

Abstract
A structure-activity relationship for symmetrical cyanine inhibitors of human tau aggregation was elaborated using a filter trap assay. Antagonist activity depended on cyanine heterocycle, polymethine bridge length, and the nature of meso- and N-substituents. One potent member of the series, 3,3’-diethyl-9-methylthiacarbocyanine iodide (compound 11), retained submicromolar potency and had calculated physical properties consistent with blood-brain barrier and cell membrane penetration. Exposure of organotypic slices prepared from JNPL3 transgenic mice (which express human tau harboring the aggregation prone P301L tauopathy mutation) to compound 11 for one week revealed a biphasic dose response relationship. Low nanomolar concentrations decreased insoluble tau aggregates to half those observed in slices treated with vehicle alone. In contrast, high concentrations (≥300 nM) augmented tau aggregation and produced abnormalities in tissue tubulin levels. These data suggest that certain symmetrical carbocyanine dyes can modulate tau aggregation in the slice biological model at concentrations well below those associated with toxicity.
Introduction
Alzheimer’s disease (AD) is defined in part by the accumulation of aggregated tau proteins within neuronal cell bodies and processes.1, 2 Because tau aggregate formation correlates with cognitive decline and neurodegeneration,3, 4 inhibitors of aggregation are under investigation as a means of controlling pathogenesis and preserving neuronal function in disease.5 Several inhibitory scaffolds capable of supporting tau aggregation antagonist activity in vitro have been identified for this purpose, including (but not limited to) phenothiazine,6 triarylmethane,7 acridine,8 phenazine,8 xanthene,8 and thiacarbocyanine9 derivatives. All are polymethine members of the broad π-delocalized lipophilic cation (DLC) family that passively cross cell membranes and accumulate intracellularly depending in part on membrane polarization.10 A Phase II clinical trial completed with one phenothiazine having aggregation antagonist activity, Methylene blue (3,7-bis(dimethylamino)phenothiazin-5-ium chloride), reported decreased cognitive decline, improved cerebral blood flow, and improved fluorodeoxyglucose uptake relative to placebo over a ~1 year treatment period.11 Thus at least one tau aggregation antagonist may have therapeutic utility for AD. However, the actions of Methylene blue in vivo are not limited to antagonism of tau aggregation. Methylene blue efficiently enters into cyclic redox reactions, during which it is reversibly reduced to Leucomethylene blue,12 and this cycling may contribute to its biological activity.13 Thus while Methylene blue is an oxidant capable of modulating the activity of both heme- and non-heme containing enzymes (including nitric oxide synthase14), Leucomethylene blue has antioxidant activity and is the active form of the dye during treatment of methemoglobinemia.15 Moreover, as a DLC, Methylene blue accumulates in mitochondria where it and its reduced form may exert antioxidant effects through interactions with resident electron transport chains.12 It is not clear whether the therapeutic benefits of Methylene blue stem from its aggregation antagonist activity, its antioxidant effects in mitochondria, its redox modulation of iron-containing proteins, or from other effects.
Recently we introduced thiacarbocyanines as structurally distinct members of the DLC family possessing tau aggregation antagonist activity.9 The scaffold has several advantages for validating tau aggregation as a therapeutic target. First, certain symmetrical thiacarbocyanines, such as N744 (compound 1), are among the most potent tau aggregation inhibitors yet described in vitro, with submicromolar IC50 under near physiological buffer conditions and tau concentrations.9 Second, because cyanines lack an electron sink analogous to the thiomorpholine ring of Methylene blue, they have much lower reduction potentials16 and do not enter readily into cycling redox reactions. Third, many carbocyanine derivatives have been prepared over the years, offering a facile route to building SARs. Finally, as with other DLCs, carbocyanine dyes are readily taken up and concentrated in cells where they can contact tau. On the other hand, carbocyanines also present special handling difficulties. First, they are the quintessential aggregation-prone dyes, and readily form face-to-face dimers and higher order aggregates at low concentration.17 Dye aggregation is a major cause of nonspecific inhibitory effects on unrelated biological targets,18 which can complicate target validation. Second, many carbocyanines form stable complexes with DNA at micromolar concentrations through groove binding19 and at high concentrations (~100 μM) through direct intercalation.20 These interactions could potentially lead to genotoxicity through frameshift mutagenesis or chromosome breakage. Finally, carbocyanines inhibit various enzymes including DNA primase21 and electron transport complex I (NADH-ubiquinone reductase) of mitochondria.22-24 Any or all of these actions may contribute to the cytoxicity reported with carbocyanine exposure, including collapse of the microtubule cytoskeleton25 and induction of apoptosis.21, 26
Here we identify the structural features of carbocyanine dyes responsible for tau aggregation inhibitory activity, and demonstrate the activity of one family member in organotypic slice culture. The results suggest potent inhibition of tau aggregation can be achieved in vitro and in the biological model at low nanomolar concentrations. Furthermore, bioactivity can be separated from dye aggregation. The results suggest that carbocyanines may have utility for validating tau aggregation as a therapeutic target for AD.
Results
In vitro Activity
Symmetrical carbocyanine dyes consist of two identical heterocycles connected by a polymethine bridge (i.e., by a system of conjugated double bonds) (Table 1). Each heterocycle contains an alkyl N-substituent, whereas the bridge can contain hydrogen or other substituents (Table 1). To determine the contribution of these four structural components to tau aggregation antagonist activity, a small library of dyes was subjected to a filter binding assay for ODS-induced fibrillization of human 2N4R tau under near physiological conditions of pH, ionic strength, and reducing conditions. The first series (2 – 5, Table 1) evaluated the influence of polymethine bridge length on inhibitory activity. Bridge length influences the photophysical properties of cyanine dyes27 and is a major determinant of molecule length. The series contained identical ethyl N-substituents, only hydrogen bridge substituents, and two benzothiazole heterocycles, whereas the polymethine bridge was systematically varied in length from zero to three methine units (i.e., the bridges varied from one to seven carbons in length) (Table 1). All four compounds inhibited tau aggregation but with weaker potencies than starting compound 1 (Figure 1A). A replot of IC50 versus polymethine bridge length revealed a clear optimum centered on a bridge length of three carbons (Figure 1B). Therefore, further studies were limited to compounds containing a polymethine bridge of this length.
Table 1.
Summary of molecular properties of cyanine salts used in SAR studies.
 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Compd | X | n | R1 | R2 | R3 | Y | tPSAa | CLogPa | MWb | IC50 (μM) |
| 1 | S | 1 | -OCH3 | -CH2CH3 | -(CH2)2OH | I- | 67.7 | 1.38 | 485.6 | 0.29 ± 0.02 |
| 2 | S | 0 | H | --- | -CH2CH3 | I- | 8.8 | 1.94 | 339.5 | ~10c |
| 3 | S | 1 | H | H | -CH2CH3 | I- | 8.8 | 2.52 | 365.5 | 0.96 ± 0.25 |
| 4 | S | 2 | H | H | -CH2CH3 | I- | 8.8 | 3.04 | 391.6 | 2.15 ± 0.47 |
| 5 | S | 3 | H | H | -CH2CH3 | I- | 8.8 | 3.56 | 417.6 | 5.00 ± 3.30c |
| 6 | O | 1 | H | H | -CH2CH3 | I- | 35.1 | 1.24 | 333.4 | ≫10c |
| 7 | (CH=CH) | 1 | H | H | -CH2CH3 | Cl- | 8.8 | 2.4 | 353.5 | 0.89 ± 0.20 |
| 8 | C(CH3)2 | 1 | H | H | -CH2CH3 | I- | 6.2 | 3.52 | 385.6 | 0.98 ± 0.35 |
| 9 | C(CH3)2 | 1 | H | H | -CH2CH2CH3 | I- | 6.2 | 4.53 | 413.6 | 2.57 ± 1.06 c |
| 10 | S | 1 | H | H | -(CH2)5CH3 | I- | 8.8 | 6.67 | 477.7 | ≫10c |
| 11 | S | 1 | H | CH3 | -CH2CH3 | I- | 8.8 | 3.07 | 379.6 | 0.28 ± 0.04 |
| 12 | S | 1 | H | CH3 | -(CH2)3SO3- | d | 123.2 | -2.22 | 565.7 | ≫10c |
| 13 | S | 1 | e | CH3 | -CH2CH3 | Br- | 8.8 | 5.39 | 479.7 | 3.29 ± 2.07c |
| 14 | C(CH3)2 | 3 | f | H | -(CH2)4SO3- | Na+ | 120.6 | 2.13 | 752.0 | --- |
tPSA and CLogP values were calculated from the website http://www.molinspiration.com/
organic component of the salt.
