

Abstract
Presbycusis – age-related hearing loss – is the number one communicative disorder and a significant chronic medical condition of the aged. Little is known about how type II diabetes, another prevalent age-related medical condition, and presbycusis interact. The present investigation aimed to comprehensively characterize the nature of hearing impairment in aged type II diabetics. Hearing tests measuring both peripheral (cochlea) and central (brainstem and cortex) auditory processing were utilized. The majority of differences between the hearing abilities of the aged diabetics and their age-matched controls were found in measures of inner ear function. For example, large differences were found in pure-tone audiograms, wideband noise and speech reception thresholds, and otoacoustic emissions. The greatest deficits tended to be at low frequencies. In addition, there was a strong tendency for diabetes to affect the right ear more than the left. One possible interpretation is that as one develops presbycusis, the right ear advantage is lost, and this decline is accelerated by diabetes. In contrast, auditory processing tests that measure both peripheral and central processing showed fewer declines between the elderly diabetics and the control group. Consequences of elevated blood sugar levels as possible underlying physiological mechanisms for the hearing loss are discussed.
Keywords: Presbycusis, Age-related hearing loss, Cochlea, Auditory system, Aging, Geriatric, Metabolic, Insulin, Type II diabetes
1. Introduction
Type II diabetes mellitus (T2DM) is a prevalent age-related metabolic disorder affecting up to 7% of the population worldwide. Diabetes alters normal levels of blood glucose and insulin and their effects on intra- and extracellular biochemical signaling pathways in multiple physiological systems of the body. Individuals with T2DM often demonstrate a marked inability of cells to utilize insulin, as well as deregulated pancreatic insulin secretion patterns (Groop, 1999; Cavaghan et al., 2000). Although early stages of T2DM may have few symptoms, abnormal carbohydrate metabolism is present causing a rise in blood glucose levels (hyperglycemia), resulting in subclinical pathological changes. In many instances diabetic changes appear to mirror natural aging inducing accelerated negative health outcomes. Societal consequences of diabetes are very costly to our health care system (Songer, 1997).
The precise etiology of T2DM is not well-understood and previous research has linked the development of T2DM to both genetic and environmental causes (Bjorbaek et al., 1995; Vaaq et al., 1995; Polonsky et al., 1996; Jung, 1997). Although all aging individuals experience similar abnormal physiological processes, such as increased oxidation, glycation, and creation of metabolic by-products during oxidative metabolism, these processes appear to be more fast-paced in diabetics.
Prior research has demonstrated detrimental neurodegenerative outcomes in T2DM such as oxidative damage, increased apoptosis and intracellular calcium excitotoxicity. Dietary restriction can slow down or prevents some of these metabolic-related problems, and may help protect neurons from oxidative damage and apoptosis (Prolla and Mattson, 2001; Mattson et al., 2001). Many studies have contributed to a detailed understanding of pathologic changes that occur as major side effects of sustained hyperglycemia; namely, coronary artery disease, peripheral vascular disease, retinopathy, neuropathy, and nephropathy.
While many reports describe distinct processes of hyperglycemia and the cascade of its relevant intracellular and extracellular physiological consequences, and in spite of the fact that the inner ear is vulnerable to metabolic and circulatory stress, rigorous characterizations of the effects of T2DM on the auditory system are limited. Yet intriguingly, Sasso et al. (1999), in a study of type II diabetics across a wide age range of subjects, discovered significantly lower otoacoustic emissions amplitudes for diabetics relative to controls. They also noted a significant effect of the duration of the T2DM on hearing loss, corroborating a previous clinical study that employed pure-tone audiogram thresholds (Tay et al., 1995). They also found longer auditory brainstem response (ABR) latencies in the diabetics, but there was no significant correlation of the ABR latency changes with decreased emissions amplitudes. Erdem et al. (2003) noted that the nature of the hearing loss associated with T2DM was still controversial, and found mixed results comparing emissions amplitude in type II diabetics with controls. Other studies utilizing limited hearing measures and/or small numbers of subjects have revealed no differences in hearing loss for type II diabetics and their off-spring relative to controls (Malpas et al., 1989; Ma et al., 1998; Ologe et al., 2005).
Age-related hearing loss – presbycusis – is the number one communication disorder of our aged population and is one of the top three chronic medical conditions of the elderly. Human and animal model investigations have revealed many of the neural bases of this age-dependent sensory deficit (Willott et al., 1985, 1987, 1995; Willott and Erway, 1998; Willott, 1986; Hunter and Willott, 1987; Caspary et al., 1990, 1995, 1999; Willott, 1991; Frisina and Frisina, 1997; Miller et al., 1998; Seidman et al., 1997; Frisina, 2001a,b; Frisina et al., 2001; Frisina and Walton, 2001; Kim et al., 2002), but there are few reports where interactions between T2DM and presbycusis have been the major focus. The current study aimed to better characterize the nature and degree of hearing loss in human aged diabetics. Our study employed not only traditional auditory measures but novel hearing evaluations to more comprehensively determine major effects of type II diabetes on both the peripheral and central auditory processing systems.
2. Methods
2.1. Subjects
Volunteers were recruited for participation in a study of presbycusis. Those with a demonstrated history of noise damage and/or audiograms signifying noise damage were excluded, as were subjects exhibiting a middle ear hearing loss. Individuals who had been treated with ototoxic medications, exhibited serious medical health problems or neurological medical conditions, diagnosed with Meniere’s disease or labyrinthitis, failed cognitive screening tests (Mini-Mental Test), current/heavy smokers, or those with poor speech discrimination scores of 80% or less, were excluded as subjects.
The current study utilized 30 type II diabetic subjects (15 female and 15 male) ranging in age from 59 to 92 years (mean age = 73 years) who met the above inclusion/exclusion criteria, and a control group of 30 non-diabetic subjects, aged 59–88 yrs of age (mean age = 73 years), who were matched to the T2DM subjects by age, sex, and medical histories.
2.2. Auditory testing
The hearing test battery comprised of absolute threshold, temporal processing and speech perception measurements, evaluated multiple functions of the auditory system.
2.3. Pure tone audiometry
Speech-frequency absolute thresholds were obtained in a sound-proof room, using a Grason-Stadler GSI 61 clinical audiometer with E-A-R insert earphones at speech-frequencies between 0.25 and 8 kHz (0.25, 0.5, 1, 2, 3, 4, 6, 8 kHz) Ultra high hearing pure tone thresholds were determined with the same audiometer, at frequencies between 8 and 14 kHz utilizing Sennheiser HDA 200 headphones. A wide-band noise signal was also used in this study as a measure of absolute threshold.
2.4. Middle ear function
Bone conduction thresholds (GSI-61) were obtained at 0.25, 0.05, 1.0, 2.0, and 4.0 kHz. Tympanometric measures were performed using a Grason- Stadler GSI 33 middle ear analyzer. Bone conduction audiometry and tympanometry were employed to rule out existence of middle ear diseases that would have precluded obtaining valid measures of the inner ear and auditory brainstem functions.
2.5. Otoacoustic emissions
The otoacoustic emissions procedures consisted of sending broad band or pure tone stimuli into the inner ear to evoke a response in the form of an echo from the outer hair cells. The ILO 88 system for transient evoked (TEOAE) emissions and the ILO 92 system for distortion product emissions (DPOAE) were used in this study. All measurements were done in a sound-proof room. A standard ILO92 DPOAE probe was positioned in the subject’s ear canal using a manufacturer-approved ear tip. The TEOAE stimulus was a click delivered at a level of 84 dB SPL and capable of evoking responses from the inner ear between 0.5 and 6.0 kHz.
