

Abstract
Background
Infection with multiple human papillomavirus (HPV) types is common. However, it is unknown whether viral DNA load is related to the coexistence of other types.
Methods
Study subjects were 802 and 303 women who were positive for HPV16 and HPV18, respectively, at enrollment into the Atypical Squamous Cells of Undetermined Significance and Low-Grade Squamous Intraepithelial Lesion Triage Study. HPV16 and HPV18 E7 copies per nanogram of cellular DNA in cervical swab samples were measured by real-time polymerase chain reaction in triplicate.
Results
Concurrent coinfection was common in this population of women with minor cervical lesions; multiple HPV types were detected in 573 (71.4%) of 802 HPV16-positive women and 227 (74.9%) of 303 HPV18-positive women. The adjusted odds ratio associating coinfection with per 1 log unit increase in HPV16 DNA load was 0.78 (95% confidence interval (CI), 0.68-0.89); it was 0.64 (95% CI, 0.52-0.79) for a similar analysis of HPV18 DNA load. Women with, compared to without, coinfection of A9 species possessed a significantly lower HPV16 DNA load (p < 0.001) whereas women with, compared to without, coinfection of A7 species types possessed a significantly lower HPV18 DNA load (p = 0.001). A trend of decrease in HPV16 DNA load with increasing number of the coexisting non-HPV16 A9 species types was statistically significant (p for trend = 0.001).
Conclusion
Coinfection with other types was associated with lower HPV16 and HPV18 DNA load. The extent of reduction was correlated to phylogenetic distance of the coexisting types to HPV16 and HPV18, respectively.
Keywords: Human Papillomavirus, Viral Load, Coinfection
Introduction
Studies of human papillomavirus (HPV) DNA load have focused mainly on the clinical consequence of higher HPV DNA load (1-9). Little is known about determinants that may alter levels of HPV DNA. Infection with multiple HPV types is common in all populations and very common in women with minor cytologic abnormalities when sensitive HPV assays are used. Knowing whether for a given type of HPV, the viral load is affected by coexistence of other types would shed new light onto the natural history of HPV infection.
Papillomavirus types are grouped as genera and species according to DNA sequence homology (10, 11). Almost all genital HPV types are members of alpha papillomavirus (A) genus. Within the genus, phylogenetically related types are grouped as species. Between species, types share between 60% and 70% nucleotide identity within the complete L1 open reading frame and within species, types share 71% to 89% (10). The majority of oncogenic HPV types belong to A9 and A7 species, of which HPV16 and HPV18 respectively are the most oncogenic representatives. The phylogenetic grouping of HPV types has been shown to correlate to oncogenicity of the virus (12). Given recent findings of some level of cross-reaction of immune responses to phylogenetically related HPV types in natural infections (13-18), we have hypothesized that coinfection with the closely related types may play a role in HPV DNA load.
To address this hypothesis, we examined baseline HPV16 and HPV18 DNA load between women with and without concurrent coinfection with other HPV types who participated in the Atypical Squamous Cells of Undetermined Significance (ASC-US) and Low-Grade Squamous Intraepithelial Lesion (LSIL) Triage Study (ALTS).
Materials and Methods
Study subjects
Study subjects were ALTS participants who were positive for HPV16 and/or HPV18 by polymerase chain reaction (PCR)-based reverse line blot assay at enrollment (19, 20). A full description of the ALTS design and the characteristics of the study participants is available elsewhere (21, 22). The ALTS protocol was approved by the institutional review boards at the National Cancer Institute and at each of the four clinical centers involved in the trial. We measured amount of HPV16 and HPV18 DNA present in enrollment samples. A total of 1,071 enrollment samples were eligible for the present study, including 759 positive for HPV16, 258 positive for HPV18, and 54 positive for both HPV16 and 18. Of them, 19 (11 HPV16-positive and 8 HPV18-positive) were unavailable, leaving 802 HPV16-positive and 304 HPV18-positive samples for viral load quantification. Data on HPV typing results, cervical cytology and histology, and characteristics of study subjects were obtained from the ALTS database. The protocol for this study was approved by the institutional review board at the University of Washington.
Quantification of HPV16 and HPV18 DNA load
HPV16 and HPV18 E7 copy number and cellular DNA amount were measured by multiplex real-time PCR, as described previously (9, 23). Briefly, the assay was set up in a reaction volume of 25 μL with the TaqMan Universal PCR Master Mix kit (Applied Biosystems, Foster City, CA). Primers and probe for human β-actin gene were commercially available (Applied Biosystems). Primers and probe for HPV16 and HPV18 E7 gene were reported previously (9, 23). Amplification was carried out on the Applied Biosystems 7900 HT Sequence Detection System with a cycling program of holding at 50°C for 2 minutes and then at 95°C for 10 minutes followed by a 2-step cycle of 10 seconds at 95°C and 1 minute at 60°C for 40 cycles.
Two logarithmic-phase five-point standard curves were implemented in each run; one for viral DNA and the other for cellular DNA. The viral load was normalized to the input amount of cellular DNA and expressed as E7 copy number per nanogram of cellular DNA. Each sample was assayed in triplicate.