Accurate estimates of SE could not be determined owing to incomplete concentration-effect relationship.
1-ethylpyridinium.
Naphthothiazole heterocyle
Naphthoindole heterocycle.
Figure 1. Polymethine bridge length modulates inhibitory activity.

Tau protein (4 μM) was incubated with ODS (50 μM) without agitation (24 h at 37°C) in the presence of either thiacarbocyanines 1 - 5 or DMSO vehicle alone, then assayed for aggregation by filter trap assay. (a) Concentration dependence of inhibition, where each point represents aggregation expressed as a normalized percentage of aggregation measured in the presence of DMSO vehicle alone (triplicate determination ± SD), and each solid line represents best fit of the data points to eq 4. All tested compounds were active, but less so than lead compound 1. (b) Replot of IC50 values determined in Panel (a) versus bridge length for compounds 2 - 5. Inhibitory potency was maximal when the polymethine bridge consisted of three carbon atoms. *, p < 0.05; **, p < 0.01 when compared with compound 3 by Student’s t test.
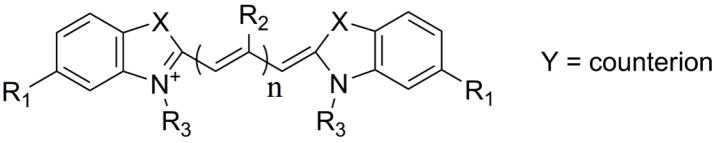
To evaluate the contribution of heterocycle composition, the second series (3, 6, 7, and 8) contained ethyl N-substituents and a hydrogen meso group in the presence of either benzothiazole, benzoxazole, dimethylindole, or quinoline heterocycles. Heterocycle composition influences compound polarizability and also ability to form H-bonds with solvent.28 All heterocycles were as potent and efficacious as benzothiazole 3 except benzoxazole 6, which had essentially no inhibitory activity at the highest concentration tested (Figure 2). These data show that inhibitory activity is not uniquely associated with the benzothiazole heterocycle, and can be extended to other heterocycles lacking sulfur.
Figure 2. Multiple heterocycles support inhibitory activity.
Tau protein (4 μM) was incubated with ODS (50 μM) without agitation (24 h at 37°C) in the presence of either 3, 6, 7, 8 (composed of benzothiazole, benzoxazole, quinoline, and dimethylindole heterocycles, respectively) or DMSO vehicle alone, then assayed for aggregation by filter trap assay. Each point represents normalized aggregation relative to the DMSO vehicle control (mean of triplicate determination ± SD), whereas each solid line represents best fit of the data points to eq 4. All heterocycles except benzoxazole supported inhibitory activity under these conditions.
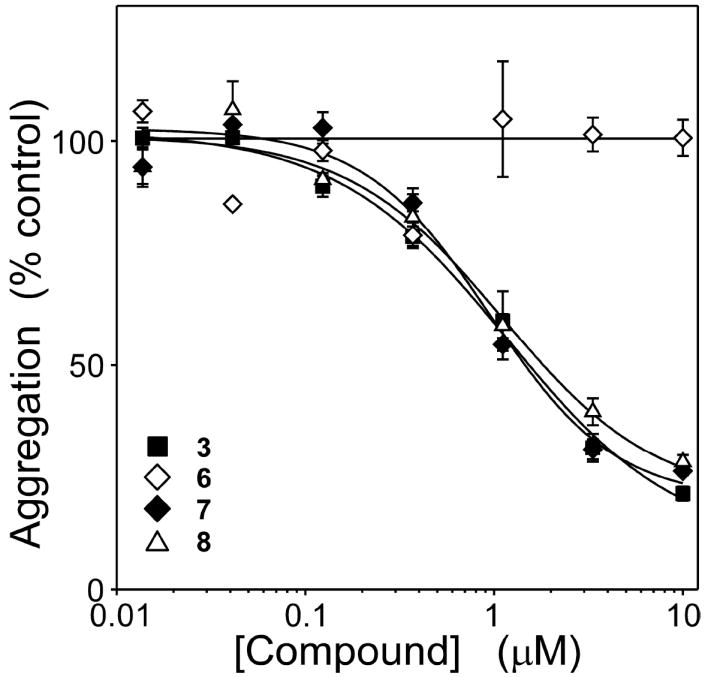
Cyanine N-substituents consist of alkyl or other groups (including ionizable groups) that do not participate in extended conjugated bond structure. Nonetheless, substituent structure influences compound pharmacokinetics by modulating binding affinity for serum albumin29 and by affecting net compound charge (and hence ability to serve as substrates for organic anion/cation transporters30). The contribution of N-substituent to inhibitory activity was evaluated in both benzothiazole and dimethylindole heterocycle-containing dyes. In the dimethylindole background, elongation of the alkyl chains from two carbons (8) to three carbons (9) reduced inhibitory potency ~3-fold (Figure 3). In a benzothiazole background, elongation of the alkyl chains from two carbons (3) to six carbons (10) completely eliminated activity at up to 10 μM concentration (Figure 3). These data indicate that short N-alkyl substituents are most compatible with inhibitory activity.
Figure 3. Meso- and N-substituents influence antagonist potency.
Tau protein (4 μM) was incubated with ODS (50 μM) without agitation (24 h at 37°C) in the presence of either thiacarbocyanines 9 - 13 or DMSO vehicle alone, then assayed for aggregation by filter trap assay. Each point represents normalized aggregation relative to the DMSO vehicle control (mean of triplicate determination ± SD), whereas each solid line represents best fit of the data points to eq 4. Data for compounds 1 and 3 are replotted from Figure 1 for comparison. Extension of the N-substituent beyond two carbons decreased potency, whereas introduction of a methyl meso substituent increased potency. The combination of benzothiazole nucleus, three carbon bridge, methyl meso substituent, and ethyl N-substituents yielded an inhibitor (compound 11) with the efficacy and potency of the starting compound 1, but with physicochemical properties more appropriate for biological experimentation.
Finally, the contribution of the meso substituent was investigated. Meso groups modulate the configuration of the polymethine bridge, with a hydrogen substituent favoring s-trans conformation whereas larger substituents favor s-cis conformation.31 The addition of a methyl meso substituent (11) retained efficacy while decreasing IC50 3-fold to ~300 nM (Figure 3). These data indicated that s-cis conformation was most compatible with inhibitory activity. Nonetheless, the beneficial effects of meso substitution were completely lost at all concentrations up to 10 μM when N-substituents were replaced with sulfonated propyl moieties (12) (Figure 3). Similarly, enlargement of the heterocycle to naphthothiazole in the presence of a methyl meso group (13) did not further increase potency above that attained with the benzothiazole heterocycle (Figure 3).