Unlike TEOAEs, DPOAE testing employs primary (f1) and secondary (f2) pure tone stimuli. Because of the inner ear’s non-linearity, the two-tone combinations presented simultaneously generate a third response called the distortion product (DP). Thus, DPOAEs reflect specific frequency responses of the inner ear as contrasted with an overall response that characterizes the TEOAE. For DPOAE measures, the ratio of f2/f1 was fixed at 1.22. The stimulus levels were held constant, at L1 = 70 dB SPL and L2 = 60 dB SPL. The 2f1 − f2 DPOAE amplitude as a function of frequency was recorded at four points per octave in the 1–6 kHz range in three steps/octave. DPs were considered to be present when they were at least 3 dB above the noise level (Lonsbury-Martin et al., 1990). Otodynamics DP software records the levels found in 12 neighboring frequencies and sets the significant levels for the DPOAE at two standard deviations above the mean noise level (for 95% confidence).
2.6. Gap detection
Temporal processing of acoustic transients was measured by determining the threshold for detecting brief silent gaps in an otherwise continuous sound (minimum gap threshold or MGT). Gaps were shaped with 1 ms cosine-squared rise-fall envelopes imbedded in 150 ms noise bursts randomly generated for each presentation (Tucker–Davis Technologies [TDT] AP2). Signal and gap durations were defined as the intervals between the half-amplitude points on the stimulus waveforms. The noise was digitized with upper cutoff frequencies of 1.0 or 4.0 kHz. The noise-bursts were transduced by a 16 bit D/A converter (TDT DD-1) attenuated (TDT PA4) to an overall level of 75 dB SPL, routed through a signal mixer (TDT SM3) and headphone buffer (TDT HB5) and then led to an insert earphone. This procedure resulted in two gap detection thresholds; one for the 1 kHz cutoff frequency and the other for the 4.0 kHz cutoff frequency.
2.7. Speech perception
A speech reception threshold (SRT) indicates the acuity with which a subject correctly perceives words of two-syllables of equal stress (Spondees). In quiet, this measure taps primarily the peripheral auditory system and indicates the lowest intensity level at which an individual is able to process words (threshold is defined as the 50% point on the psychometric function). The SRT, which correlates highly with the pure tone average of 0.5, 1.0, and 2.0 kHz, also served as a validating criterion for pure tone absolute threshold measurements.
2.8. Sentence perception
Sentences and the standardized spectrally shaped speech noise from the hearing in noise test (HINT, Nilsson et al., 1994) were digitized with a TDT AP-2 Array processor, transduced (TDT D/A Converter) and attenuated (TDT PA 4 and Grason-Stadler GSI -16) for use in the speech-in-quiet and speech-in-noise tasks. The HINT speech stimuli consisted of four lists of 20 sentences and three practice lists of 10 sentences.
The subject was seated one meter equidistant from three loudspeakers in (a) double-walled sound booth (Acoustical Systems RE243). Speech was presented at 0° azimuth in quiet (Q) and in each of the following three noise conditions: (1) in 65 dBA of noise located at 0° azimuth (N0), (2) at 90° azimuth (N90), and (3) at 270° azimuth (N270). An adaptive procedure (Levitt, 1971) without feedback was used to determine the 50% point on the psychometric function required for speech recognition thresholds. In the noise conditions noise onset preceded each sentence by 1 s and was turned o. 1 s after each sentence was completed. The calculation of the sentence speech reception threshold in quiet or signal-to-noise ratio (S/R) necessary for 50% sentence recognition was based on averaging the presentation levels of sentences 4 through 20 for each test list and 4 though 10 for the practice lists.
2.9. Medical histories
Health histories included attention to past and current medical diagnoses; past and current medications; surgical history; diet, smoking and exercise histories; work and recreational noise exposures; past and present auditory problems; ototoxic substance screening; and family history of hearing loss and major illnesses.
2.10. Statistics and data reduction
The HINT test, conducted in the free field (binaural/spatial processing) relied on both ears. The gap detection temporal processing task utilized the better ear. All other tests were monaural and presented to each ear independently, and data for the two ears were analyzed separately. For statistical analysis, individual frequency responses for some hearing tests, such as audiograms (PTA-1, -2, -3, -4) and DPOAEs (DP1-low, DP2- high), were grouped together for determining overall affects between subject groups. Data were analyzed with two-way analysis of variance (ANOVA), with subject groups as one factor, and dimension of the hearing test or ear as the other main factor. Statistical significance of the main effects and interactions were obtained, and post-hoc Bonferroni pairwise comparisons, power corrected for repeated pairwise testing, were performed to assess significance of differences between two groups (GraphPad Prism statistical software). Unless otherwise noted, error bars in the figures represent SEMs.
3. Results
Significant differences were found between the T2DM group and the control group in several of the auditory measures. The hearing deficits associated with T2DM were found principally in the peripheral auditory complex with fewer effects located at the more central levels of the auditory system. The results also demonstrated a consistent trend toward greater T2DM auditory processing deficits in the right ear as compared to the left ear.
3.1. Pure tone audiograms
Four pure tone average (PTA) groups were computed: PTA-1 (0.5, 1, 2 kHz), PTA-2 (1, 2, 4 kHz), PTA-3 (4, 8, 9 kHz), and PTA-4 (10, 11, 12, 14 kHz). For all PTAs, diabetics consistently needed more sound intensity for threshold detection (Table 1). Differences in average thresholds ranged from 7.9 to 12.3 decibels (dB) in the right ear, and from 2.7 to 10.7 dB in the left ear. The differences were most pronounced in the lower frequencies. Elevated thresholds of the right ear in diabetics were significantly higher than the left ear differences (Fig. 1, Table 1).
Table 1.
Pure-tone threshold averages (dB HL)
| Group | PTA 1 0.5/1/2 kHz |
PTA 2 1/2/4 kHz |
PTA 3 4/8/9 kHz |
PTA 4 10/11/12/14 kHz |
||||
|---|---|---|---|---|---|---|---|---|
| R | L | R | L | R | L | R | L | |
| Type 2 diabetics | 31.9 | 29.8 | 39.9 | 38.2 | 59.4 | 55.9 | 75.0 | 73.0 |
| Non-diabetics | 21.6 | 21.0 | 27.6 | 27.5 | 50.2 | 51.8 | 67.1 | 70.3 |
| Differences | 10.3 | 8.8 | 12.3 | 10.7 | 9.2 | 4.1 | 7.9 | 2.7 |
R: right ear, L: left ear.
Fig. 1.

For all frequencies tested, diabetics had higher tone thresholds than non-diabetic controls. Note that differences were greater at low frequencies, and were greater for the right ear compared to the left. See Table 1 for PTA frequency ranges. Right ear main effect: p < 0.0001, F = 30.8, df = 1; right ear post-hoc for PTA1: p < 0.05, t = 2.88; PTA2: p < 0.01, t = 3.44; PTA3: p < 0.05, t = 2.57, df = 1. Left ear main effect: p < 0.001, F = 13.8, df = 1; left ear post-hoc for PTA2: p < 0.05, t = 3.03, df = 1. Error bars are SEM.
3.2. Wide-band noise thresholds
Right and left ear wide-band noise thresholds demonstrated a highly significant difference between the T2DM subjects and the control group (Table 2). Noise threshold increases of the diabetic group were 19.9 dB in the right ear and 18.5 dB in the left. Post-hoc pair-wise analysis of thresholds for each ear also exhibited significant differences for wide-band noise thresholds between the two groups (Fig. 2). These results link consistently with the PTA findings presented above where the right ear was more affected by T2DM than the left ear.
Table 2.
Wide-band noise thresholds (dB HL)
| Group | R | L |
|---|---|---|
| Type 2 diabetics | 12.9 | 11.5 |
| Non-diabetics | −7.0 | −7.0 |
| Differences | 19.9 | 18.5 |
R: right ear, L: left ear.
Fig. 2.