We excluded one sample that was positive for HPV18 E7 but negative for cellular DNA, leaving 303 women in the analysis of HPV18 DNA load. E7 DNA was undetectable by real-time PCR in 64 HPV16-positive and 22 HPV18-positve samples. The assay used in the present study was able to reliably measure a minimum number of 10 viral copies (data not shown). Considering that the negative result might be due to a tiny amount of viral DNA in the sample, a value of one viral copy per nanogram of cellular DNA was assigned to each of these samples.
To address whether a closely related type could potentially compete for any reagent or interfere with amplification of the target gene, we assessed viral copy number in 10 samples positive for HPV16 alone with and without arbitrarily added natural non-HPV16 A9 species types. Each sample was aliquoted to 2 parts with an equal volume (2μl). One part was individually mixed with an equal volume of 10 samples that were positive for at least one A9 species type, the other mixed with an equal volume of HPV-negative samples. HPV16 E7 copy number was independently measured 2 times in triplicate.
Statistical analyses
The normalized viral DNA load was log10-transformed and treated as a continuous variable. The mean of log10-transformed values from the triplicate measurements was used for analyses. Odds ratios (ORs) and 95% confidence intervals (CIs) for the association between viral load and coinfection were estimated using polynomial logistic regression (24). Coinfection was measured by either yes/no, number (1, 2, 3, or ≥4) or species (A9, A7, or other) of the co-infecting types. As defined previously (10, 12), A9 species (HPV16-related) includes HPV16, 31, 33, 35, 52, 58, and 67 and A7 species (HPV18-related) includes HPV18, 39, 45, 59, 68, 70, and 85. The other 24 types (HPV6, 11, 26, 40, 42, 51, 53, 54, 55, 56, 57, 61, 62, 64, 66, 69, 71, 72, 73, 81, 82, 83, 84, and 91) detected by PCR-based reverse line blot assay were grouped together as a category called “other species.” Women with coinfection of both A7 and A9 species types were treated as a separate group in the initial analyses to avoid a potential bias in species assignment. Women with coinfection of A7 and/or A9 species might have or might not have other species types. The ORs were adjusted for cervical cytology at enrollment (normal cytology, ASC-US, LSIL, and HSIL) and current smoking status (yes or no).
Linear regression models (25) were used to compare HPV16 and HPV18 DNA load between women with and without coinfection with types that belong to the same species (i.e., A9 species for analyses of HPV16 DNA load and A7 species for analyses of HPV18 DNA load). P-values were adjusted for cervical cytology at enrollment and current smoking status for cytology-combined analyses and current smoking status for cytology-stratified analyses. A trend test was used to examine the association between HPV16 DNA load and number of the co-infecting A9 species types (none, 1 type, or ≥2 types).
We used student t test or one way ANOVA, whichever was appropriate, to compare viral load by age at enrollment (18-24 versus ≥ 25 years), race (white versus nonwhite), current use of hormonal contraceptives (yes or no), lifetime number of sex partners (0–5 versus ≥6), HPV variant (European or non-European), current smoking status, and cervical cytology at enrollment. The differences in HPV16 DNA load between samples with and without arbitrarily added non-HPV16 A9 species types were tested using paired-sample t test. All statistical tests were at the 5% two-sided significance level.
Results
The geometric means of E7 copy number per 1 nanogram of cellular DNA was 6.10 × 102 (95% CI, 4.92 × 102-7.55 × 102) for 802 women with HPV16 and 7.37 × 103 (95% CI, 4.78 × 103-1.13 × 104) for 303 women with HPV18. The mean value of log10-transformed HPV16 E7 copy number per 1 nanogram of cellular DNA for women with normal cytology, ASC-US, LSIL and HSIL was 1.76, 2.71, 3.11 and 3.39, respectively (P for trend <0.001). The corresponding values for HPV18 DNA load were 2.82, 3.87, 4.19 and 4.49, respectively (P for trend <0.001). The viral load was statistically significantly higher among current smokers than among nonsmokers (2.91 versus 2.68, p =0.02 for HPV16; 4.13 versus 3.69, p = 0.02 for HPV18). HPV16 DNA load (but not HPV18 DNA load) was higher in white, compared to nonwhite, women (2.86 versus 2.59, p = 0.01). Neither HPV16 nor HPV18 DNA load was appreciably related to age at enrollment in this predominantly young study population, limited range of lifetime number of sex partners, current use of hormonal contraceptives, or lineage of the variants (data not shown).
Coinfection with other HPV types was detected in 573 (71.4%) of 802 women with HPV16 infection and 227 (74.9%) of 303 women with HPV18 infection. With adjustment for concurrent cervical cytology and smoking status, the OR associating coinfection with per 1 log unit increase in viral load was 0.78 (95% CI, 0.68-0.89) for HPV16 and 0.64 (95% CI, 0.52-0.79) for HPV18 (Table 1). A diagnosis of cervical intraepithelial neoplasia grades 2-3 (CIN2-3) was histologically confirmed at enrollment in 274 HPV16-positive women and 57 HPV18-positive women. The results remained similar when the analysis was restricted to women without CIN2-3 at enrollment (OR = 0.74, 95% CI, 0.62-0.87 for HPV16; OR = 0.67, 95% CI, 0.54-0.83 for HPV18). Additional adjustment of analysis of HPV16 DNA load for subject race did not materially alter the magnitude of the association (data not shown). The number of the infecting types ranged from 1 to 11 with an average of 2.1 for women with HPV16 infection and ranged from 1 to 9 with an average of 2.4 for those with HPV18 infection. Coinfection with other A9 species types was detected in 243 HPV16-positive women and coinfection with other A7 species types in 77 HPV18-positive women.