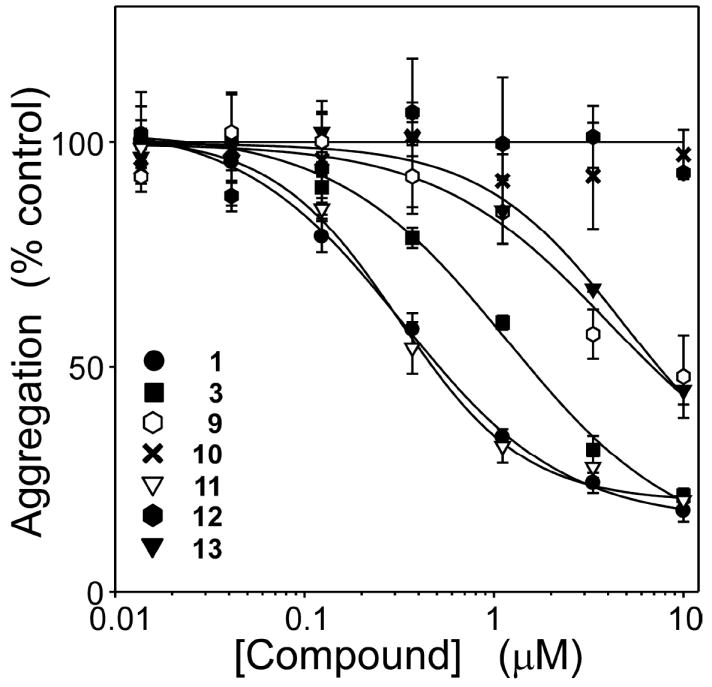
The SAR for symmetrical cyanine dyes summarized above indicates that it is possible to retain the potency and efficacy of parent compound 1, but with molecular weight, calculated octanol:water partition coefficient (ClogP), and topological polar surface area (tPSA) values more consistent with potential CNS penetration32 (Table 1). To confirm these findings, the most potent compound in the cyanine series (11) was assayed by an independent method (quantitative electron microscopy) and compared to established inhibitor Methylene blue. Results showed that the efficacy and potency of 11 was consistent in both filter trap and electron microscopy assay formats (Figure 4). In comparison, Methylene blue behaved as a partial antagonist with an IC50 of 610 ± 50 nM. These data indicate that cyanine inhibitors with superior in silico physical properties are available that exceed the potency and efficacy of Methylene blue in vitro.
Figure 4. Thiacarbocyanine 11 is a stronger aggregation antagonist than Methylene blue.
Tau protein (4 μM) was incubated with ODS (50 μM) without agitation (24 h at 37°C) in the presence of either 11, Methylene blue, or DMSO vehicle alone, then assayed for aggregation. Each point represents normalized aggregation relative to the DMSO vehicle control (mean of triplicate determination ± SD), whereas each line represents best fit of the data points to eq 4. Compound 11 was equally efficacious when assayed by either filter trap assay (○) or direct measurement of total filament length by quantitative electron microscopy assays (●), confirming that inhibitory activity did not depend on assay modality. However, 11 was significantly more potent and efficacious than Methylene blue (■) (electron microscopy assay format).
Dye Aggregation
Many planar heterocycles aggregate depending on pH, ionic strength, temperature, solvent polarity, and compound concentration.33 Symmetrical carbocyanine dyes are especially aggregation prone in water owing to strong dispersion forces between their nearly planar faces34 and H-bond formation with solvent.28 Shifts in absorbance spectra accompany thiacarbocyanine aggregation depending on the quaternary structure of the aggregate formed. Hypsochromic shifts to shorter wavelengths are generally referred to as H-bands.35 In solution, these correspond primarily to small aggregates (e.g., dimer) although much larger sizes can be attained at high concentration or in the presence of certain surfaces.36 In previous studies, formation of large H-aggregates correlated with loss of 1 inhibitory activity.17 However, substantial amounts of H-dimer were present at inhibitory concentrations suggesting these were candidate inhibitory species. To test this hypothesis, the concentration dependent aggregation of the carbocyanine dye series shown in Table 1 was assessed by absorbance spectroscopy and compared to the SAR for tau aggregation inhibition. In neat methanol (which does not support dye oligomerization27), 11 absorbance appeared as a major band at 544 nm with a weak vibrational shoulder at ~510 nm (Figure 5A). At low micromolar concentration in aqueous solution (i.e., assembly buffer), monomer appeared centered at 541 nm, consistent with the solvatochromic behavior of many thiacarbocyanine derivatives.37 As 11 concentration increased, a second absorbance maximum centered at 502 nm also became apparent. On the basis of absorption spectra of other thiacarbocyanine dyes,27 the 502 nm band corresponds to an H-dimer (Figure 5A). To confirm this prediction, the amount of monomer as a function of bulk 11 concentration was determined by absorbance spectroscopy and used to calculate the concentration of putative dimer by use of eq 1. A double log plot of monomer versus dimer concentration was linear with a slope of 2.3 ± 0.1, confirming that the reaction was consistent with dimerization (Figure 6). On the basis of eq 3, the dissociation equilibrium constant for dimerization (Kdim) was 20.9 ± 0.7μM. Thus, 11 readily dimerizes at low micromolar concentrations under near physiological buffer conditions, but at 10.0 ± 0.2-fold higher concentrations than 1 under identical conditions.38 These data suggest that dimerization can be dissociated from inhibitory activity. Indeed, certain cyanines with weak or no inhibitory activity, such as 5 (Kdim = 0.68 ± 0.04 μM) and 12 (Kdim = 7.5 ± 0.4 μM) dimerized more efficiently than 11 (Figs. 5B and 6). Conversely, other compounds, such as dimethylindocarbocyanine 8, inhibited tau aggregation at micromolar concentrations (Table 1) but did not dimerize at any concentration tested up to 96 μM (Figure 5C). Overall, there was no discernible correlation between dimerization propensity of compounds listed in Table 1 and inhibitory potency, suggesting that the active inhibitory species in the low concentration regime is the cyanine monomer.
Figure 5. Inhibitory potency does not correlate with cyanine aggregation propensity.
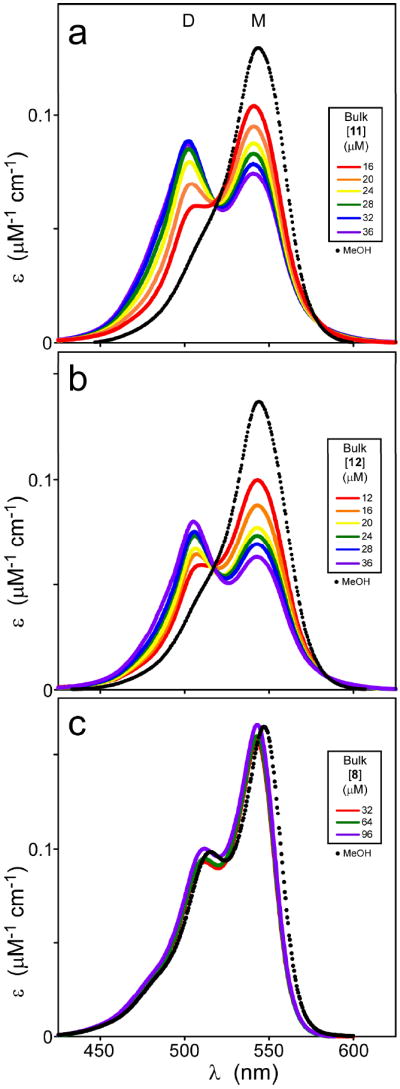
When dissolved in methanol at 5 μM concentration, compounds 11 (a), 12 (b), and 8 (c) yield simple spectra (dotted lines) consistent with monomeric structure (M). In assembly buffer (solid lines), increasing concentrations of compounds 11 and 12, but not 8, revealed the presence of both monomeric (M) and aggregated (D) species, with the latter more pronounced at high concentrations. These data suggest that 11 and 12, but not 8, underwent aggregation in aqueous solution at the concentrations tested.
Figure 6. The principal cyanine aggregate formed in solution is a dimer.