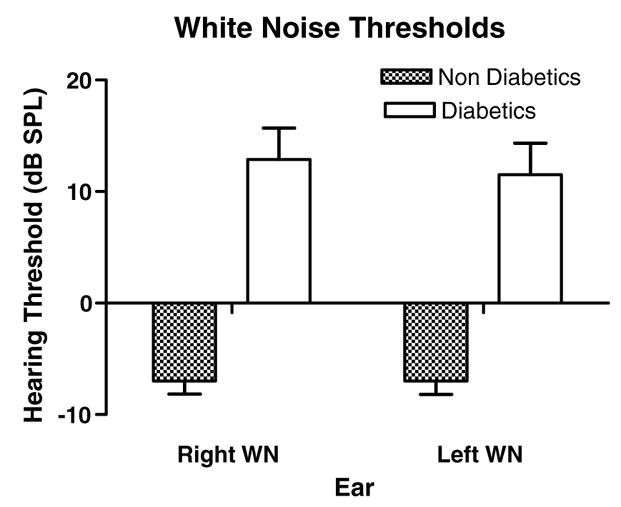
Diabetics showed a dramatic increase in white noise (WN) thresholds for both ears. Like pure tones, a greater difference was found for the right ear. ANOVA showed significant main effect for subject groups (p < 0.0001, F = 76.7, df = 1). Pairwise comparisons for each ear were also highly significant (right: p < 0.001, t = 6.42, df = 1; left: p < 0.001, t = 5.97, df = 1). Interactions were not significant. Error bars are SEM.
3.3. Distortion product otoacoustic emissions
Distortion-product otoacoustic emissions reflect cochlear mechanisms at the level of the outer hair cells. Right and left ears were tested independently using a series of tone pairs that generated distortion products whose geometric mean frequencies were: 1001, 1257, 1587, 2002, 2500, 3174, 4004, 5042, and 6748 Hz. Large amplitude responses indicate larger outer hair cell nonlinear responses. For data analyses, DPOAE frequencies were divided into two groups DP1 (1001, 1257, 1587, and 2002) and DP2 (3174, 4004, 5042, and 6748). A more pro-found damaging effect of T2DM was found for outer hair cells in the right ear as compared to the left (Fig. 3). Right and left ears displayed a significant decrease in DPOAE amplitudes. Amplitude differences between the T2DM group and control group were DP1: −5.5(R) and −3.2(L) dB, and DP2: −4.0(R) to −3.7(L) (Table 3).
Fig. 3.

At all frequencies, DPOAEs were smaller for diabetics relative to non-diabetics. Like the threshold measures presented in the previous figures, the right ear was more affected than the left. ANOVA showed significant main effects of subject group (right: p < 0.0001, F = 31.1, df = 1; left: p < 0.0001, F = 15.2, df = 1). Interactions and subject group Bonferroni post-hocs were not significant, except for the right ear at 2 kHz: p < 0.05, t = 2.82, df = 1. GM = geometric mean of f1 and f2. Error bars are SEM.
Table 3.
DPOAE amplitudes (dB SPL)
| Group | DP1: 1.0–2.0 kHz |
DP2: 3.2–6.8 kHz |
||
|---|---|---|---|---|
| R | L | R | L | |
| Type 2 diabetics | −7.0 | −5.2 | −14.5 | −14.1 |
| Non-diabetics | −1.5 | −2.0 | −10.5 | −10.4 |
| Differences | −5.5 | −3.2 | −4.0 | −3.7 |
R: right ear, L: left ear.
3.4. Transient otoacoustic emissions test
The transient otoacoustic emissions test (TEOAE) click stimulus stimulates a wide range of frequencies (similar to wide-band noise). Responses tend to be smaller in amplitude than DPOAEs because the energy of the signal is dispersed over the entire frequency range of the cochlea. Only 16 of the diabetic subjects showed amplitudes above the recording noise floor based on pass/fail criteria, as compared to 23 of non-diabetic subjects. However, trends were found between TEOAE amplitudes of diabetic and non-diabetic groups (Fig. 4), but these were not statistically significant (p = 0.07).
Fig. 4.
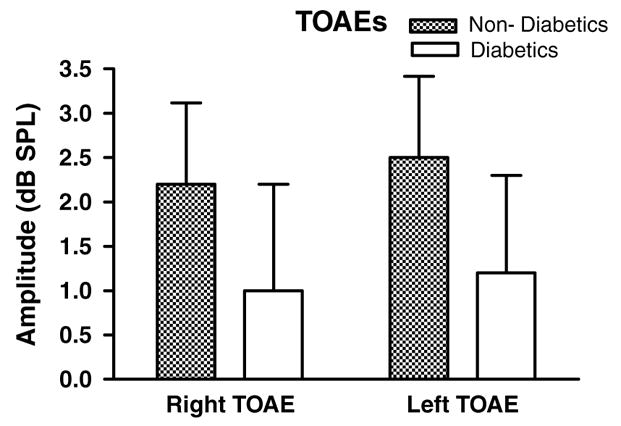
Similar to the DPOAEs, diabetics showed smaller TEOAEs amplitudes than non-diabetics. The differences were not statistically significant. Error bars are SEM.
3.5. Speech reception threshold test
Speech reception threshold test (SRT) utilizes two-syllable words (Spondees) as stimuli. This is a standard threshold test of sensitivity of the peripheral auditory system’s ability to process speech-frequency sounds. Fig. 5 illustrates a significant difference between diabetics and controls, and the post-hoc test was significant for the right ear as well. With a 10.2 dB increase in threshold for the right ear and 6.3 dB in the left ear, a continuation of the right vs. left theme was evident.
Fig. 5.
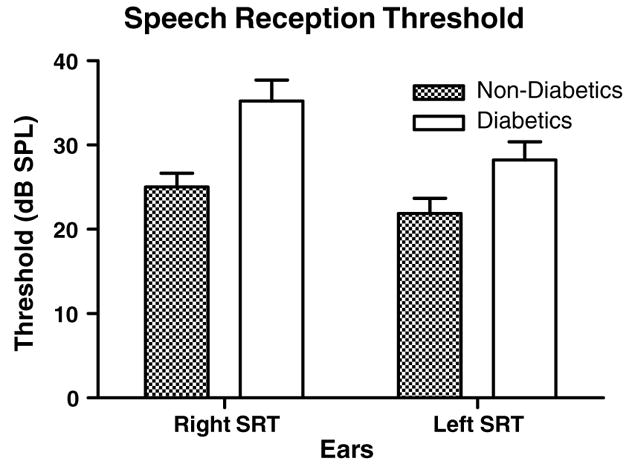
Although not as dramatic as the white noise threshold differences, speech reception thresholds were significantly lower for non-diabetic controls relative to diabetics. ANOVA showed significant main effect for subject groups (p < 0.0001, F = 16.5, df = 1). The pairwise comparison for the right ear was also significant (p < 0.05, t = 3.56, df = 1). Interactions and left ear differences were not significant. SRT: speech reception threshold. Error bars are SEM.
3.6. Hearing-in-noise-test (HINT)
The HINT is conducted in the free field and measures sentence processing when spoken in quiet and/or when the challenge of background noise emanating from different spatial locations is added. In quiet, diabetics had higher thresholds than controls (Table 4, p < 0.02, t = 2.52, df = 54). Noise added to speech challenges not only the peripheral system, but also the brainstem auditory system’s binaural neural networks used to distinguish sounds at different spatial locations. The more negative the S/N, the better the subject’s ability to understand speech in background noise. Normally, subjects are able to perform this test better when the speech and noise come from different spatial locations, especially when noise is presented to the left ear. This accounts for the significant main effect of noise location. For diabetics, this task becomes significantly more difficult than for controls (Fig. 6). The threshold of speech understanding under noisy conditions increased for diabetics by 1.6, 1.9, and 2.5 dB for 0°, 90°, and 270°, respectively (Table 4). Although these threshold differences appear small, each decibel improvement in S/N threshold represents a critical increase in speech recognition of up to 20% (Frisina and Frisina, 1997).
Table 4.
Hearing in noise thresholds (HINT) (dB S/N)
| Group | Quiet (dBA) | 0° | 90° | 270° |
|---|---|---|---|---|
| Type 2 diabetics | 43.3 | +0.7 | −1.0 | −1.7 |
| Non-diabetics | 37.2 | −0.9 | −2.9 | −4.2 |
| Differences | 6.1 | 1.6 | 1.9 | 2.5 |
Fig. 6.
