280:5145–5153. doi: 10.1074/jbc.M409234200. [DOI] [PubMed] [Google Scholar]
- Atarashi et al., 2007.Atarashi R, Moore RA, Sim VL, Hughson AG, Dorward DW, Onwubiko HA, Priola SA, Caughey B. Ultrasensitive detection of scrapie prion protein using seeded conversion of recombinant prion protein. Nat Methods. 2007;4:645–650. doi: 10.1038/nmeth1066. [DOI] [PubMed] [Google Scholar]
- Bessen et al., 1995.Bessen RA, Kocisko DA, Raymond GJ, Nandan S, Lansbury PT, Caughey B. Non-genetic propagation of strain-specific properties of scrapie prion protein. Nature. 1995;375:698–700. doi: 10.1038/375698a0. [DOI] [PubMed] [Google Scholar]
- Bocharova et al., 2006.Bocharova OV, Makarava N, Breydo L, Anderson M, Salnikov VV, Baskakov IV. Annealing prion protein amyloid fibrils at high temperature results in extension of a proteinase K-resistant core. J Biol Chem. 2006;281:2373–2379. doi: 10.1074/jbc.M510840200. [DOI] [PubMed] [Google Scholar]
- Bounhar et al., 2001.Bounhar Y, Zhang Y, Goodyer CG, LeBlanc A. Prion protein protects human neurons against Bax-mediated apoptosis. J Biol Chem. 2001;276:39145–39149. doi: 10.1074/jbc.C100443200. [DOI] [PubMed] [Google Scholar]
- Breydo et al., 2005.Breydo L, Bocharova OV, Makarava N, Salnikov VV, Anderson M, Baskakov IV. Methionine oxidation interferes with conversion of the prion protein into the fibrillar proteinase K-resistant conformation. Biochemistry. 2005;44:15534–15543. doi: 10.1021/bi051369+. [DOI] [PubMed] [Google Scholar]
- Caughey, 1993.Caughey B. Scrapie associated PrP accumulation and its prevention: insights from cell culture. Br Med Bull. 1993;49:860–872. doi: 10.1093/oxfordjournals.bmb.a072651. [DOI] [PubMed] [Google Scholar]
- Chiesa et al., 1998.Chiesa R, Piccardo P, Ghetti B, Harris DA. Neurological illness in transgenic mice expressing a prion protein with an insertional mutation. Neuron. 1998;21:1339–1351. doi: 10.1016/s0896-6273(00)80653-4. [DOI] [PubMed] [Google Scholar]
- Deleault et al., 2003.Deleault NR, Lucassen RW, Supattapone S. RNA molecules stimulate prion protein conversion. Nature. 2003;425:717–720. doi: 10.1038/nature01979. [DOI] [PubMed] [Google Scholar]
- Deleault et al., 2005.Deleault NR, Geoghegan JC, Nishina K, Kascsak R, Williamson RA, Supattapone S. Protease-resistant prion protein amplification reconstituted with partially purified substrates and synthetic polyanions. J Biol Chem. 2005;280:26873–26879. doi: 10.1074/jbc.M503973200. [DOI] [PubMed] [Google Scholar]
- Deleault et al., 2007.Deleault NR, Harris BT, Rees JR, Supattapone S. Formation of native prions from minimal components in vitro. Proc Natl Acad Sci U S A. 2007;104:9741–9746. doi: 10.1073/pnas.0702662104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donne et al., 1997.Donne DG, Viles JH, Groth D, Mehlhorn I, James TL, Cohen FE, Prusiner SB, Wright PE, Dyson HJ. Structure of the recombinant full-length hamster prion protein PrP(29–231): the N terminus is highly flexible. Proc Natl Acad Sci U S A. 1997;94:13452–13457. doi: 10.1073/pnas.94.25.13452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiwara et al., 2002.Fujiwara H, Hasegawa M, Dohmae N, Kawashima A, Masliah E, Goldberg MS, Shen J, Takio K, Iwatsubo T. alpha-Synuclein is phosphorylated in synucleinopathy lesions. Nat Cell Biol. 2002;4:160–164. doi: 10.1038/ncb748. [DOI] [PubMed] [Google Scholar]