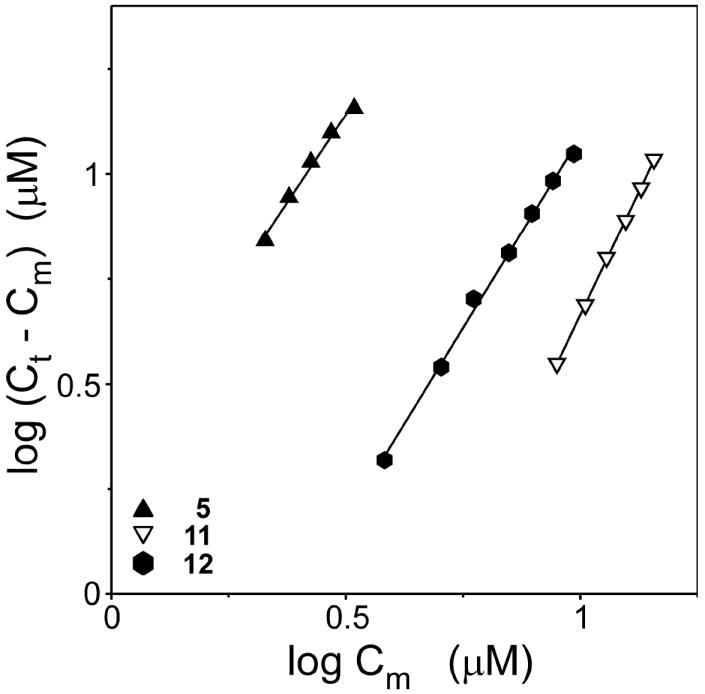
Concentrations of monomeric (Cm) and aggregate (Ct-Cm) forms of 11, 12, and 5 were calculated from absorbance spectra and replotted on double logarithmic scales. Each point represents a spectrum collected at a different bulk compound concentration, whereas each line represents best fit of the data points to a linear regression. Regression slopes for 11, 12, and 5 were 2.3 ± 0.1, 1.8 ± 0.1, and 1.7 ± 0.1, respectively, consistent with dimerization being mostly responsible for the spectral shifts of all three compounds.
Biological Activity
The in vitro SAR described above suggests that cyanine dyes should inhibit tau aggregation in biological models at sub-micromolar concentrations. To test this prediction, organotypic slices prepared from JNPL3 transgenic mice were exposed to a broad concentration range (0.1 nM – 1 μM) of 11 over a period of seven days after which levels of sarkosyl-insoluble tau were estimated. Compound 11 was chosen for study because it was the most potent member of the carbocyanine series. The JNPL3 model was selected as host because it accumulates tau aggregates in the diencephalon, brainstem, and cerebellar nuclei and is well characterized.39-43 The model is driven by expression of tau above normal physiological levels, which leads to increased nucleation and extension rates,44 and by insertion of the P301L missense mutation, which increases both the rate and extent of aggregation still further compared to wild-type tau protein.45 The effects of 11 in this model were biphasic (Figure 7AB). In the low concentration regime (1 – 10 nM) the compound decreased tau aggregation to ~50% of that observed in the presence of DMSO vehicle alone. Inhibitory activity was lost at 100 nM, whereas tau aggregation was induced at 300 nM and above.
Figure 7. Compound 11 inhibits tau aggregation in organotypic slices.

Slice cultures prepared from JNPL3 transgenic mice were incubated with either DMSO vehicle control or varying concentrations of 11 for seven days, then extracted and analyzed for levels of sarkosyl-insoluble tau (a measure of tau aggregation). (a) Two representative immunoblots collected after incubation with DMSO vehicle control (C) or thiacarbocyanine 11 (T) are shown. The presence of 1 nM 11 reduced the amount of tau aggregation in the slices relative to DMSO vehicle control, whereas 1 μM 11 exacerbated aggregation. (b) Sarkosyl-insoluble tau immunoreactivity was quantified by densitometry (n = three observations) and plotted as a normalized percentage of aggregation measured in the presence of DMSO vehicle alone. Tau aggregation was suppressed at 11 concentrations below 100 nM, but was aggravated at higher concentrations (≥300 nm). *, p < 0.05; **, p < 0.01 when compared to slices treated with vehicle alone by Student’s t test.
In cell culture models, >300 nM 11 reportedly initiates apoptosis21, 26 and collapse of the microtubule cytoskeleton.25 To determine whether 11 induced similar toxicity in neurons, organotypic slices exposed to 11 were assayed for changes in levels of cleaved PARP1 (an enzyme that is proteolytically inactivated by caspases during apoptosis)46 and α-tubulin (the structural component of microtubules). Levels of cleaved PARP1 were low in the presence of vehicle alone and did not increase at any tested concentration of 11, suggesting that proapoptotic caspases were not activated under these conditions (Fig. 8). Similarly, levels of α-tubulin were unchanged in cultures incubated with concentrations of 11 (1 nM) that inhibited tau aggregation (Fig. 8). However, at high 11 concentrations (1 μM), depletion of α-tubulin from extracts was detected, suggesting misfunction of the microtubule cytoskeleton under these conditions (Fig. 8). Together these data indicate that a cyanine identified on the basis of in vitro methods can modulate tau aggregation in a biological model, and is effective at concentrations well below those associated with toxicity in the form of induction of apoptosis or misfunction of the microtubule cytoskeleton.
Figure 8. Compound 11 does not induce apoptosis in organotypic slices.

Slices prepared from JNPL3 transgenic mice were incubated with either DMSO vehicle control or varying concentrations of 11 for seven days, then extracted and analyzed for levels of α-tubulin and cleaved PARP1. (a) Representative immunoblots collected after incubation with DMSO vehicle control (C) or 11 (T) are shown. (b) Levels of cleaved PARP1 (hollow bar) and α-tubulin (solid bar) immunoreactivity were quantified by densitometry (n = three observations) and plotted as a normalized percentage relative to treatment with vehicle alone. Cleaved PARP1 (CP) levels were unaffected after exposure to 11, suggesting that executioner caspase activity was not activated even at the highest concentration tested. Similarly, efficacious concentrations of 11 (1 nM) did not modulate levels of α-tubulin immunoreactivity in tissue extracts. However, high concentrations of 11 associated with induction of tau aggregation (1 μM) led to marked decreases in α-tubulin immunoreactivity. *, p < 0.05; **, p < 0.01 when compared to slices treated with vehicle alone by Student’s t test.
Discussion
Despite promising effects reported for Methylene blue on AD progression, the significance of tau aggregation as a therapeutic target requires further validation. The SAR summarized above suggests that cyanine dyes may have utility for this purpose. In particular, the availability of active and inactive cyanines sharing very similar structure will help clarify whether direct (herein) or indirect47 mechanisms mediate clearance of tau aggregates in biological models.
Both cyanines and Methylene blue consist of strong electron donor and acceptor substituents separated by a conjugated bridge composed of an odd number of carbon atoms. This organization supports the ideal polymethine state characterized by alternating π-electron density distribution along the molecular chain and by high polarizability.48 These properties may be important for tau aggregation inhibitory activity. Consistent with this hypothesis, the benzoxazole heterocycle, which had the lowest polarizability in the series (owing to the electronegative oxygen atom at position X; Table 1) lacked inhibitory activity. Polarizability also gives rise to strong dispersion forces that support efficient compound self association.48 However, as reported previously for a macrocyclic thiacarbocyanine,49 and through SAR here, the propensity for face-to-face association can be separated from inhibitory activity. Dimethylindocyanines, which contain geminal methyl groups that interfere with ordered stacking even in the presence of templates such as DNA,50 offer a scaffold that is particularly dimerization resistant. Elimination of stacking interactions with itself or with DNA may be important for limiting potential toxicity associated with chronic cyanine exposure.