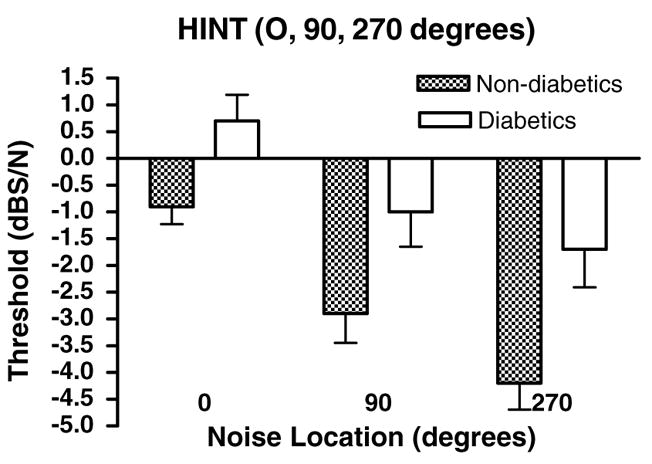
HINT speech thresholds were lower for non-diabetics than diabetics for all background noise speaker locations. HINT speech recognition in quiet was better for non-diabetics (not shown). ANOVA showed significant main effects of subject group (p < 0.0001, F = 20.5, df = 1) and background noise location (p < 0.0001, F = 14.3, df = 2). Bonferroni post-hoc tests for subject groups were significant for 90 (p < 0.05, t = 2.49, df = 1) and 270 (p < 0.01, t = 3.27, df = 1) degree background noise locations. Error bars are SEM.
3.7. Supra-threshold gap detection
The suprathreshold gap detection threshold test involves both peripheral and central auditory processing abilities with specificity to temporal processing. Individuals who score well on this test are able to discriminate extremely small time intervals (gaps) between two sounds. Findings with diabetic and non-diabetic subjects consistently showed shorter gap thresholds for controls relative to diabetics (Fig. 7).
Fig. 7.
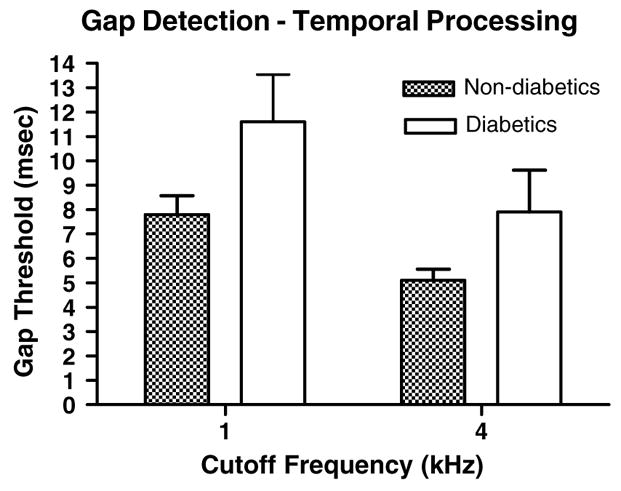
Diabetics had longer gap thresholds than controls (p < 0.02, F = 6.32, df = 1) and 4 kHz thresholds were shorter than 1 kHz thresholds (p < 0.02, F = 5.94, df = 1). Error bars are SEM.
4. Discussion
4.1. Type II diabetes and presbycusis
Significant differences between the hearing abilities of the aged T2DM subjects and their age-matched controls in the present investigation were found in auditory tests that measured inner ear function: pure-tone audiograms, wideband noise thresholds, speech reception thresholds, HINT-Quiet and otoacoustic emissions. The greatest deficits tended to be at low frequencies, where presbycusic high-frequency hearing losses were not yet affecting the control subjects, allowing for greater differences with the diabetics. Additionally, there was a strong tendency for diabetes to affect the right ear more than the left. One possible interpretation is that as one ages and develops presbycusis, the right ear advantage is lost (Tadros et al., 2005), and this age-related decline in the right ear advantage is accelerated by T2DM. Since T2DM damages vascular endothelial walls, and there are asymmetries in the blood supply to the right and left cochlea, it may be that in T2DM the vasculature to the right ear is more compromised with age.
The auditory perception tests that measure both peripheral and central auditory processing also showed differences between the elderly diabetics and controls. For example, speech processing in background noise (HINT) and gap detection thresholds displayed significant differences between diabetics and non-diabetics, suggesting that the CNS, like the inner ear, is susceptible to the damaging effects of the hyperglycemic conditions of diabetes.
4.2. Diabetic biochemical cascades – possible physiological bases of hearing impairment
Hyperglycemia initiates a complex cascade of biochemical changes within our body’s metabolic systems. Three main consequences of elevated blood sugar will be discussed: non-enzymatic glycation, activation of the polyol pathway and generation of reactive oxygen species (ROS). Glycation of low-density lipoproteins (LDLs) causes atherosclerosis. As glycation continues, advanced glycation end products (AGE) are formed, which modifies the functional morphology of blood vessels. Activation of the polyol pathway leads to accumulation of toxic intracellular substances, harming cell structures and metabolic processing, along with increased levels of ROS (Gries et al., 2003). Metabolic processes disrupted in the presence of diabetes include; energy production, abnormal accumulation of metabolic by-products (Stevens et al., 2000), nitric oxide deregulation (Williams et al., 1997), glycation (Matsumura et al., 2000), lipid balance abnormalities (Bucala et al., 1993), and protein synthesis dysfunction (Vlassara et al., 1995). Hyperglycemia causes widespread tissue damage, most specifically injuring endothelial (Vaughan, 2003), neural (Cameron et al., 1991; Stevens et al., 1996), extracellular matrix (Boyd-White and Williams, 1996) and collagen tissues (Charonis and Tsilbary, 1992; Haitoglou et al., 1992). The remainder of this discussion will highlight possible biological mechanisms that may be responsible for the auditory deficits demonstrated here.
The auditory system requires glucose and high-energy utilization for its complex signal processing. This suggests that the cochlea may also be a target organ for the ill effects of hyperglycemia. Increased glucose exposure, even for short periods, initiates a metabolic cascade that could disrupt the cochlea both anatomically and physiologically (see Fig. 8).
Fig. 8.

Flow diagram of possible biochemical pathways involved in the detrimental effects of type II diabetes on sensory end-organs such as the cochlea. See Section 4 for detailed explanations of these pathways and their likely effects on the auditory system.
4.3. Microangiopathy
Possibly, one of the most serious negative outcomes of hyperglycemia for auditory system functionality is that of microangiopathy. Research and clinical observations have documented important changes that occur in the visual system, including retinopathy (Brownlee et al., 1988; McCance et al., 1993). Hyperglycemia sequelae include increased metabolic by-products such a diacylglycerol (DAG) that activate the protein kinase C (PKC) gene family, affecting intracellular signal transduction pathways (Shiba et al., 1993; Nishizuka, 1995). PKC activity is increased in various diabetic animal model tissues including the heart, endothelium, vascular smooth muscle, glomerulus, retina, and sensory-motor endoneurial vasculature (Ways and Sheetz, 2000). Increased basement membrane thickening and porosity of the endothelium (Chapman et al., 1999) is a result of upregulation of vascular endothelial growth factor (VEGF) (Fong et al., 2003; Thieme et al., 1995; Williams et al., 1997), and increased vascular porosity due to increased vascular permeability factor (VPF). The cochlea, particularly its stria vascularis, is a highly micro-vascular-dependent organ. Increased permeability of the endothelium may lead to changes in auditory electrolyte homeostasis within the endolymph that interfere with hair cell transduction and signal transmission.