- Geoghegan et al., 2007.Geoghegan JC, Valdes PA, Orem NR, Deleault NR, Williamson RA, Harris BT, Supattapone S. Selective incorporation of polyanionic molecules into hamster prions. J Biol Chem. 2007;282:36341–36353. doi: 10.1074/jbc.M704447200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilch et al., 2003.Gilch S, Wopfner F, Renner-Müller I, Kremmer E, Bauer C, Wolf E, Brem G, Groschup MH, Schätzl HM. Polyclonal anti-PrP auto-antibodies induced with dimeric PrP interfere efficiently with PrPSc propagation in prion-infected cells. J Biol Chem. 2003;278:18524–18531. doi: 10.1074/jbc.M210723200. [DOI] [PubMed] [Google Scholar]
- Grasbon-Frodl et al., 2004.Grasbon-Frodl E, Lorenz H, Mann U, Nitsch RM, Windl O, Kretzschmar HA. Loss of glycosylation associated with the T183A mutation in human prion disease. Acta Neuropathol. 2004;108:476–484. doi: 10.1007/s00401-004-0913-4. [DOI] [PubMed] [Google Scholar]
- Hsiao et al., 1990.Hsiao KK, Scott M, Foster D, Groth DF, DeArmond SJ, Prusiner SB. Spontaneous neurodegeneration in transgenic mice with mutant prion protein. Science. 1990;250:1587–1590. doi: 10.1126/science.1980379. [DOI] [PubMed] [Google Scholar]
- Jackson et al., 1999.Jackson GS, Hosszu LL, Power A, Hill AF, Kenney J, Saibil H, Craven CJ, Waltho JP, Clarke AR, Collinge J. Reversible conversion of monomeric human prion protein between native and fibrilogenic conformations. Science. 1999;283:1935–1937. doi: 10.1126/science.283.5409.1935. [DOI] [PubMed] [Google Scholar]
- Jodoin et al., 2007.Jodoin J, Laroche-Pierre S, Goodyer CG, LeBlanc AC. Defective retrotranslocation causes loss of anti-Bax function in human familial prion protein mutants. J Neurosci. 2007;27:5081–5091. doi: 10.1523/JNEUROSCI.0957-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kascsak et al., 1987.Kascsak RJ, Rubenstein R, Merz PA, Tonna-DeMasi M, Fersko R, Carp RI, Wisniewski HM, Diringer H. Mouse polyclonal and monoclonal antibody to scrapie-associated fibril proteins. J Virol. 1987;61:3688–3693. doi: 10.1128/jvi.61.12.3688-3693.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kocisko et al., 1994.Kocisko DA, Come JH, Priola SA, Chesebro B, Raymond GJ, Lansbury PT, Caughey B. Cell-free formation of protease-resistant prion protein. Nature. 1994;370:471–474. doi: 10.1038/370471a0. [DOI] [PubMed] [Google Scholar]
- Legname et al., 2004.Legname G, Baskakov IV, Nguyen HO, Riesner D, Cohen FE, DeArmond SJ, Prusiner SB. Synthetic mammalian prions. Science. 2004;305:673–676. doi: 10.1126/science.1100195. [DOI] [PubMed] [Google Scholar]
- Li et al., 2007.Li T, Chalifour LE, Paudel HK. Phosphorylation of protein phosphatase 1 by cyclin-dependent protein kinase 5 during nerve growth factor-induced PC12 cell differentiation. J Biol Chem. 2007;282:6619–6628. doi: 10.1074/jbc.M606347200. [DOI] [PubMed] [Google Scholar]
- Litersky and Johnson, 1992.Litersky JM, Johnson GV. Phosphorylation by cAMP-dependent protein kinase inhibits the degradation of tau by calpain. J Biol Chem. 1992;267:1563–1568. [PubMed] [Google Scholar]
- Lührs et al., 2006.Lührs T, Zahn R, Wüthrich K. Amyloid formation by recombinant full-length prion proteins in phospholipid bicelle solutions. J Mol Biol. 2006;357:833–841. doi: 10.1016/j.jmb.2006.01.016. [DOI] [PubMed] [Google Scholar]
- Ma and Lindquist, 2002.Ma J, Lindquist S. Conversion of PrP to a self-perpetuating PrPSc-like conformation in the cytosol. Science. 2002;298:1785–1788. doi: 10.1126/science.1073619. [DOI] [PubMed] [Google Scholar]
- Ma et al., 2002.Ma J, Wollmann R, Lindquist S. Neurotoxicity and neurodegeneration when PrP accumulates in the cytosol. Science. 2002;298:1781–1785. doi: 10.1126/science.1073725. [DOI] [PubMed] [Google Scholar]