One symmetrical carbocyanine, indocyanine green (14; Table 1), is approved for diagnostic applications,51, 52 but its molecular properties are not appropriate for tau aggregation inhibitor activity. In particular, its high molecular weight precludes efficient uptake into CNS, and its extended polymethine bridge, long sulfonated N-substituents, and hydrophobic benzindole heterocycles, confer tight binding to serum albumin. As a result, 14 resides primarily in blood plasma after administration.53 In contrast, the molecular properties associated with cyanine aggregation antagonists are predicted to support better CNS bioavailability than currently possible with 14. Nonetheless, it will be necessary to address the short half-life of cyanines in plasma. For example, compound 14 is rapidly cleared unchanged through the hepatobiliary route.54 It enters hepatocytes through organic anion-transporting polypeptides in the sinusoidal (basolateral) membrane, and is excreted through the canalicular (apical) membrane through the P-glycoprotein efflux transporter.55 Other cyanines, such as 6, share affinity for this efflux transporter,56 but not necessarily the passive transporters in the sinusoidal membrane owing to differences in net charge. Chemical strategies for decreasing the rate of 14 hepatic clearance are under investigation57 and may be applicable to tau aggregation inhibitors as well.
Compound 11 inhibited tau aggregation in organotypic slices at media concentrations as low as 1 nM. This high apparent potency may arise from several considerations. First, DLCs including cyanine dyes are readily taken up and concentrated in cells and tissues. For example, cultured K562 cells (a human erythroleukemic cell line established from a patient with chronic myelogenous leukemia in blast transformation) reportedly accumulate intracellular indo-, thia-, and oxo-carbocyanines two orders of magnitude over applied concentrations in media.58 These cells accumulate DLCs because their mitochondrial membrane potentials are hyperpolarized relative to non-transformed cells10, 59 and because they lack the P-glycoprotein efflux transporter.60 Second, cytoplasmic DLCs accumulate near membranes, which are putative sites of tau filament nucleation.61, 62 Thus, compound concentrations at local intracellular sites of action may be far higher than implied solely on the basis of bulk concentrations in media.
Compound uptake into organotypic brain slices may follow similar principles. In brain, the P-glycoprotein efflux transporter is predominantly located on the luminal membrane of endothelial cells lining brain microvessels associated with the blood-brain barrier. The slice preparation bypasses this barrier and facilitates direct exposure of neurons undergoing tau aggregation to compounds. Because neurons do not express P-glycoprotein under basal conditions,63 the slice preparation is particularly useful for assessing the activity of cyanines in neurons until analogs with improved pharmacokinetics are available.
In summary, symmetrical carbocyanines are more potent tau aggregation antagonists than Methylene blue. Their structures are consistent with favorable in silico pharmacokinetic predictions, whereas confounding activities such as dye aggregation, DNA intercalation, and high affinity serum albumin binding can be attenuated without destroying inhibitory potency. Therefore, these compounds may have utility for target validation. Organotypic slices prepared from transgenic mouse models will help this effort. In particular, their ability to support robust aggregation over a period of weeks while preserving CNS organization is ideally suited to development of intracellular protein aggregation antagonists.
Experimental Section
Materials
Recombinant His-tagged wild-type 2N4R tau (i.e., the longest human central nervous system isoform of tau64) was prepared as described previously.65 Mouse monoclonal antibodies Tau566 and CP2767 were gifts from L.I. Binder (Northwestern University, IL) and P. Davies (Albert Einstein College of Medicine, NY), respectively, whereas rabbit polyclonal anti-cleaved PARP1 and mouse monoclonal anti-α-tubulin antibodies were from Cell Signaling (Danvers, MA) and Sigma-Aldrich (St. Louis, MO), respectively. Nitrocellulose membrane (0.2 μM porosity) was from Bio-Rad Laboratories (Hercules, CA). Protease inhibitors (1× complete contained: 1 mM 4-(2-aminoethyl)-bezenesulfonylfluoride, 0.85 μM aprotinin, 40 μM bestatin, 20 μM leupeptin, 15 μM pepstatin A, 14 μM trans-Epoxysuccinyl-L-leucylamido[4- guanidino]butane) were from Sigma-Aldrich. Formvar/carbon-coated copper grids, glutaraldehyde, and uranyl acetate were obtained from Electron Microscopy Sciences (Fort Washington, PA). Aggregation inducer ODS was obtained from Lancaster Synthesis (Pelham, NH) and was dissolved in 1:1 water:isopropanol before use. Screened compounds included 1 9, 17, cyanines 2 - 13 (Sigma-Aldrich), and Methylene blue (Acros Organics, Morris Plains, NJ). All compounds, which were at least 95% pure on the basis of argentometric titration (5), high-performance capillary electrophoresis (6), thin-layer chromatography (8, 9, 10), or HPLC analysis (all others), were dissolved in DMSO prior to use.
Tau Aggregation in vitro
Tau preparations were incubated (37°C) without agitation in assembly buffer (10 mM HEPES, pH 7.4, 100 mM NaCl, 5 mM dithiothreitol) for 24 h in the presence or absence of fibrillization inducer ODS (50 μM) and candidate inhibitors. Control reactions contained DMSO vehicle, which was limited to 1% (v/v) final concentration in all aggregation reactions. After incubation, reactions were immediately assayed by either filter trap assay or electron microscopy as described below.
Filter Trap Assay
This was performed as described previously.45 Briefly, reactions were diluted in 2% SDS before vacuum filtration in triplicate through a 96-well dot blot apparatus onto nitrocellulose membranes. Membranes were washed twice with 2% SDS, blocked with 4% nonfat dry milk in blocking buffer (100 mM Tris-HCl, pH 7.4, and 150 mM NaCl) for 2 h, and incubated with Tau5 primary antibody for 1.5 h. They were then washed twice in blocking buffer and then incubated with HRP-linked secondary antibody for 1.5 h. The membranes were washed again in blocking buffer and developed with the ECL (Enhanced Chemiluminescence) Western Blotting Analysis System (GE Healthcare, Buckinghamshire, UK). Chemiluminescence was recorded on an Omega 12iC Molecular Imaging System and quantified using UltraQuant software (UltraLum, Claremont, CA) and is reported ± SD.
Electron Microscopy
Reaction aliquots were removed, treated with 2% glutaraldehyde (final concentration), mounted on formvar/carbon-coated 300 mesh grids, and negatively stained with 2% uranyl acetate as described previously.68 Random fields were viewed with a Tecnai G2 Spirit BioTWIN transmission electron microscope (FEI, Hillsboro, OR) operated at 80kV and 23,000-49,000× magnification. At least three viewing fields were captured for each reaction condition and filaments >10 nm in length were counted and quantified with ImageJ software (National Institutes of Health, Bethesda, MD). Total filament length is defined as the sum of the lengths of all resolved filaments per field and is reported as ± SD.
Dye Aggregation
Compounds were incubated (1 h at 37°C) in assembly buffer, then subjected to absorbance spectroscopy (Varian Cary 50 Bio) over the wavelength range 400 - 700 nm. Concentrations of dye monomer (Cm) were estimated as described previously.27, 38 Briefly, the extinction coefficient for dye monomer was established in methanol, and then used to estimate monomer concentration in aqueous solutions on the basis of the Beer-Lambert law:
| (1) |
where A is absorbance, ε is the molar extinction coefficient, and l is the path length of the sample. Dimer concentration (Cd) was estimated from total dye concentration (Ct) assuming:27
| (2) |
The dissociation constant for dimerization (Kdim) was then calculated from the function:27
| (3) |
Kdim values were derived from at least five spectra and reported as mean ± S.D.