Nitric oxide resides in the organ of Corti, and plays an important role in regulation of vascular endothelium by ATP-induced increases in cochlear blood flow (Ren et al., 1997), anti-thrombotic activity, and regulation of vascular tone and cellular growth (Vane et al., 1990). Nitric oxide has been located in key cochlear blood vessels including the spiral modiolar, basilar membrane and spiral osseous lamina vessels (Shi and Nuttall, 2002; Si et al., 2002) as well as vessels in proximity to the spiral ganglion, and inner and outer hair cells (Gosepath et al., 2000). A critical balance of nitric oxide is crucial for optimal cochlear sensory and support cell health in the long term. For example, vasodilatory effects of nitrous oxide are impaired when hyperglycemia and its related AGEs trigger PKC activation and reduce the production of nitric oxide synthase (NOS) (Harrison et al., 1996; Kuboki et al., 2000). Auditory vascular perfusion may become limited in certain conditions requiring flexibility of blood vessels to provide adequate blood flow. Tabuchi et al. (2001) have reported that following in vitro ischemia in the guinea pig cochlea, DPOAE amplitudes decreased. When reperfused, the DPOAEs had a short-lived recovery with subsequent decrease in amplitude after 20 min. When nitric oxide inhibitors were administered this impairment was not seen. If these conditions are long-lasting, auditory sensory cell apoptosis may occur. Once ischemia is present a cascade of neural protective measures initiates increases in NOS1, NOS2, and NOS3. Increases in NOS1 result in elevated intracellular calcium which can cause excitotoxicity. Ischemia also initiates the increased production of superoxide, which combines with nitric oxide to form peroxynitrite a highly toxic anion thought to cause single strand DNA damage.
4.4. Cellular effects
Deposits of advanced glycation end-products (AGEs) occur in many cellular structures and diverse tissues. Changes resulting from hyperglycemia and its activation of the PKC enzyme involve AGE deposits into collagen, including Type IV (Mizisin and Powell, 2003). Type IV collagen is found in many areas of the peripheral auditory system including the tectorial membrane, basement membranes, stria vascularis and myelinated auditory nerve fibers, spiral ligament, spiral prominence, spiral limbus, scala media and epithelial cells (Satoh et al., 1998; Tsuprun and Santi, 2001). AGE deposits into affected collagen result in abnormal post-translational protein modifications including increased protein cross-linking. It can be hypothesized, that AGE deposits in the tectorial membrane, containing prominent collagen IV matrices, may become more fibrous and less flexible causing inarticulation with the outer hair cells, and deficits in sound transduction.
These cellular changes cause degraded cellular communication both within the cell itself and cell-to-cell. In vitro studies of the retina (Antcli. and Marshall, 1999; Oku et al., 2001) show that gap and tight junctions become displaced. If similar changes occur in the diabetic cochlea, the tight cellular junctions which create anatomical barriers between the perilymph and endolymph in the reticular lamina could be disrupted, jeopardizing cochlear ionic homeostasis.
In a study done by Lisowska et al. (2001) DPOAE amplitude was found to be significantly decreased in type I diabetics compared to controls. Interestingly, this decrease was seen regardless of the presence of microangiopathy, perhaps related to damage to other cellular structural elements such as tubulin. The macromolecule tubulin, responsible for microtubule formation is often affected by glycation causing reduced microtubule formation. Tubulin is present in inner and outer hair cells. Glycation of tubulin (Stitt and Vlassara, 2000) may cause anatomical and functional changes, processes that may render the hair cells less able to carry out sensory transduction.
Chronic hyperglycemic environments elicit pathophysiological changes to the nervous system as a result of endoneurial hypoxia, oxidative stress, nerve energy deficits, decreased Na/K/ATPase activity and decreased neurotrophism (Greene et al., 1993). Previous studies have shown that damage to myelin sheaths and other nerve components as a result of hyperglycemia occurs prominently in the peripheral nervous system (Mizisin and Powell, 2003), but also has been demonstrated in the spinal cord (Sato et al., 1999), cranial, optic (Amano et al., 2001) and vestibular nerves. One possible cause for this difference may be related to how myelin is glycosylated. In the peripheral nervous system major myelin proteins are glycosylated, perhaps making them more prone to the effects of AGE, while in the CNS major myelin proteins are not glycosylated. The glial margin demarcates the boundary of the peripheral and central auditory systems, and is located about halfway between the cochlea and the cochlear nucleus. These findings may explain why the peripheral auditory function was more affected than the central auditory nervous system in the present study.
4.5. Metabolic changes
Increases in glucose metabolic by-products can easily lead to toxicity, which is likely why approximately 50% of all type I diabetics are affected by neuropathy (Feldman et al., 2003). In addition, the enzyme Na/K/ATPase, present on cochlear cell membranes, is a sodium-potassium pump regulating intracellular ionic balance by expelling Na+ and importing K+. In diabetes, PKC is up regulated and binds with PX2 receptors on mammalian hair cells; activating a down-regulation of Na/K/ATPase causing elevated extracellular K+ and intracellular Na+ and Ca++ and excitotoxicity (Sweeney and Klip, 1998; Boue-Grabot et al., 2000).
Another important metabolic consideration in T2DM results from abnormal metabolism of glucose resulting in deregulation of the polyol pathway and generation of reactive oxygen species. As glucose is metabolized by using NADPH (Lee and Chung, 1999), sorbitol and fructose are formed (Brownlee, 1999). Fructose is a known powerful glycator (Szwergold et al., 1990), and activates several metabolic cascades including the efflux of taurine (Wright et al., 1986; Cameron and Cotter, 1997), which has anti-oxidant, free radical scavenger, osmoregualtor, neuromodualtor, and inhibitory neurotransmitter roles in many tissues throughout the body (Greene et al., 1992). Sorbitol oxidation by NAD+ changes the ratio of NADH to NAD+, therefore increasing the level of triose phosphate; a precursor to the reactive diacylglycerol and methylgloxal molecules (Williamson et al., 1993). Elevated intracellular sorbitol also increases oxidation of NADPH to NADP+. This biochemical pathway reduces the level of glutathione, a potent antioxidant (Lee and Chung, 1999) leaving the cell vulnerable to ROS damage.
5. Conclusion
Diabetes is a complex, systemic disease that can impact widespread body tissues and physiological functions, on molecular and biochemical levels. Opportunities exist for the further exploration of diabetes’ effects on sensory organs and their central neural processing pathways. As demonstrated in the present investigation, deleterious outcomes can occur in the ear and brain as diabetics age, suggesting that a more complete understanding of hyperglycemic effects on sensory organs could lead to earlier clinical involvement and the development of clinical testing that benefits earlier intervention and prevention of deleterious complications of T2DM diabetes for the aged.
Acknowledgments
Supported by NIH Grants P01 AG09524 from the Nat. Inst. on Aging, P30 DC05409 from the Nat. Inst. on Deafness & Communication Disorders, and the International Center for Hearing & Speech Research, Rochester, NY.