Table 1.
Odds ratio for the association of HPV16 and HPV18 DNA load with concurrent coinfection with other HPV types
| Log10 HPV16 E7 copies per 1 nanogram of cellular DNA | Log10 HPV18 E7 copies per 1 nanogram of cellular DNA | |||||
|---|---|---|---|---|---|---|
| HPV at enrollment* | No. | Mean (SD) | OR† | No. | Mean (SD) | OR† |
| Without coinfection | 229 | 2.95 (±1.31) | 1.00 | 76 | 4.41 (±1.64) | 1.00 |
| With coinfection | 573 | 2.72 (±1.35) | 0.78 (0.68-0.89) | 227 | 3.68 (±1.63) | 0.64 (0.52-0.79) |
| Coinfection with | ||||||
| other species | 185 | 2.78 (±1.37) | 1.00 | 64 | 4.00 (±1.35) | 1.00 |
| A7 species | 145 | 2.92 (±1.33) | 1.04 (0.87-1.25) | 34 | 3.35 (±1.91) | 0.76 (0.58-1.00) |
| A9 species | 162 | 2.60 (±1.37) | 0.87 (0.73-1.03) | 86 | 3.70 (±1.76) | 0.81 (0.65-1.02) |
| A7 and A9 species | 81 | 2.46 (±1.27) | 0.77 (0.62-0.96) | 43 | 3.45 (±1.48) | 0.75 (0.58-0.97) |
| Coinfection with | ||||||
| 1 type | 239 | 2.84 (±1.33) | 1.00 | 85 | 3.65 (±1.59) | 1.00 |
| 2 types | 172 | 2.59 (±1.36) | 0.86 (0.74-1.02) | 63 | 3.74 (±1.57) | 1.07 (0.86-1.33) |
| 3 types | 95 | 2.78 (±1.31) | 0.99 (0.81-1.21) | 36 | 4.09 (±1.48) | 1.10 (0.85-1.43) |
| ≥4 types | 67 | 2.52 (±1.36) | 0.81 (0.65-1.00) | 43 | 3.33 (±1.88) | 0.84 (0.67-1.07) |
HPV16 was not counted in species A9 for analysis of HPV16 DNA load; HPV18 was not counted in species A7 for analysis of HPV18 DNA load. Whether there was a concurrent coinfection with other species types was not distinguished among women with coinfection of species A7 and/or species A9 types.
Adjusted for cytologic diagnoses at enrollment and current smoking status
Among women with coinfection, viral load was not statistically significantly associated with the number of the co-infecting HPV types, though the association was marginally significant for 2 co-infecting types for HPV16 infection and for ≥ 4 co-infecting types for women with HPV16 or HPV18 infection (Table 1). Relative to coinfection with other species types, lower HPV16 DNA load was significantly associated with coexistence of A9 and A7 species types (OR = 0.77, 95% CI, 0.62-0.96), marginally associated with existence of A9 species types (OR = 0.87, 95% CI, 0.73-1.03), but unrelated to existence of A7 species types. The distinctions were not quite as clear among women with HPV18 infection; the OR was 0.75 (95% CI, 0.58-0.97) for coexistence of A9 and A7 species types and 0.76 (95% CI, 0.58-1.00) for existence of A7 species types. However, HPV18 DNA load was also marginally lower when there was coinfection with A9 species relative to other (non A7, non A9) species.
Given the species-related association, we categorized women as with or without coinfection of A9 species types for analysis of HPV16 DNA load and with or without coinfection of A7 species types for analysis of HPV18 DNA load. As shown in Table 2, women with, as compared to without, coinfection of A9 species types possessed a significantly lower mean HPV16 DNA load (log10-transformed E7 copy number per 1 nanogram of cellular DNA of 2.55 versus 2.89, p < 0.001). Women with, as compared to without, coinfection of A7 species types possessed a significantly lower HPV18 DNA load (3.40 versus 4.03, p = 0.001). The results remained similar when the analysis was restricted to women without CIN2-3 at enrollment (2.30 versus 2.70 for HPV16, p = 0.003; 3.37 versus 4.11 for HPV18, p = 0.003). This pattern was constant across concurrent cytologic diagnoses although the estimated reduction of HPV16 DNA load for women with normal cytology or ASC-US and of HPV18 DNA load for women with ASC-US was not statistically significant. The coinfection-related reduction of viral load remained similar when the analysis was stratified by current smoking status (data not shown).
Table 2.