Organotypic Slice Culture
Brains were harvested from JNPL3 mice40 at postnatal day ten. After removal of cerebellum and brainstem, the cerebral cortex and hippocampus were sectioned at 400 μm as described previously.69 Slices from each hemisphere were then separated in ice cold buffer (pH 7.1), transferred to membrane inserts containing 0.4 μm pores (Laboratory Disposable Products; Wayne, NJ), and then maintained in culture for 14 days in media containing 25% serum (media was changed every two days). After the culture period, slices prepared from one hemisphere were treated with varying concentrations of compound for seven days, whereas slices prepared from the contralateral hemisphere were treated with DMSO vehicle alone (0.008% final concentration of DMSO in media). Thus, the vehicle-treated hemispheres served as within-animal controls for each treatment condition.
Tissue fractionation
Treated and control slices were separately pooled and homogenized by sonication in cold RIPA buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1 mM EDTA, 1 mM NaF, 1 mM Na3VO4, 1× protease inhibitors, 1μg/mL phosphatase inhibitors).70 Homogenates were then centrifuged at 20,000g at 4°C for 20 min. The resulting supernatant fractions (crude extracts) were collected and assayed for α-tubulin and cleaved PARP1 immunoreactivity using a Fujifilm LAS3000 imaging system.
To assess tau aggregation, extracts were diluted in RIPA buffer as necessary to achieve equivalent total protein levels. An aliquot of low-speed supernatant was incubated in 1% sarkosyl for 30 min at room temperature. Samples were then centrifuged for 1 h at 100,000g at 20°C. Supernatant was discarded and the pellet, termed the sarkosyl-insoluble fraction, was then resuspended in 20 μL O+ buffer (a modified O buffer71 composed of 62.5 mM Tris-HCl, pH 6.8, 10% glycerol, 5% 2-mercaptoethanol, 2.3% SDS, 1 mM EGTA, 1 mM EDTA, 1 mM PMSF, 1 mM Na3VO4, 1 mM NaF, 1× protease inhibitors), boiled for 3 min, then subjected to immunoblot analysis using monoclonal antibody CP27.
Analytical Methods
Concentration effect data from either filter or electron microscopy assays were normalized to DMSO vehicle control reactions and fit to the function:
| (4) |
where y and ymax represent the minimum and maximum aggregation measured in the presence and absence of inhibitor (at concentration x), respectively, n is the Hill coefficient, and IC50 is the concentration of inhibitor that results in 50% of maximal inhibition. IC50 values are reported ± standard error of the estimate.
Acknowledgments
We thank Dr. Jeremy D. Pettigrew, Ohio State University Department of Chemistry, for helpful discussions. This work was supported by National Institutes of Health grants NS047447 (K.E.D.) and AG14452 (J.K.).
Abbreviations used
- AD
Alzheimer’s disease
- CNS
central nervous system
- DLC
π-delocalized lipophilic cation
- ODS
octadecyl sulfate
- PARP1
Poly (ADP-ribose) polymerase-1
- RIPA
radioimmunoprecipitation assay
- SAR
structure-activity relationship
References
- 1.Ballatore C, Lee VM, Trojanowski JQ. Tau-mediated neurodegeneration in Alzheimer’s disease and related disorders. Nat Rev Neurosci. 2007;8:663–672. doi: 10.1038/nrn2194. [DOI] [PubMed] [Google Scholar]
- 2.Buee L, Bussiere T, Buee-Scherrer V, Delacourte A, Hof PR. Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res Brain Res Rev. 2000;33:95–130. doi: 10.1016/s0165-0173(00)00019-9. [DOI] [PubMed] [Google Scholar]
- 3.Congdon EE, Duff KE. Is tau aggregation toxic or protective? J Alzheimers Dis. 2008;14:453–457. doi: 10.3233/jad-2008-14415. [DOI] [PubMed] [Google Scholar]
- 4.Honson NS, Kuret J. Tau aggregation and toxicity in tauopathic neurodegenerative diseases. J Alzheimers Dis. 2008;14:417–422. doi: 10.3233/jad-2008-14409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kuret J. Detection and Reduction of Neurofibrillary Lesions. In: Smith HJ, Sewell RDE, Simons C, editors. Protein Folding Diseases: Enzyme Inhibitors and Other Agents as Prospective Therapies. Vol. 5. CRC Press, Taylor & Francis Books; Boca Raton, FL: 2007. pp. 287–324. [Google Scholar]
- 6.Wischik CM, Edwards PC, Lai RY, Roth M, Harrington CR. Selective inhibition of Alzheimer disease-like tau aggregation by phenothiazines. Proc Natl Acad Sci U S A. 1996;93:11213–11218. doi: 10.1073/pnas.93.20.11213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Honson NS, Johnson RL, Huang W, Inglese J, Austin CP, Kuret J. Differentiating Alzheimer disease-associated aggregates with small molecules. Neurobiol Dis. 2007;363:229–234. doi: 10.1016/j.nbd.2007.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wischik CM, Edwards PC, Harrington CR, Roth M, Klug A. Inhibition of tau-tau association. 6,953,794. U S Patent. 2005
- 9.Chirita CN, Necula M, Kuret J. Ligand-Dependent Inhibition and Reversal of Tau Filament Formation. Biochemistry. 2004;43:2879–2887. doi: 10.1021/bi036094h. [DOI] [PubMed] [Google Scholar]
- 10.Modica-Napolitano JS, Aprille JR. Delocalized lipophilic cations selectively target the mitochondria of carcinoma cells. Adv Drug Deliv Rev. 2001;49:63–70. doi: 10.1016/s0169-409x(01)00125-9. [DOI] [PubMed] [Google Scholar]
- 11.Wischik CM, Bentham P, Wischik DJ, Seng KM. Tau aggregation inhibitor (TAI) therapy with rember(tm) arrests disease progression in mild and moderate Alzheimer’s disease over 50 weeks. 11th International Conference on Alzheimer’s Disease; Chicago, IL. 2008. [Google Scholar]
- 12.Atamna H, Nguyen A, Schultz C, Boyle K, Newberry J, Kato H, Ames BN. Methylene blue delays cellular senescence and enhances key mitochondrial biochemical pathways. FASEB J. 2008;22:703–712. doi: 10.1096/fj.07-9610com. [DOI] [PubMed] [Google Scholar]
- 13.Buchholz K, Schirmer RH, Eubel JK, Akoachere MB, Dandekar T, Becker K, Gromer S. Interactions of methylene blue with human disulfide reductases and their orthologues from Plasmodium falciparum. Antimicrob Agents Chemother. 2008;52:183–191. doi: 10.1128/AAC.00773-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mayer B, Brunner F, Schmidt K. Inhibition of nitric oxide synthesis by methylene blue. Biochem Pharmacol. 1993;45:367–374. doi: 10.1016/0006-2952(93)90072-5. [DOI] [PubMed] [Google Scholar]
- 15.Yusim Y, Livingstone D, Sidi A. Blue dyes, blue people: the systemic effects of blue dyes when administered via different routes. J Clin Anesth. 2007;19:315–321. doi: 10.1016/j.jclinane.2007.01.006. [DOI] [PubMed] [Google Scholar]
- 16.Zigman S, Gilman P., Jr Inhibition of cell division and growth by a redox series of cyanine dyes. Science. 1980;208:188–191. doi: 10.1126/science.7361115. [DOI] [PubMed] [Google Scholar]