Abbreviations
- ABR
auditory brainstem response
- AGE
advanced glycation end-products of cellular respiration and metabolism
- ANOVA
analysis of variance
- Ca++
calcium ion
- df
degrees of freedom
- DPOAE
distortion product otoacoustic emission
- HINT
hearing in noise test, standardized
- K+
potassium ion
- MGT
minimum gap threshold
- Na+
sodium ion
- NOS
nitric oxide synthase
- PKC
protein kinase C
- PTA
pure tone average threshold
- SEM
standard error of the mean
- SRT
speech reception threshold
- T2DM
type II diabetes mellitus
- TEOAE
transient otoacoustic emission
- VEGF
vascular endothelial growth factor
- VPF
vascular permeability factor
- WN
wideband, or white noise
References
- Amano S, Kaji Y, Oshika T, Oka T, Machinami R, Nagai R, Horiuchi S. Advanced glycation end products in human optic nerve head. Br J Ophthalmol. 2001;85:52–55. doi: 10.1136/bjo.85.1.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antcliff RJ, Marshall J. The pathogenesis of edema in diabetic maculopathy. Sem Ophthalmol. 1999;14:223–232. doi: 10.3109/08820539909069541. [DOI] [PubMed] [Google Scholar]
- Bjorbaek C, Vik TA, Echwald SM, Yang PY, Vestergaard H, Wang JP, Webb GC, Richmond K, Hansen T, Erikson RL, et al. Cloning of human insulin stimulated protein kinase (ISPK-1) gene and analysis of coding regions and mRNA levels of the ISPK-1 and protein phospahte-1 genes in muscle from NIDDM patients. Diabetes. 1995;44:90–97. doi: 10.2337/diab.44.1.90. [DOI] [PubMed] [Google Scholar]
- Boue-Grabot E, Archambault V, Seguela P. A protein kinase C site highly conserved in P2X subunits controls the desensitization kinetics of P2XX(2) ATO-gated channels. J Biol Chem. 2000;275:10190–10195. doi: 10.1074/jbc.275.14.10190. [DOI] [PubMed] [Google Scholar]
- Boyd-White J, Williams JC., Jr Effects of cross-linking on matrix permeability: a model for AGE-modified basement membranes. Diabetes. 1996;45:348. doi: 10.2337/diab.45.3.348. [DOI] [PubMed] [Google Scholar]
- Brownlee M. Mechanisms of hyperglycemic damage in diabetes. In: Kahn CR, editor. Atlas of Clinical Endocrinology. Blackwell Science; New York: 1999. [Google Scholar]
- Brownlee M, Cerami A, Vlassara H. Advanced glycosylation end products in tissue and the biochemical basis of diabetic complications. N Engl J Med. 1988;318:1315. doi: 10.1056/NEJM198805193182007. [DOI] [PubMed] [Google Scholar]
- Bucala R, Makita Z, Koschinsky T, Cerami A, Vlassara H. Lipid advanced glycosylation: pathway for lipid oxidation in vivo. Proc Natl Acad Sci. 1993;90:6434–6438. doi: 10.1073/pnas.90.14.6434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron NE, Cotter MA. Metabolic and vascular factors in the pathogenesis of diabetic neuropathy. Diabetes. 1997;46:31S. doi: 10.2337/diab.46.2.s31. [DOI] [PubMed] [Google Scholar]
- Cameron NE, Cotter MA, Low PA. Nerve blood flow in early diabetes in rats: relation to conduction deficits. Am J Physiol. 1991;261:e1–e8. doi: 10.1152/ajpendo.1991.261.1.E1. [DOI] [PubMed] [Google Scholar]
- Caspary DM, Holder TM, Hughes LF, Milbrandt JC, McKernan RM, Naritoku DK. Age-related changes in GABAa receptor subunit composition and function in rat auditory system. Neuroscience. 1999;93:307–312. doi: 10.1016/s0306-4522(99)00121-9. [DOI] [PubMed] [Google Scholar]
- Caspary DM, Milbrandt JC, Helfert RH. Central auditory aging: GABA changes in the inferior colliculus. Exp Gerontol. 1995;30:349–360. doi: 10.1016/0531-5565(94)00052-5. [DOI] [PubMed] [Google Scholar]
- Caspary DM, Raza A, Armour BAL, Pippin J, Arneric SP. Immunocytochemical and neurochemical evidence for age-related loss of GABA in the inferior colliculus: implications for neural presbycusis. J Neurosci. 1990;10:2363–2372. doi: 10.1523/JNEUROSCI.10-07-02363.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavaghan MK, Ehrmann DA, Polonsky KS. Interactions between insulin resistance and insulin secretion in the development of glucose intolerance. J Clin Invest. 2000;106:329–333. doi: 10.1172/JCI10761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman T, McQueen MD, Baxter A, Smith T, Raynor E, Yoon SM, Prazma J, Pillsbury HC. Non-insulin-dependent diabetic microangiopathy in the inner ear. J Laryngol Otol. 1999;113:13–18. doi: 10.1017/s0022215100143051. [DOI] [PubMed] [Google Scholar]
- Charonis AS, Tsilbary EC. Structural and functional changes of laminin and type IV collagen after nonenzymatic glycation. Diabetes. 1992;41 (Suppl 2):49–51. doi: 10.2337/diab.41.2.s49. [DOI] [PubMed] [Google Scholar]
- Erdem T, Ozturan O, Miman MC, Ozturk C, Karatas E. Exploration of the early auditory effects of hyperlipoproteinemia and diabetes mellitus using otoacoustic emissions. Eur Arch Oto-Rhino-Laryngol. 2003;260:62–66. doi: 10.1007/s00405-002-0519-1. [DOI] [PubMed] [Google Scholar]
- Feldman EL, Stevens MJ, Russell JW, Greene DA. Somatosensory neuropathy. In: Porte D, Sherwin RS, Baron A, editors. Ellenberg and Rifkin’s Diabetes Mellitus. 6. McGraw-Hill; New York: 2003. pp. 185–187. [Google Scholar]
- Fong DS, Aiello L, Gardner TW, King GL, Blankenship G, Cavallerano JD, Ferris FL, Klein R. Diabetic retinopathy. Diabetes Care. 2003;26:226–229. doi: 10.2337/diacare.26.1.226. [DOI] [PubMed] [Google Scholar]
- Frisina RD. Anatomical and neurochemical bases of presbycusis. In: Hof PR, Mobbs CV, editors. Functional Neurobiology of Aging. Academic Press; San Diego: 2001a. pp. 531–547. Chapter 37. [Google Scholar]
- Frisina RD. Possible neurochemical and neuroanatomical bases of age-related hearing loss-presbycusis. Sem Hear: Innovations Aging Auditory Res. 2001b;22:213–225. [Google Scholar]
- Frisina DR, Frisina RD. Speech recognition in noise and presbycusis: relations to possible neural sites. Hear Res. 1997;106:95–104. doi: 10.1016/s0378-5955(97)00006-3. [DOI] [PubMed] [Google Scholar]
- Frisina RD, Walton JP. Aging of the mouse central auditory system. In: Willott JP, editor. Handbook of Mouse Auditory Res: From Behavior to Molecular Biology. CRC Press; New York: 2001. pp. 339–379. Chapter 24. [Google Scholar]
- Frisina DR, Frisina RD, Snell KB, Burkard R, Walton JP, Ison JR. Auditory temporal processing during aging. In: Hof PR, Mobbs CV, editors. Functional Neurobiology of Aging. Academic Press; San Diego: 2001. pp. 565–579. Chapter 39. [Google Scholar]
- Gosepath K, Heinrich UR, Ecke U, Maurer J, Amedee R, Mann WJ. Possible roles of nitric oxide in physiology and pathophysiology of the guinea pig cochlea. Eur Arch Oto-Rhino-Laryngol. 2000;257:418–424. doi: 10.1007/s004050000261. [DOI] [PubMed] [Google Scholar]
- Greene DA, Sima AA, Stevens MJ. Complications: neuropathy, pathogenetic considerations. Diabetes Care. 1992;15:1902–1925. doi: 10.2337/diacare.15.12.1902. [DOI] [PubMed] [Google Scholar]