HPV16 and HPV18 DNA load between women with and without concurrent coinfection of the same species types, stratified by cervical cytology
| Log10 HPV16 E7 copies per 1 nanogram of cellular DNA in women | Log10 HPV18 E7 copies per 1 nanogram of cellular DNA in women | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Cervical cytology* | without other A9 species types | with other A9 species types | without other A7 species types | with other A7 species types | ||||||
| No. | Mean (SD) | No. | Mean (SD) | P-value† | No. | Mean (SD) | No. | Mean (SD) | P-value† | |
| All diagnoses | 559 | 2.89 (±1.33) | 243 | 2.55 (±1.33) | <0.001 | 226 | 4.03 (±1.63) | 77 | 3.40 (±1.68) | 0.001 |
| Normal | 110 | 1.86 (±1.32) | 40 | 1.50 (±1.19) | 0.11 | 43 | 3.08 (±1.48) | 13 | 1.88 (±2.15) | 0.01 |
| ASC-US | 162 | 2.79 (±1.17) | 71 | 2.54 (±1.39) | 0.16 | 63 | 3.90 (±1.60) | 19 | 3.77 (±0.98) | 0.86 |
| LSIL | 175 | 3.27 (±1.26) | 87 | 2.78 (±1.23) | 0.002 | 88 | 4.39 (±1.55) | 39 | 3.76 (±1.63) | 0.04 |
| HSIL | 107 | 3.52 (±0.99) | 45 | 3.08 (±1.06) | 0.05 | 28 | 4.77 (±1.52) | 6 | 3.19 (±0.38) | 0.03 |
Five women were excluded from cytology-stratified analyses because their enrollment samples were inadequate for cytologic evaluation.
Adjusted for current smoking status for cytology-stratified analyses and cytologic diagnoses and current smoking status for cytology-combined analyses
No appreciable difference was seen in comparing HPV16 DNA load between coinfection with HPV31 (the type mostly closely related to HPV16) and other A9 species types or comparing HPV18 DNA load between coinfection with HPV45 (the type most closely related to HPV18) and other A7 species types (data not shown).
Although the analyses of viral load by the number of all co-infecting types yielded null or borderline associations, we found a statistically significant trend of decrease in HPV16 DNA load with increasing number of A9 species types (Table 3, p trend = 0.001). The trend was statistically significant among women with LSIL or HSIL and marginally significant among those with normal cytology or ASC-US. Only 10 HPV18-postive women had a coinfection with ≥ 2 A7 species types and thus, an analysis of the association with number of the same species types was not performed.
Table 3.
Trend of decrease in HPV16 DNA load with increasing number of the co-infecting A9 species types
| Log10 HPV16 E7 copies per 1 nanogram of cellular DNA in women with | |||||||
|---|---|---|---|---|---|---|---|
| Cervical cytology* | no other A9 species types | 1 A9 species type | ≥2 A9 species types | ||||
| No. | Mean (SD) | No. | Mean (SD) | No. | Mean (SD) | Pfor trend | |
| All diagnoses | 559 | 2.89 (±1.33) | 202 | 2.60 (±1.34) | 41 | 2.32 (±1.28) | 0.001 |
| Normal | 110 | 1.86 (±1.32) | 34 | 1.52 (±1.22) | 6 | 1.33 (±1.07) | 0.12 |
| ASC-US | 162 | 2.79 (±1.17) | 53 | 2.57 (±1.41) | 18 | 2.44 (±1.36) | 0.15 |
| LSIL | 175 | 3.27 (±1.26) | 75 | 2.83 (±1.22) | 12 | 2.42 (±1.27) | 0.002 |
| HSIL | 107 | 3.52 (±0.99) | 40 | 3.10 (±1.09) | 5 | 2.86 (±0.84) | 0.02 |
Five women were excluded from cytology-stratified analyses because their enrollment samples were inadequate for cytologic evaluation.
Figure 1 shows the estimate of HPV16 DNA copy number in each pair of samples with or without arbitrarily added non-HPV16 A9 species types. Of 10 samples positive for HPV16 alone, 8 were individually mixed with samples containing 1 non-HPV16 A9 species type, 1 with 2 non-HPV16 A9 species types, and 1 with 3 non-HPV16 A9 species types. As a comparison group, the same 10 samples were individually mixed with HPV-negative samples. The estimates of copy number in each sample pair were not meaningfully different. The difference in mean value of log10-transformed HPV16 E7 copy number between samples with and without arbitrarily added non-HPV16 A9 species types was 0.029 (95% CI, -0.097-0.156; P = 0.61).
Figure 1.
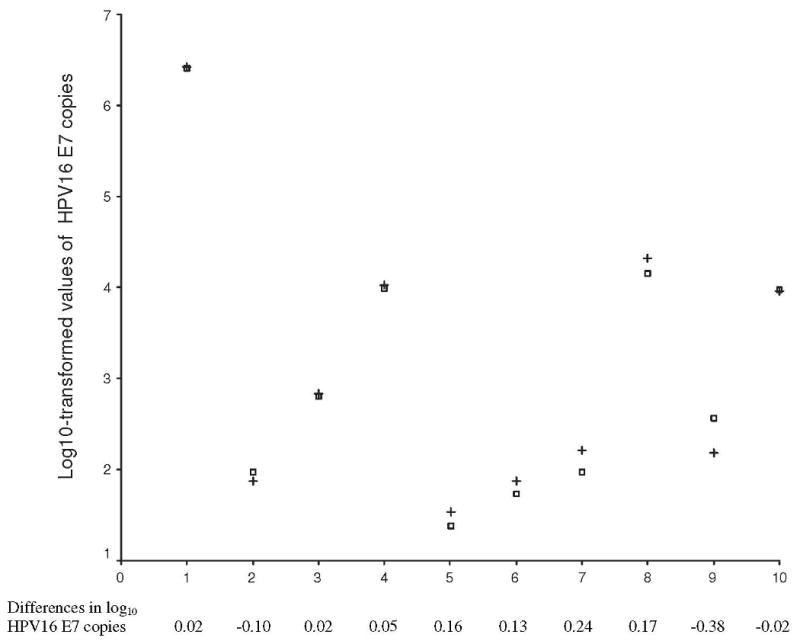
Estimates of HPV16 copy number in 10 pair of samples with or without arbitrarily added A9 species types +.