- 17.Congdon EE, Necula M, Blackstone RD, Kuret J. Potency of a tau fibrillization inhibitor is influenced by its aggregation state. Arch Biochem Biophys. 2007;465:127–135. doi: 10.1016/j.abb.2007.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Feng BY, Simeonov A, Jadhav A, Babaoglu K, Inglese J, Shoichet BK, Austin CP. A High-Throughput Screen for Aggregation-Based Inhibition in a Large Compound Library. J Med Chem. 2007;50:1385–2390. doi: 10.1021/jm061317y. [DOI] [PubMed] [Google Scholar]
- 19.Hannah KC, Armitage BA. DNA-templated assembly of helical cyanine dye aggregates: a supramolecular chain polymerization. Acc Chem Res. 2004;37:845–853. doi: 10.1021/ar030257c. [DOI] [PubMed] [Google Scholar]
- 20.Biver T, De Biasi A, Secco F, Venturini M, Yarmoluk S. Cyanine dyes as intercalating agents: kinetic and thermodynamic studies on the DNA/Cyan40 and DNA/CCyan2 systems. Biophys J. 2005;89:374–383. doi: 10.1529/biophysj.105.059790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li ZM, Liu ZC, Guan ZZ, Zhu XF, Zhou JM, Xie BF, Feng GK, Zhu ZY, Jiang WQ. Inhibition of DNA primase and induction of apoptosis by 3,3’-diethyl-9-methylthia-carbocyanine iodide in hepatocellular carcinoma BEL-7402 cells. World J Gastroenterol. 2004;10:514–520. doi: 10.3748/wjg.v10.i4.514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Anderson WM, Chambers BB, Wood JM, Benninger L. Inhibitory effects of two structurally related carbocyanine laser dyes on the activity of bovine heart mitochondrial and Paracoccus denitrificans NADH-ubiquinone reductase. Evidence for a rotenone-type mechanism. Biochem Pharmacol. 1991;41:677–684. doi: 10.1016/0006-2952(91)90066-e. [DOI] [PubMed] [Google Scholar]
- 23.Anderson WM, Delinck DL, Benninger L, Wood JM, Smiley ST, Chen LB. Cytotoxic effect of thiacarbocyanine dyes on human colon carcinoma cells and inhibition of bovine heart mitochondrial NADH-ubiquinone reductase activity via a rotenone-type mechanism by two of the dyes. Biochem Pharmacol. 1993;45:691–696. doi: 10.1016/0006-2952(93)90144-l. [DOI] [PubMed] [Google Scholar]
- 24.Anderson WM, Wood JM, Anderson AC. Inhibition of mitochondrial and Paracoccus denitrificans NADH-ubiquinone reductase by oxacarbocyanine dyes. A structure- activity study. Biochem Pharmacol. 1993;45:2115–2122. doi: 10.1016/0006-2952(93)90024-q. [DOI] [PubMed] [Google Scholar]
- 25.Lee C, Wu SS, Chen LB. Photosensitization by 3,3’-dihexyloxacarbocyanine iodide: specific disruption of microtubules and inactivation of organelle motility. Cancer Res. 1995;55:2063–2069. [PubMed] [Google Scholar]
- 26.Li ZM, Jiang WQ, Guan ZZ, Zhu XF, Zhou JM, Xie BF, Feng GK, Zhu ZY, Liu ZC. Apoptosis induced by DNA primase inhibitor 3,3’-diethyl-9-methylthia-carbocyanine iodide in human leukemia HL-60 cells. Acta Pharmaceutica Sinica. 2006;41:978–984. [PubMed] [Google Scholar]
- 27.West W, Pearce S. The dimeric state of cyanine dyes. J Phys Chem. 1965;69:1894–1903. [Google Scholar]
- 28.Vranchev DP, Andreev GN, Staneva TG, Krustev TA. Association processes of some polymethine compounds, studied in relation to their structural peculiarities. Bulg J Phys. 1978;5:607–612. [Google Scholar]
- 29.Tatikolov AS, Costa SM. Complexation of polymethine dyes with human serum albumin: a spectroscopic study. Biophys Chem. 2004;107:33–49. doi: 10.1016/S0301-4622(03)00218-7. [DOI] [PubMed] [Google Scholar]
- 30.Klaassen CD, Lu H. Xenobiotic transporters: ascribing function from gene knockout and mutation studies. Toxicol Sci. 2008;101:186–196. doi: 10.1093/toxsci/kfm214. [DOI] [PubMed] [Google Scholar]
- 31.West W, Pearce S, Grum F. Steroisomerism in Cyanine Dyes-Meso-Substituted Thiacarbocyanines. J Phys Chem. 1967;71:1316–1326. [Google Scholar]
- 32.Hou TJ, Xu XJ. ADME evaluation in drug discovery. 3. Modeling blood-brain barrier partitioning using simple molecular descriptors. J Chem Inf Comput Sci. 2003;43:2137–2152. doi: 10.1021/ci034134i. [DOI] [PubMed] [Google Scholar]
- 33.Murakami K. Thermodynamic and kinetic aspects of self-association of dyes in aqueous solution. Dyes and Pigments. 2002;53:31–43. [Google Scholar]
- 34.Herz AH. Adsorption of sensititzing dyes to silver halides. In: James TH, editor. Theory of the photographic Process. Fourth Edition. Macmillan, Inc; New York, NY: 1977. pp. 235–250. [Google Scholar]
- 35.Herz AH. Dye-Dye interactions of cyanines in solution and at silver bromide surfaces. Photogr Sci Eng. 1974;18:323–335. [Google Scholar]
- 36.Maskasky JE. Molecular-orientation of individual J aggregates on gelatin-grown AgBr tabular microcrystals. Langmuir. 1991;7:407–421. [Google Scholar]
- 37.Seifert JL, Connor RE, Kushon SA, Wang M, Armitage BA. Spontaneous assembly of helical cyanine dye aggregates on DNA nanotemplates. J Am Chem Soc. 1999;121:2987–2995. [Google Scholar]
- 38.Necula M, Chirita CN, Kuret J. Cyanine dye n744 inhibits tau fibrillization by blocking filament extension: implications for the treatment of tauopathic neurodegenerative diseases. Biochemistry. 2005;44:10227–10237. doi: 10.1021/bi050387o. [DOI] [PubMed] [Google Scholar]
- 39.Arendash GW, Lewis J, Leighty RE, McGowan E, Cracchiolo JR, Hutton M, Garcia MF. Multi-metric behavioral comparison of APPsw and P301L models for Alzheimer’s disease: linkage of poorer cognitive performance to tau pathology in forebrain. Brain Res. 2004;1012:29–41. doi: 10.1016/j.brainres.2004.02.081. [DOI] [PubMed] [Google Scholar]
- 40.Lewis J, McGowan E, Rockwood J, Melrose H, Nacharaju P, Van Slegtenhorst M, Gwinn-Hardy K, Paul Murphy M, Baker M, Yu X, Duff K, Hardy J, Corral A, Lin WL, Yen SH, Dickson DW, Davies P, Hutton M. Neurofibrillary tangles, amyotrophy and progressive motor disturbance in mice expressing mutant (P301L) tau protein. Nat Genet. 2000;25:402–405. doi: 10.1038/78078. [DOI] [PubMed] [Google Scholar]
- 41.Lin WL, Lewis J, Yen SH, Hutton M, Dickson DW. Ultrastructural neuronal pathology in transgenic mice expressing mutant (P301L) human tau. J Neurocytol. 2003;32:1091–1105. doi: 10.1023/B:NEUR.0000021904.61387.95. [DOI] [PubMed] [Google Scholar]
- 42.Lin WL, Lewis J, Yen SH, Hutton M, Dickson DW. Filamentous tau in oligodendrocytes and astrocytes of transgenic mice expressing the human tau isoform with the P301L mutation. Am J Pathol. 2003;162:213–218. doi: 10.1016/S0002-9440(10)63812-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lin WL, Zehr C, Lewis J, Hutton M, Yen SH, Dickson DW. Progressive white matter pathology in the spinal cord of transgenic mice expressing mutant (P301L) human tau. J Neurocytol. 2005;34:397–410. doi: 10.1007/s11068-006-8726-0. [DOI] [PubMed] [Google Scholar]