- Greene DA, Sima AA, Stevens MJ, et al. Aldose reductase inhibitors: an approach to the treatment of diabetic nerve damage. Diabetes Met Rev. 1993;9:189–217. doi: 10.1002/dmr.5610090304. [DOI] [PubMed] [Google Scholar]
- Gries A, Herr A, Kirsch S, Gunther C, Weber S, Szabo G, Holzmann A, Bottiger BW, Martin E. Inhaled nitric oxide inhibits platelet-leukocyte interactions in patients with acute respiratory distress syndrome. Crit Care Med. 2003;31:1697–1704. doi: 10.1097/01.CCM.0000063446.19696.D3. [DOI] [PubMed] [Google Scholar]
- Groop LC. Insulin resistance: the fundamental trigger of T2 diabetes. Diabetes Obesity Metabol. 1999;1:S1–S7. doi: 10.1046/j.1463-1326.1999.0010s1001.x. [DOI] [PubMed] [Google Scholar]
- Haitoglou CS, Tsilbary EC, Brownlee M, Charonis AS. Altered cellular interactions between endothelial cells and non-enzymatically glycosylated laminin/type IV collagen. J Biol Chem. 1992;267:12404–12407. [PubMed] [Google Scholar]
- Harrison DG, Inoue N, Ohara Y, Fukai T. Modulation of endothelial cell nitric oxide synthase expression. Japan Circ J. 1996;60:815–821. doi: 10.1253/jcj.60.815. [DOI] [PubMed] [Google Scholar]
- Hunter KP, Willott JF. Aging and the auditory brainstem response in mice with severe or mild presbycusis. Hear Res. 1987;30:207–218. doi: 10.1016/0378-5955(87)90137-7. [DOI] [PubMed] [Google Scholar]
- Jung RT. Obesity and nutritional factors in the pathogenesis of non-insulin dependent diabetes mellitus. In: Pickup J, Williams G, editors. Textbook of Diabetes. 2. Blackwell Science; Oxford, UK: 1997. pp. 19.1–19.3. [Google Scholar]
- Kim SH, Frisina DR, Frisina RD. Effects of age on contralateral suppression of distortion-product otoacoustic emissions in human listeners with normal hearing. Audiol Neuro-Otol. 2002;7:348–357. doi: 10.1159/000066159. [DOI] [PubMed] [Google Scholar]
- Kuboki K, Jiang ZY, Takahara N, Ha SW, Igarashi M, Yamauchi T, Feener EP, Herbert TP, Rhodes CJ, King GL. Regulation of endothelial constitutive nitric oxide synthase gene expression in endothelial cells and in vivo: a specific vascular action of insulin. Circulation. 2000;101:676–681. doi: 10.1161/01.cir.101.6.676. [DOI] [PubMed] [Google Scholar]
- Lee AY, Chung SS. Contributions of polyol pathway to oxidative stress in diabetic cataract. FASEB J. 1999;13:23. doi: 10.1096/fasebj.13.1.23. [DOI] [PubMed] [Google Scholar]
- Levitt H. Transformed up-down methods in psychoacoustics. J Acoust Soc Am. 1971;49:476–477. [PubMed] [Google Scholar]
- Lisowska G, Namyslowski G, Morawski K, Strojek K. Cochlear dysfunction and diabetic microangiopathy. Scand Audiol. 2001;52:199–203. doi: 10.1080/010503901300007524. [DOI] [PubMed] [Google Scholar]
- Lonsbury-Martin BL, Harris FP, Stagner BB, Hawkins MD, Martin GK. Distortion product emissions in humans. I. Basic properties in normally hearing subjects. Ann Otol Rhinol Laryngol. 1990;(Suppl 147):3–14. [PubMed] [Google Scholar]
- Ma F, Gomez-Martin O, Lee DJ, Balkany T. Diabetes and hearing impairment in Mexican American adults: a population-based study. J Laryngol Otol. 1998;112:835–839. doi: 10.1017/s0022215100141842. [DOI] [PubMed] [Google Scholar]
- Malpas S, Blake P, Bishop R, Robinson B, Johnson R. Does autonomic neuropathy in diabetes cause hearing deficits? New Zeal Med J. 1989;102:434–435. [PubMed] [Google Scholar]
- Matsumura T, Yamagishi S, Brownlee M. Diabetes Mellitus: A Fundamental and Clinical Text. Lippincott Williams and Wilkins; Philadelphia, PA: 2000. Advanced glycation end-products and the pathogenesis of diabetic complications; pp. 983–990. [Google Scholar]
- Mattson MP, Duan W, Lee J, Guo Z. Suppression of brain aging and neurodegenerative disorders by dietary restriction and environmental enrichment: molecular mechanisms. Mech Aging Dev. 2001;122:757–778. doi: 10.1016/s0047-6374(01)00226-3. [DOI] [PubMed] [Google Scholar]
- McCance DR, Dyer DG, Dunn JA, Bailie KE, Thorpe SR, Baynes JW, Lyons TJ. Maillard reaction products and their relation to complications in insulin-dependent diabetes mellitus. J Clin Invest. 1993;91:2470–2478. doi: 10.1172/JCI116482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JM, Dolan DF, Raphael Y, Altschuler RA. Interactive effects of aging with noise induced hearing loss. Scan Audiol. 1998;27 (Suppl 48):53–61. [PubMed] [Google Scholar]
- Mizisin AP, Powell HC. Pathogenesis and pathology of diabetic neuropathy. In: Gries FA, Cameron NE, Low PA, Ziegler D, editors. Textbook of Diabetic Neuropathy. Theime; Stuttgart: 2003. p. 86. [Google Scholar]
- Nilsson M, Soli SD, Sullivan JA. Development of the hearing in noise test for the measurement of speech reception thresholds in quiet and in noise. J Acoust Soc Am. 1994;95 (2):1085–1099. doi: 10.1121/1.408469. [DOI] [PubMed] [Google Scholar]
- Nishizuka Y. Protein kinase C and lipid signaling for sustained cellular responses. FASEB J. 1995;9:484. [PubMed] [Google Scholar]
- Oku H, Kodama T, Sakagami K, Puro DG. Diabetes-induced disruption of gap junction pathways within the retinal microvasculature. 2001;42:1915–1920. [PubMed] [Google Scholar]
- Ologe FE, Okoro EO, Oyejola BA. Hearing function in Nigerian children with a family history of type II diabetes. Int J Ped Otorhinolaryngol. 2005;69:387–391. doi: 10.1016/j.ijporl.2004.11.009. [DOI] [PubMed] [Google Scholar]
- Polonsky KS, Sturis J, Bell GI. Non-insulin dependent diabetes mellitus-a genetically programmed failure of the beta cell to compensate for insulin resistance. N Engl J Med. 1996;334:777–783. doi: 10.1056/NEJM199603213341207. [DOI] [PubMed] [Google Scholar]
- Prolla TA, Mattson MP. Molecular mechanisms of brain aging and neurodegenerative disorders: lessons from dietary restriction. TINS. 2001;24:S21–S31. doi: 10.1016/s0166-2236(00)01957-3. [DOI] [PubMed] [Google Scholar]
- Ren T, Nuttall AL, Miller JM. ATP-induced cochlear blood flow changes involve the nitric oxide pathway. Hear Res. 1997;112:87–94. doi: 10.1016/s0378-5955(97)00109-3. [DOI] [PubMed] [Google Scholar]
- Sasso FC, Salvatore T, Tranchino G, Cozzolino D, Caruso AA, Persico M, Gentile S, Torella D, Torella R. Cochlear dysfunction in type 2 diabetes: a complication independent of neuropathy and acute hyperglycemia. Metabol Clin Exp. 1999;48:1346–1350. doi: 10.1016/s0026-0495(99)90141-5. [DOI] [PubMed] [Google Scholar]
- Sato Y, Kirmura M, Yasuda C, Nakano Y, Tomita M, Kobata A, Endo T. Evidence for the presence of major peripheral myelin glycoprotein PO in mammalian spinal cord and a change of its glycosylation state during aging. Glycobiology. 1999;9:655–660. doi: 10.1093/glycob/9.7.655. [DOI] [PubMed] [Google Scholar]
- Satoh H, Kawasaki K, Kihara I, Nakano Y. Importance of type IV collagen, laminin, and heparin sulfate proteoglycan in the regulation of labyrinthine fluid in the rat cochlear duct. Eur Arch Oto-Rhino-Laryngol. 1998;255:285–288. doi: 10.1007/s004050050060. [DOI] [PubMed] [Google Scholar]