Samples 1-10, individually mixed with 10 samples containing HPV52, HPV31, HPV31/51, HPV52/62, HPV33/73, HPV35/51/52, HPV31/39/83, HPV18/53/67, HPV51/52/59/61, HPV33/39/51/52/59, respectably
□. Samples 1-10, individually mixed with HPV-negative samples * Difference = log10 units (of sample with A9 types) minus log10 units (of sample with HPV-negative sample)
Discussion
We observed statistically significant association between lower HPV16 and HPV18 DNA load and coinfection with other HPV types. Interestingly, among women with HPV coinfection, phylogenetic grouping of the types was correlated to the extent of viral load reduction, i.e., coinfection with A9 species types being associated with a substantial reduction of HPV16 DNA load and coinfection with A7 species types with reduction of HPV18 DNA load. An immediate question to these results is whether there was a competition of a related type for PCR reagents, thereby lowering the quantity of HPV16 and HPV18 DNA. Theoretically, the competition is expected to be minimal if the primers and probe are specific and the nucleotides and other components such as enzymes in the reaction are in excess. The unchanged estimates of HPV16 copy number between samples with and without arbitrarily added other A9 species types (i.e., HPV31, 33, 35, 52 and/or 67) are reassuring for this. We noted that approximately 7-8% of samples initially tested positive for HPV16 or HPV18 by PCR-based reverse line blot assay were negative for by real-time PCR. We presumed that the amount of viral DNA was too small to be detectable and assigned a value of 1 viral copy per nanogram cellular DNA to each sample for primary analysis. It is also possible that the discrepancy might result from a false positive result by the reverse line blot assay or a difference in regions tested. Whereas the reverse line blot assay targeted the L1 region, the real-time PCR targeted the E7 region. The discrepancy might have occurred if not a whole viral genome had been retained in the sample. However, the results remained similar when these samples were excluded from the analysis (data not shown). Our analyses indicated that the association was not confounded or modified by abnormalities of cervical cytology or current smoking status, the factors that were significantly associated with higher viral load. The association was also not explained by presence of underlying CIN2-3 which, like cytology, was thought to correlate with HPV DNA load (26). Also, the association was not distorted by biopsy or therapy, the procedure that may physically remove the virus, because the samples tested were collected prior to any of these procedures.
The finding of coinfection-related reduction of viral load disagrees with a previous report (26) in which HPV DNA load was found to be significantly higher in women infected with multiple types than in those with a single type. However, the viral load measured in that study was an aggregate of DNA amount from all types captured by Hybrid Capture. The underlying mechanism for the coinfection-related reduction of HPV16 and HPV18 DNA load is not clear. One interpretation is that if types are in the same cell, they may compete for replication, transcription, translation, and/or persistence factors. HPVs utilize cellular DNA machinery for replication of the virus. It is therefore possible that insufficient availability of cellular regulators may lead to a decrease in replication of HPV16 and HPV18 DNA and/or production of infectious virus. This interpretation, however, is not well supported by findings from an in vitro study where in all cases, HPV18 dominated when HPV31, 33, 39, and 45 were introduced into the same cell (27). Also, this explanation cannot adequately address why the magnitude of reduction of HPV16 and HPV18 DNA load varied by species of the co-infecting types.
Alternatively, coinfection with other types may be somehow dampening HPV16 and HPV18 DNA replication, possibly mediated through cross-reactivity of immune response. It is well recognized that most of HPV infections are transient and that cellular, but not humoral, immune responses may play a role in the clearance of infection. Williams et al. have shown that natural infection-induced T cell response to HPV11 in healthy adults cross-reacted with a range of HPV types (17). In addition, the extent of cellular immune response to HPV16 L1 VLP vaccine in healthy volunteers was found to correlate with phylogenetic distance of HPV types (18). Because of higher sequence homology and similar genome structure, HPV types, particularly those within the same species may share some biologic properties that may translate to similar epitopes to elicit cross-reactive immune responses (28). Indeed, a shared T cell epitope between HPV18 and HPV45 was reported previously (29). It is plausible that cellular responses induced by natural infection with other types might help with eliminating the infected cells and/or restraining the virus for replication of viral genome or production of progeny, thereby leading to reduction of HPV16 and HPV18 DNA load.
Consistent with this possibility, some, although not all (30), HPV vaccine trials have shown that both bivalent vaccine against HPV16 and HPV18 (31, 32) and quadrivalent vaccine against HPV6, HPV11, HPV16 and HPV18 (33, 34) afford partial cross-protection against a 6-month persistent infection with some but not all phylogenetically-related HPV types. HPV16 L1 VLP vaccine has been shown to induce L1-specific T-cell responses detected by proliferation of both CD4 and CD8 T cells and in vitro production of Th1 and Th2 type cytokines (35). Given the polyclonal nature of the immune response to vaccine, the acceleration of the clearance of phylogenetically related nonvaccine types is thought to result from anti-HPV16 and -HPV18 cellular immune responses. There has been, however, no evidence from natural history studies that coinfection promotes clearance of HPV infection. In contrast, coinfection was found to be either independent of (36, 37) or associated with persistent infection (38, 39). Thus, in addition to the potential influence of cross-reaction of cellular responses, factors associated multiple infections may also play a role in clearance of viral infection (39).