- 44.Congdon EE, Kim S, Bonchak J, Songrug T, Matzavinos A, Kuret J. Nucleation-dependent tau filament formation: the importance of dimerization and an estimation of elementary rate constants. J Biol Chem. 2008;283:13806–13816. doi: 10.1074/jbc.M800247200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chang E, Kim S, Yin H, Nagaraja HN, Kuret J. Pathogenic missense MAPT mutations differentially modulate tau aggregation propensity at nucleation and extension steps. J Neurochem. 2008;107:1113–1123. doi: 10.1111/j.1471-4159.2008.05692.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Oliver FJ, de la Rubia G, Rolli V, Ruiz-Ruiz MC, de Murcia G, Murcia JM. Importance of poly(ADP-ribose) polymerase and its cleavage in apoptosis. Lesson from an uncleavable mutant. J Biol Chem. 1998;273:33533–33539. doi: 10.1074/jbc.273.50.33533. [DOI] [PubMed] [Google Scholar]
- 47.Honson NS, Jensen JR, Abraha A, Hall GF, Kuret J. Small-molecule mediated neuroprotection in an in situ model of tauopathy. Neurotox Res. 2009;15:274–283. doi: 10.1007/s12640-009-9028-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dähne S. Color and Constitution: One Hundred Years of Research. Science. 1978;199:1163–1167. doi: 10.1126/science.199.4334.1163. [DOI] [PubMed] [Google Scholar]
- 49.Honson NS, Jensen JR, Darby MV, Kuret J. Potent inhibition of tau fibrillization with a multivalent ligand. Biochem Biophys Res Commun. 2007;363:229–234. doi: 10.1016/j.bbrc.2007.08.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Garoff RA, Litzinger EA, Connor RE, Fishman I, Armitage BA. Helical aggregation of cyanine dyes on DNA templates: Effect of dye structure on formation of homo- and heteroaggregates. Langmuir. 2002;18:6330–6337. [Google Scholar]
- 51.Brancato R, Trabucchi G. Fluorescein and indocyanine green angiography in vascular chorioretinal diseases. Semin Ophthalmol. 1998;13:189–198. doi: 10.3109/08820539809056052. [DOI] [PubMed] [Google Scholar]
- 52.Caesar J, Shaldon S, Chiandussi L, Guevara L, Sherlock S. The use of indocyanine green in the measurement of hepatic blood flow and as a test of hepatic function. Clin Sci. 1961;21:43–57. [PubMed] [Google Scholar]
- 53.Meijer DK, Weert B, Vermeer GA. Pharmacokinetics of biliary excretion in man. VI. Indocyanine green. Eur J Clin Pharmacol. 1988;35:295–303. doi: 10.1007/BF00558268. [DOI] [PubMed] [Google Scholar]
- 54.Cherrick GR, Stein SW, Leevy CM, Davidson CS. Indocyanine green: observations on its physical properties, plasma decay, and hepatic extraction. J Clin Invest. 1960;39:592–600. doi: 10.1172/JCI104072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Huang L, Vore M. Multidrug resistance p-glycoprotein 2 is essential for the biliary excretion of indocyanine green. Drug Metab Dispos. 2001;29:634–637. [PubMed] [Google Scholar]
- 56.Li M, Yuan H, Li N, Song G, Zheng Y, Baratta M, Hua F, Thurston A, Wang J, Lai Y. Identification of interspecies difference in efflux transporters of hepatocytes from dog, rat, monkey and human. Eur J Pharm Sci. 2008;35:114–126. doi: 10.1016/j.ejps.2008.06.008. [DOI] [PubMed] [Google Scholar]
- 57.Licha K, Riefke B, Ntziachristos V, Becker A, Chance B, Semmler W. Hydrophilic cyanine dyes as contrast agents for near-infrared tumor imaging: synthesis, photophysical properties and spectroscopic in vivo characterization. Photochem Photobiol. 2000;72:392–398. doi: 10.1562/0031-8655(2000)072<0392:hcdaca>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 58.Sima PD, Kanofsky JR. Cyanine dyes as protectors of K562 cells from photosensitized cell damage. Photochem Photobiol. 2000;71:413–421. doi: 10.1562/0031-8655(2000)071<0413:cdapok>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 59.Modica-Napolitano JS, Kulawiec M, Singh KK. Mitochondria and human cancer. Curr Mol Med. 2007;7:121–131. doi: 10.2174/156652407779940495. [DOI] [PubMed] [Google Scholar]
- 60.Marques-Santos LF, Oliveira JG, Maia RC, Rumjanek VM. Mitotracker green is a P-glycoprotein substrate. Biosci Rep. 2003;23:199–212. doi: 10.1023/b:bire.0000007693.33521.18. [DOI] [PubMed] [Google Scholar]
- 61.Galvan M, David JP, Delacourte A, Luna J, Mena R. Sequence of neurofibrillary changes in aging and Alzheimer’s disease: A confocal study with phospho-tau antibody, AD2. J Alzheimers Dis. 2001;3:417–425. doi: 10.3233/jad-2001-3409. [DOI] [PubMed] [Google Scholar]
- 62.Gray EG, Paula-Barbosa M, Roher A. Alzheimer’s disease: paired helical filaments and cytomembranes. Neuropathol Appl Neurobiol. 1987;13:91–110. doi: 10.1111/j.1365-2990.1987.tb00174.x. [DOI] [PubMed] [Google Scholar]
- 63.Volk HA, Burkhardt K, Potschka H, Chen J, Becker A, Loscher W. Neuronal expression of the drug efflux transporter P-glycoprotein in the rat hippocampus after limbic seizures. Neuroscience. 2004;123:751–759. doi: 10.1016/j.neuroscience.2003.10.012. [DOI] [PubMed] [Google Scholar]
- 64.Goedert M, Spillantini MG, Jakes R, Rutherford D, Crowther RA. Multiple isoforms of human microtubule-associated protein tau: sequences and localization in neurofibrillary tangles of Alzheimer’s disease. Neuron. 1989;3:519–526. doi: 10.1016/0896-6273(89)90210-9. [DOI] [PubMed] [Google Scholar]
- 65.Carmel G, Mager EM, Binder LI, Kuret J. The structural basis of monoclonal antibody Alz50’s selectivity for Alzheimer’s disease pathology. J Biol Chem. 1996;271:32789–32795. doi: 10.1074/jbc.271.51.32789. [DOI] [PubMed] [Google Scholar]
- 66.LoPresti P, Szuchet S, Papasozomenos SC, Zinkowski RP, Binder LI. Functional implications for the microtubule-associated protein tau: localization in oligodendrocytes. Proc Natl Acad Sci U S A. 1995;92:10369–10373. doi: 10.1073/pnas.92.22.10369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Espinoza M, de Silva R, Dickson DW, Davies P. Differential incorporation of tau isoforms in Alzheimer’s disease. J Alzheimers Dis. 2008;14:1–16. doi: 10.3233/jad-2008-14101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Necula M, Kuret J. Electron microscopy as a quantitative method for investigating tau fibrillization. Anal Biochem. 2004;329:238–246. doi: 10.1016/j.ab.2004.02.023. [DOI] [PubMed] [Google Scholar]
- 69.Duff K, Noble W, Gaynor K, Matsuoka Y. Organotypic slice cultures from transgenic mice as disease model systems. J Mol Neurosci. 2002;19:317–320. doi: 10.1385/JMN:19:3:317. [DOI] [PubMed] [Google Scholar]
- 70.Greenberg SG, Davies P. A preparation of Alzheimer paired helical filaments that displays distinct tau proteins by polyacrylamide gel electrophoresis. Proc Natl Acad Sci U S A. 1990;87:5827–5831. doi: 10.1073/pnas.87.15.5827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.O’Farrell PH. High resolution two-dimensional electrophoresis of proteins. J Biol Chem. 1975;250:4007–4021. [PMC free article] [PubMed] [Google Scholar]