- Seidman MD, Bai U, Khan MJ, Quirk WS. Mitochondrial DNA deletions associated with aging and presbycusis. Arch Otolaryngol- Head Neck Surg. 1997;123:1039–1045. doi: 10.1001/archotol.1997.01900100009001. [DOI] [PubMed] [Google Scholar]
- Shi X, Nuttall AL. The demonstration of nitric oxide in cochlear blood vessels in vivo and in vitro: the role of endothelial nitric oxide on venial permeability. Hear Res. 2002;172:73–80. doi: 10.1016/s0378-5955(02)00513-0. [DOI] [PubMed] [Google Scholar]
- Shiba T, Inoguchi T, Spartman JR, Heath WF, Bursell S, King GL. Correlation of diacylglycerol level and protein kinase C activity in rat retina to retinal circulation. Am J Physiol. 1993;265:E783–E793. doi: 10.1152/ajpendo.1993.265.5.E783. [DOI] [PubMed] [Google Scholar]
- Si JQ, Zhao H, Yang Y, Jiang ZG, Nuttall AL. Nitric oxide induces hyperpolarization by opening ATP-sensitive K+ channels in guinea pig spiral modiolar artery. Hear Res. 2002;171:167–176. doi: 10.1016/s0378-5955(02)00497-5. [DOI] [PubMed] [Google Scholar]
- Songer TJ. The economics of diabetes care: USA. In: Alberti KGMM, Zimmer P, DeFronzo RA, editors. International Textbook of Diabetes Mellitus. Vol. 2. John Wiley and Sons; Chichester: 1997. pp. 1762–1777. [Google Scholar]
- Stevens MJ, Lattimer SA, Feldman EL, et al. Acetyl-L carnitine deficiency as a cause of altered nerve myo-inositol content, Na+/K+/ATPase and motor conduction velocity in the streptozocin diabetic rat. Metabol Clin Exp. 1996;45:865–872. doi: 10.1016/s0026-0495(96)90161-4. [DOI] [PubMed] [Google Scholar]
- Stevens MJ, Obrosova I, Feldman EL, Greene DA. The sorbitol-osmotic and sorbitol-redox hypotheses. In: LeRoith D, Taylor SI, Olefsky JM, editors. Diabetes Mellitus: A Fundamental and Clinical Text. Lippincott Williams & Wilkins; Philadelphia PA: 2000. pp. 972–983. [Google Scholar]
- Stitt AW, Vlassara H. Advanced glycation end-products: impact on diabetic complications. In: Betteridge DJ, editor. Diabetes: Current Perspectives. Dunitz; London: 2000. pp. 67–68. [Google Scholar]
- Sweeney G, Klip A. Regulation of the Na+/K+/ATPase by insulin: why and how? Mol Cell Biochem. 1998;182:121–133. [PubMed] [Google Scholar]
- Szwergold BS, Kappler F, Brown TR. Identification of fructose-3-phosphatein the lens of diabetic rats. Science. 1990;247:451. doi: 10.1126/science.2300805. [DOI] [PubMed] [Google Scholar]
- Tabuchi K, Okubo H, Fujihira K, Tsuji S, Hara A, Kusakari J. Protection of outer hair cells from reperfusion injury by an iron chelator and nitric oxide synthase inhibitor in the guinea pig cochlea. Neurosci Lett. 2001;307:29–32. doi: 10.1016/s0304-3940(01)01919-x. [DOI] [PubMed] [Google Scholar]
- Tadros S, Frisina ST, Mapes F, Kim SH, Frisina DR, Frisina RD. Loss of peripheral right ear advantage in age-related hearing loss. Audiol Neuro-Otol. 2005;10:44–52. doi: 10.1159/000082307. [DOI] [PubMed] [Google Scholar]
- Tay HL, Ray N, Ohri R, Frootko NJ. Diabetes mellitus and hearing loss. Clin Otolaryngol Allied Sci. 1995;20:130–134. doi: 10.1111/j.1365-2273.1995.tb00029.x. [DOI] [PubMed] [Google Scholar]
- Thieme H, Aiello LP, Takagi H, Ferrara N, King GL. Comparative analysis of vascular endothelial growth factor receptors on retinal and aortic vascular endothelial cells. Diabetes. 1995;44:98–103. doi: 10.2337/diab.44.1.98. [DOI] [PubMed] [Google Scholar]
- Tsuprun V, Santi P. Proteoglycan arrays in the cochlear basement membrane. Hear Res. 2001;157:65–76. doi: 10.1016/s0378-5955(01)00278-7. [DOI] [PubMed] [Google Scholar]
- Vaaq A, Henriiksen JE, Madsbad S, Holm N, Beck-Nielsen H. Insulin secretion, insulin action and hepatic glucose production in identical twins discordant for non-insulin dependent diabetes mellitus. J Clin Invest. 1995;95:690–698. doi: 10.1172/JCI117715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vane JR, Anggard EE, Botting RM. Regulatory functions of the vascular endothelium. N Engl J Med. 1990;323:27–36. doi: 10.1056/NEJM199007053230106. [DOI] [PubMed] [Google Scholar]
- Vaughan DE. Endothelial dysfunction and vascular thrombosis in diabetes. In: Porte D, Sherwin RS, Baron A, editors. Ellenberg and Rifkin’s Diabetes Mellitus. 6. McGraw-Hill; New York: 2003. pp. 175–180. [Google Scholar]
- Vlassara H, Fuh H, Donnelly T, Cybulsky M. Advancedglycation end-products promote adhesion molecule (VCAM-1, ICAM-1) expression and atheroma formation in normal rabbits. Mol Med. 1995;1:447–456. [PMC free article] [PubMed] [Google Scholar]
- Ways DK, Sheetz MJ. The role of protein kinase C in the development of the complications of diabetes. Vitamins Hormones. 2000;60:149–193. doi: 10.1016/s0083-6729(00)60019-5. [DOI] [PubMed] [Google Scholar]
- Williams B, Gallacher B, Patel H, Orme C. Glucose-induced protein kinase C activation regulates vascular permeability factor mRNA expression of peptide production by vascular smooth muscles in cells in vitro. Diabetes. 1997;46:1497–1503. doi: 10.2337/diab.46.9.1497. [DOI] [PubMed] [Google Scholar]
- Williamson JR, Chang K, Frangos M, et al. Hyperglycemic pseudohypoxia and diabetic complications. Diabetes. 1993;42:801–813. doi: 10.2337/diab.42.6.801. [DOI] [PubMed] [Google Scholar]
- Willott JF. Effects of aging, hearing loss, and anatomical location on thresholds of inferior colliculus neurons in C57BL/6 and CBA mice. J Neurophysiol. 1986;56:391–408. doi: 10.1152/jn.1986.56.2.391. [DOI] [PubMed] [Google Scholar]
- Willott JF. Aging and the Auditory System. Singular; San Diego: 1991. Aging and the inner ear of animals; pp. 56–80. [Google Scholar]
- Willott JF, Erway LC. Genetics of age-related hearing loss in mice. IV Cochlear pathology and hearing loss in 25 BXD recombinant inbred mouse strains. Hear Res. 1998;119:27–36. doi: 10.1016/s0378-5955(98)00029-x. [DOI] [PubMed] [Google Scholar]
- Willott JF, Erway LC, Archer JR, Harrison D. Genetics of age-related hearing loss in mice. II Strain differences and effects of caloric restriction on cochlear pathology and evoked response thresholds. Hear Res. 1995;88:143–155. doi: 10.1016/0378-5955(95)00107-f. [DOI] [PubMed] [Google Scholar]
- Willott JF, Jackson LM, Hunter KP. Morphometric study of the anteroventral cochlear nucleus of two mouse models of presbycusis. J Comp Neurol. 1987;260:472–480. doi: 10.1002/cne.902600312. [DOI] [PubMed] [Google Scholar]
- Willott JF, Pankow D, Hunter KP, Kordyban M. Projections from the anterior ventral cochlear nucleus to the central nucleus of the inferior colliculus in young and aging C57Bl/6 mice. J Comp Neurol. 1985;237:545–551. doi: 10.1002/cne.902370410. [DOI] [PubMed] [Google Scholar]
- Wright CE, Tallan HH, Lin YY, et al. Taurine: biological update. Ann Rev Biochem. 1986;55:427–453. doi: 10.1146/annurev.bi.55.070186.002235. [DOI] [PubMed] [Google Scholar]