Among women with coinfection, the association between HPV16 and HPV18 DNA load and number of the co-infecting types was inconsistent. However, when the analysis was restricted to A9 species types, a trend of decrease in HPV16 DNA load with increasing number of the co-infecting types was evident. We also noted that as compared to other species, coinfection with A9 species types was related to lower HPV18 DNA load, although the magnitude of the reduction was not as substantial as coinfection with A7 species types. A possible interpretation for this is that the immune response induced by some A9 species types may also react with HPV18, but with the extent less than that by A7 species types.
Several limitations should be addressed. The design of this study is cross-sectional in nature. Thus, HPV16 and HPV18 detected could be a newly acquired or a persistent infection. The co-infecting types, although prevalent at the same enrollment visit, might have been acquired concurrently or sequentially. Also, women might have obtained and cleared different HPV types prior to the time of enrollment. Clearly, further studies are needed to examine whether coinfection-related reduction of viral load is related to the timing of acquisition. Clarification of this may help with understanding of natural history of HPV infection and may have both screening and vaccination implications. We are aware that cellular DNAs used for normalization were a mixture of the infected and non-infected cells. Viral load per infected cell would give a different copy number. Thus, it remained undetermined whether the coinfection-related lower viral load was due to a decrease in number of the infected cells or number of viral copies per infected cell or whether this was due to reduction of viral DNA replication or production of viral progeny. Discrimination between viral DNA replication and production of viral progeny may be an interesting topic for future studies because productive infections (i.e., production of progeny) lead to increased number of infected sites with their own potential to progress within the host. Lastly, women who were eligible for participation in ALTS were required to have a Pap smear showing ASC-US or LSIL within 6 months of screening. As infection with multiple HPV types is a risk factor for abnormal cytology (40), the proportion of coinfection in this study might be more than that in the general population. However, this lack of generalizability does not affect the validity of relative comparisons of viral load between women with and without coinfection because the coinfection-related reduction of viral load was similar across cytologic diagnoses. In spite of these limitations, to our knowledge, this is one of the first, if not the first report, to address impacts of concurrent coinfection with species-specific types on HPV16 and HPV18 DNA load. The findings of coinfection-related reduction of viral load are important and likely to suggest the presence of cross-reactivity of cellular immune responses in the interplay between the coexisting HPV types. In addition, the species-specific association found in this study provides a further support for the notion that the phylogenetic classification of HPVs correlates with biologic properties of the virus.
In summary, lower HPV16 and HPV18 DNA load was associated with coinfection with other HPV types, particularly those that belong to the same species. The species-specific association may in part reflect differences in the extent of cross-reaction of cellular immune response induced by other types.
Acknowledgments
This study was part of the project ancillary to the ALTS clinical trial but does not represent the ALTS Group. The authors would like to thank the ALTS Group for providing the biological specimens and HPV typing results and Dr Kathrin Jansen for providing the HPV16 and HPV18 DNA standard.
Grant supports: Public Health Service grant CA 84396 (L. F. Xi).
Footnotes
Notes: The authors have no commercial or other associations that might pose a conflict of interest.
References
- 1.Kovacic MB, Castle PE, Herrero R, et al. Relationships of human papillomavirus type, qualitative viral load, and age with cytologic abnormality. Cancer Res. 2006;66:10112–9. doi: 10.1158/0008-5472.CAN-06-1812. [DOI] [PubMed] [Google Scholar]
- 2.Fiander AN, Hart KW, Hibbitts SJ, et al. Variation in human papillomavirus type-16 viral load within different histological grades of cervical neoplasia. J Med Virol. 2007;79:1366–9. doi: 10.1002/jmv.20875. [DOI] [PubMed] [Google Scholar]
- 3.Fontaine J, Hankins C, Mayrand MH, et al. High levels of HPV-16 DNA are associated with high-grade cervical lesions in women at risk or infected with HIV. Aids. 2005;19:785–94. doi: 10.1097/01.aids.0000168972.65304.6b. [DOI] [PubMed] [Google Scholar]