- Mastrianni et al., 2001.Mastrianni JA, Capellari S, Telling GC, Han D, Bosque P, Prusiner SB, DeArmond SJ. Inherited prion disease caused by the V210I mutation: transmission to transgenic mice. Neurology. 2001;57:2198–2205. doi: 10.1212/wnl.57.12.2198. [DOI] [PubMed] [Google Scholar]
- Monari et al., 1994.Monari L, Chen SG, Brown P, Parchi P, Petersen RB, Mikol J, Gray F, Cortelli P, Montagna P, Ghetti B, et al. Fatal familial insomnia and familial Creutzfeldt-Jakob disease: different prion proteins determined by a DNA polymorphism. Proc Natl Acad Sci U S A. 1994;91:2839–2842. doi: 10.1073/pnas.91.7.2839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neary et al., 1991.Neary K, Caughey B, Ernst D, Race RE, Chesebro B. Protease sensitivity and nuclease resistance of the scrapie agent propagated in vitro in neuroblastoma cells. J Virol. 1991;65:1031–1034. doi: 10.1128/jvi.65.2.1031-1034.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negro et al., 2000.Negro A, Meggio F, Bertoli A, Battistutta R, Sorgato MC, Pinna LA. Susceptibility of the prion protein to enzymic phosphorylation. Biochem Biophys Res Commun. 2000;271:337–341. doi: 10.1006/bbrc.2000.2628. [DOI] [PubMed] [Google Scholar]
- Neumann et al., 2002.Neumann M, Kahle PJ, Giasson BI, Ozmen L, Borroni E, Spooren W, Müller V, Odoy S, Fujiwara H, Hasegawa M, Iwatsubo T, Trojanowski JQ, Kretzschmar HA, Haass C. Misfolded proteinase K-resistant hyperphosphorylated alpha-synuclein in aged transgenic mice with locomotor deterioration and in human alpha-synucleinopathies. J Clin Invest. 2002;110:1429–1439. doi: 10.1172/JCI15777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okochi et al., 2000.Okochi M, Walter J, Koyama A, Nakajo S, Baba M, Iwatsubo T, Meijer L, Kahle PJ, Haass C. Constitutive phosphorylation of the Parkinson's disease associated alpha-synuclein. J Biol Chem. 2000;275:390–397. doi: 10.1074/jbc.275.1.390. [DOI] [PubMed] [Google Scholar]
- Paudel et al., 1993.Paudel HK, Lew J, Ali Z, Wang JH. Brain proline-directed protein kinase phosphorylates tau on sites that are abnormally phosphorylated in tau associated with Alzheimer's paired helical filaments. J Biol Chem. 1993;268:23512–23518. [PubMed] [Google Scholar]
- Piccardo et al., 2001.Piccardo P, Liepnieks JJ, William A, Dlouhy SR, Farlow MR, Young K, Nochlin D, Bird TD, Nixon RR, Ball MJ, DeCarli C, Bugiani O, Tagliavini F, Benson MD, Ghetti B. Prion proteins with different conformations accumulate in Gerstmann-Straussler-Scheinker disease caused by A117V and F198S mutations. Am J Pathol. 2001;158:2201–2207. doi: 10.1016/S0002-9440(10)64692-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prusiner, 1982.Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science. 1982;216:136–144. doi: 10.1126/science.6801762. [DOI] [PubMed] [Google Scholar]
- Race et al., 1987.Race RE, Fadness LH, Chesebro B. Characterization of scrapie infection in mouse neuroblastoma cells. J Gen Virol. 1987;68:1391–1399. doi: 10.1099/0022-1317-68-5-1391. [DOI] [PubMed] [Google Scholar]
- Ricchelli et al., 2006.Ricchelli F, Buggio R, Drago D, Salmona M, Forloni G, Negro A, Tognon G, Zatta P. Aggregation/fibrillogenesis of recombinant human prion protein and Gerstmann-Straussler-Scheinker disease peptides in the presence of metal ions. Biochemistry. 2006;45:6724–6732. doi: 10.1021/bi0601454. [DOI] [PubMed] [Google Scholar]
- Roucou et al., 2003.Roucou X, Guo Q, Zhang Y, Goodyer CG, LeBlanc AC. Cytosolic prion protein is not toxic and protects against Bax-mediated cell death in human primary neurons. J Biol Chem. 2003;278:40877–40881. doi: 10.1074/jbc.M306177200. [DOI] [PubMed] [Google Scholar]