- 4.Lorincz AT, Castle PE, Sherman ME, et al. Viral load of human papillomavirus and risk of CIN3 or cervical cancer. Lancet. 2002;360:228–9. doi: 10.1016/S0140-6736(02)09463-1. [DOI] [PubMed] [Google Scholar]
- 5.Schlecht NF, Trevisan A, Duarte-Franco E, et al. Viral load as a predictor of the risk of cervical intraepithelial neoplasia. Int J Cancer. 2003;103:519–24. doi: 10.1002/ijc.10846. [DOI] [PubMed] [Google Scholar]
- 6.Josefsson AM, Magnusson PK, Ylitalo N, et al. Viral load of human papilloma virus 16 as a determinant for development of cervical carcinoma in situ: a nested case-control study. Lancet. 2000;355:2189–93. doi: 10.1016/S0140-6736(00)02401-6. [DOI] [PubMed] [Google Scholar]
- 7.Ylitalo N, Sorensen P, Josefsson AM, et al. Consistent high viral load of human papillomavirus 16 and risk of cervical carcinoma in situ: a nested case-control study. Lancet. 2000;355:2194–8. doi: 10.1016/S0140-6736(00)02402-8. [DOI] [PubMed] [Google Scholar]
- 8.Castle PE, Schiffman M, Scott DR, et al. Semiquantitative human papillomavirus type 16 viral load and the prospective risk of cervical precancer and cancer. Cancer Epidemiol Biomarkers Prev. 2005;14:1311–4. doi: 10.1158/1055-9965.EPI-04-0799. [DOI] [PubMed] [Google Scholar]
- 9.Xi LF, Kiviat NB, Galloway DA, Zhou XH, Ho J, Koutsky LA. Effect of cervical cytologic status on the association between human papillomavirus type 16 DNA load and the risk of cervical intraepithelial neoplasia grade 3. J Infect Dis. 2008;198:324–31. doi: 10.1086/589715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.de Villiers EM, Fauquet C, Broker TR, Bernard HU, zur Hausen H. Classification of papillomaviruses. Virology. 2004;324:17–27. doi: 10.1016/j.virol.2004.03.033. [DOI] [PubMed] [Google Scholar]
- 11.Bernard HU. The clinical importance of the nomenclature, evolution and taxonomy of human papillomaviruses. J Clin Virol. 2005;32(Suppl 1):S1–6. doi: 10.1016/j.jcv.2004.10.021. [DOI] [PubMed] [Google Scholar]
- 12.Schiffman M, Herrero R, Desalle R, et al. The carcinogenicity of human papillomavirus types reflects viral evolution. Virology. 2005;337:76–84. doi: 10.1016/j.virol.2005.04.002. [DOI] [PubMed] [Google Scholar]
- 13.Combita AL, Bravo MM, Touze A, Orozco O, Coursaget P. Serologic response to human oncogenic papillomavirus types 16, 18, 31, 33, 39, 58 and 59 virus-like particles in colombian women with invasive cervical cancer. Int J Cancer. 2002;97:796–803. doi: 10.1002/ijc.10153. [DOI] [PubMed] [Google Scholar]
- 14.Wang SS, Schiffman M, Shields TS, et al. Seroprevalence of human papillomavirus-16, -18, -31, and -45 in a population-based cohort of 10000 women in Costa Rica. Br J Cancer. 2003;89:1248–54. doi: 10.1038/sj.bjc.6601272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wideroff L, Schiffman M, Haderer P, et al. Seroreactivity to human papillomavirus types 16, 18, 31, and 45 virus-like particles in a case-control study of cervical squamous intraepithelial lesions. J Infect Dis. 1999;180:1424–8. doi: 10.1086/315055. [DOI] [PubMed] [Google Scholar]
- 16.Marais D, Rose RC, Lane C, Aspinall S, Bos P, Williamson AL. Seroresponses to virus-like particles of human papillomavirus types 16, 18, 31, 33, and 45 in San people of Southern Africa. J Med Virol. 2000;60:331–6. doi: 10.1002/(sici)1096-9071(200003)60:3<331::aid-jmv12>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 17.Williams OM, Hart KW, Wang EC, Gelder CM. Analysis of CD4(+) T-cell responses to human papillomavirus (HPV) type 11 L1 in healthy adults reveals a high degree of responsiveness and cross-reactivity with other HPV types. J Virol. 2002;76:7418–29. doi: 10.1128/JVI.76.15.7418-7429.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pinto LA, Viscidi R, Harro CD, et al. Cellular immune responses to HPV-18, -31, and -53 in healthy volunteers immunized with recombinant HPV-16 L1 virus-like particles. Virology. 2006;353:451–62. doi: 10.1016/j.virol.2006.06.021. [DOI] [PubMed] [Google Scholar]
- 19.Gravitt PE, Peyton CL, Alessi TQ, et al. Improved amplification of genital human papillomaviruses. J Clin Microbiol. 2000;38:357–61. doi: 10.1128/jcm.38.1.357-361.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Peyton CL, Gravitt PE, Hunt WC, et al. Determinants of genital human papillomavirus detection in a US population. J Infect Dis. 2001;183:1554–64. doi: 10.1086/320696. [DOI] [PubMed] [Google Scholar]
- 21.Schiffman M, Adrianza ME. ASCUS-LSIL Triage Study. Design, methods and characteristics of trial participants. Acta Cytol. 2000;44:726–42. doi: 10.1159/000328554. [DOI] [PubMed] [Google Scholar]
- 22.Results of a randomized trial on the management of cytology interpretations of atypical squamous cells of undetermined significance. Am J Obstet Gynecol. 2003;188:1383–92. doi: 10.1067/mob.2003.457. [DOI] [PubMed] [Google Scholar]
- 23.Xi LF, Koutsky LA, Castle PE, et al. Human papillomavirus type 18 DNA load and 2-year cumulative diagnoses of cervical intraepithelial neoplasia grades 2-3. J Natl Cancer Inst. 2009;101:153–61. doi: 10.1093/jnci/djn461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ananth CV, Kleinbaum DG. Regression models for ordinal responses: a review of methods and applications. Int J Epidemiol. 1997;26:1323–33. doi: 10.1093/ije/26.6.1323. [DOI] [PubMed] [Google Scholar]
- 25.Slinker BK, Glantz SA. Multiple linear regression is a useful alternative to traditional analyses of variance. Am J Physiol. 1988;255:R353–67. doi: 10.1152/ajpregu.1988.255.3.R353. [DOI] [PubMed] [Google Scholar]
- 26.Sherman ME, Wang SS, Wheeler CM, et al. Determinants of human papillomavirus load among women with histological cervical intraepithelial neoplasia 3: dominant impact of surrounding low-grade lesions. Cancer Epidemiol Biomarkers Prev. 2003;12:1038–44. [PubMed] [Google Scholar]
- 27.McLaughlin-Drubin ME, Meyers C. Evidence for the coexistence of two genital HPV types within the same host cell in vitro. Virology. 2004;321:173–80. doi: 10.1016/j.virol.2003.12.019. [DOI] [PubMed] [Google Scholar]
- 28.Combita AL, Touze A, Bousarghin L, Christensen ND, Coursaget P. Identification of two cross-neutralizing linear epitopes within the L1 major capsid protein of human papillomaviruses. J Virol. 2002;76:6480–6. doi: 10.1128/JVI.76.13.6480-6486.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McCarthy C, Youde SJ, Man S. Definition of an HPV18/45 cross-reactive human T-cell epitope after DNA immunisation of HLA-A2/KB transgenic mice. Int J Cancer. 2006;118:2514–21. doi: 10.1002/ijc.21643. [DOI] [PubMed] [Google Scholar]
- 30.Hildesheim A, Herrero R, Wacholder S, et al. Effect of human papillomavirus 16/18 L1 viruslike particle vaccine among young women with preexisting infection: a randomized trial. Jama. 2007;298:743–53. doi: 10.1001/jama.298.7.743. [DOI] [PubMed] [Google Scholar]
- 31.Paavonen J, Jenkins D, Bosch FX, et al. Efficacy of a prophylactic adjuvanted bivalent L1 virus-like-particle vaccine against infection with human papillomavirus types 16 and 18 in young women: an interim analysis of a phase III double-blind, randomised controlled trial. Lancet. 2007;369:2161–70. doi: 10.1016/S0140-6736(07)60946-5. [DOI] [PubMed] [Google Scholar]
- 32.Harper DM, Franco EL, Wheeler CM, et al. Sustained efficacy up to 4.5 years of a bivalent L1 virus-like particle vaccine against human papillomavirus types 16 and 18: follow-up from a randomised control trial. Lancet. 2006;367:1247–55. doi: 10.1016/S0140-6736(06)68439-0. [DOI] [PubMed] [Google Scholar]
- 33.Brown DR, Kjaer SK, Sigurdsson K, et al. The Impact of Quadrivalent Human Papillomavirus (HPV; Types 6, 11, 16, and 18) L1 Virus-Like Particle Vaccine on Infection and Disease Due to Oncogenic Nonvaccine HPV Types in Generally HPV-Naive Women Aged 16-26 Years. J Infect Dis. 2009;199:926–35. doi: 10.1086/597307. [DOI] [PubMed] [Google Scholar]
- 34.Wheeler CM, Kjaer SK, Sigurdsson K, et al. The Impact of Quadrivalent Human Papillomavirus (HPV; Types 6, 11, 16, and 18) L1 Virus-Like Particle Vaccine on Infection and Disease Due to Oncogenic Nonvaccine HPV Types in Sexually Active Women Aged 16-26 Years. J Infect Dis. 2009;199:936–44. doi: 10.1086/597309. [DOI] [PubMed] [Google Scholar]
- 35.Pinto LA, Edwards J, Castle PE, et al. Cellular immune responses to human papillomavirus (HPV)-16 L1 in healthy volunteers immunized with recombinant HPV-16 L1 virus-like particles. J Infect Dis. 2003;188:327–38. doi: 10.1086/376505. [DOI] [PubMed] [Google Scholar]
- 36.Plummer M, Schiffman M, Castle PE, Maucort-Boulch D, Wheeler CM. A 2-year prospective study of human papillomavirus persistence among women with a cytological diagnosis of atypical squamous cells of undetermined significance or low-grade squamous intraepithelial lesion. J Infect Dis. 2007;195:1582–9. doi: 10.1086/516784. [DOI] [PubMed] [Google Scholar]
- 37.Liaw KL, Hildesheim A, Burk RD, et al. A prospective study of human papillomavirus (HPV) type 16 DNA detection by polymerase chain reaction and its association with acquisition and persistence of other HPV types. J Infect Dis. 2001;183:8–15. doi: 10.1086/317638. [DOI] [PubMed] [Google Scholar]
- 38.Ho GY, Bierman R, Beardsley L, Chang CJ, Burk RD. Natural history of cervicovaginal papillomavirus infection in young women. N Engl J Med. 1998;338:423–8. doi: 10.1056/NEJM199802123380703. [DOI] [PubMed] [Google Scholar]
- 39.Trottier H, Mahmud S, Prado JC, et al. Type-specific duration of human papillomavirus infection: implications for human papillomavirus screening and vaccination. J Infect Dis. 2008;197:1436–47. doi: 10.1086/587698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Trottier H, Mahmud S, Costa MC, et al. Human papillomavirus infections with multiple types and risk of cervical neoplasia. Cancer Epidemiol Biomarkers Prev. 2006;15:1274–80. doi: 10.1158/1055-9965.EPI-06-0129. [DOI] [PubMed] [Google Scholar]