- Saborio et al., 2001.Saborio GP, Permanne B, Soto C. Sensitive detection of pathological prion protein by cyclic amplification of protein misfolding. Nature. 2001;411:810–813. doi: 10.1038/35081095. [DOI] [PubMed] [Google Scholar]
- Sokolowski et al., 2003.Sokolowski F, Modler AJ, Masuch R, Zirwer D, Baier M, Lutsch G, Moss DA, Gast K, Naumann D. Formation of critical oligomers is a key event during conformational transition of recombinant syrian hamster prion protein. J Biol Chem. 2003;278:40481–40492. doi: 10.1074/jbc.M304391200. [DOI] [PubMed] [Google Scholar]
- Supattapone, 2004.Supattapone S. Prion protein conversion in vitro. J Mol Med. 2004;82:348–356. doi: 10.1007/s00109-004-0534-3. [DOI] [PubMed] [Google Scholar]
- Swietnicki et al., 1997.Swietnicki W, Petersen R, Gambetti P, Surewicz WK. pH-dependent stability and conformation of the recombinant human prion protein PrP(90–231) J Biol Chem. 1997;272:27517–27520. doi: 10.1074/jbc.272.44.27517. [DOI] [PubMed] [Google Scholar]
- Swietnicki et al., 2000.Swietnicki W, Morillas M, Chen SG, Gambetti P, Surewicz WK. Aggregation and fibrillization of the recombinant human prion protein huPrP90–231. Biochemistry. 2000;39:424–431. doi: 10.1021/bi991967m. [DOI] [PubMed] [Google Scholar]
- Tateishi and Kitamoto, 1995.Tateishi J, Kitamoto T. Inherited prion diseases and transmission to rodents. Brain Pathol. 1995;5:53–59. doi: 10.1111/j.1750-3639.1995.tb00577.x. [DOI] [PubMed] [Google Scholar]
- Tholey et al., 1999.Tholey A, Lindemann A, Kinzel V, Reed J. Direct effects of phosphorylation on the preferred backbone conformation of peptides: a nuclear magnetic resonance study. Biophys J. 1999;76:76–87. doi: 10.1016/S0006-3495(99)77179-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torrent et al., 2004.Torrent J, Marchal S, Tortora P, Lange R, Balny C. High pressure, an alternative approach to understand protein misfolding diseases. Cell Mol Biol (Noisy-le-grand) 2004;50:377–385. [PubMed] [Google Scholar]
- Wang et al., 2007.Wang F, Yang F, Hu Y, Wang X, Wang X, Jin C, Ma J. Lipid interaction converts prion protein to a PrPSc-like proteinase K-resistant conformation under physiological conditions. Biochemistry. 2007;46:7045–7053. doi: 10.1021/bi700299h. [DOI] [PubMed] [Google Scholar]
- Xiong et al., 2001.Xiong LW, Raymond LD, Hayes SF, Raymond GJ, Caughey B. Conformational change, aggregation and fibril formation induced by detergent treatments of cellular prion protein. J Neurochem. 2001;79:669–678. doi: 10.1046/j.1471-4159.2001.00606.x. [DOI] [PubMed] [Google Scholar]
- Zahn et al., 2000.Zahn R, Liu A, Lührs T, Riek R, von Schroetter C, López García F, Billeter M, Calzolai L, Wider G, Wüthrich K. NMR solution structure of the human prion protein. Proc Natl Acad Sci U S A. 2000;97:145–150. doi: 10.1073/pnas.97.1.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang et al., 2008.Zhang J, Cicero SA, Wang L, Romito-Digiacomo RR, Yang Y, Herrup K. Nuclear localization of Cdk5 is a key determinant in the postmitotic state of neurons. Proc Natl Acad Sci U S A. 2008;105:8772–8777. doi: 10.1073/pnas.0711355105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang et al., 2002.Zhang Y, McLaughlin R, Goodyer C, LeBlanc A. Selective cytotoxicity of intracellular amyloid beta peptide1–42 through p53 and Bax in cultured primary human neurons. J Cell Biol. 2002;156:519–529. doi: 10.1083/jcb.200110119. [DOI] [PMC free article] [PubMed] [Google Scholar]