

Abstract
In contrast to many eukaryotic organisms in which kinetochores are assembled on localized centromeres of monocentric chromosomes, Caenorhabditis elegans has diffuse kinetochores, termed holo-kinetochores, which are assembled along the entire length of the mitotic chromosome. Despite this cytologically distinct chromosomal architecture, holo-kinetochores of C. elegans and kinetochores of other eukaryotes share structurally and functionally conserved properties. The amphitelic attachment of sister kinetochores to microtubules can be achieved by proper chromosomal organization, which relies on spatiotemporally orchestrated functions of conserved protein complexes such as the cohesin, condensin, and chromosomal passenger complexes during mitosis and meiosis in C. elegans. Moreover, the structure of spindle assembly checkpoint components and their safeguard function are also well conserved in C. elegans. Extensive efforts in the last few years to elucidate the molecular mechanisms of the C. elegans spindle assembly checkpoint have revealed its unique features. In this review, I will focus on the conservation and diversity of proteins that are required to maintain chromosome transmission fidelity during mitosis and meiosis in C. elegans.
Keywords: C. elegans, Kinetochore, Spindle assembly checkpoint
2. INTRODUCTION
The kinetochore is a protein complex with highly ordered structure that is assembled on a chromosomal region called the centromere, which includes unique nucleosomes containing the histone H3 variant CENP-A. The centromere serves as the platform that mediates and monitors microtubule-chromosome attachment to ensure proper chromosome segregation. The kinetochore's major functions are to provide the interface for spindle microtubules, to generate the force and tension required for chromosome movement, to sense the association of microtubules, and to regulate the timing of anaphase onset via spindle assembly checkpoint.
Unlike many eukaryotic organisms which have monocentric chromosomes, where localized kinetochores are assembled on centromeres that are specialized by unique sequences and chromatin structure, Caenorhabditis elegans has holocentric chromosomes, where kinetochores are assembled along the entire length of the chromosomes in embryonic cells undergoing mitosis (Figure 1). Despite this cytologically distinct chromosomal architecture, C. elegans shares the structurally and functionally conserved kinetochore properties with those organisms that have monocentric kinetochores. In this review, I will focus on the history of identifying and characterizing each kinetochore component, the spindle assembly checkpoint signaling pathway and proteins required for maintaining the chromosome transmission fidelity in C. elegans and describe their structures and functions from the aspects of conservation and diversity.
Figure 1. C. elegans has holo-kinetochores.

The conserved spindle assembly checkpoint protein BUB-1 kinase localizes to the diffuse C. elegans holo-kinetochore. Immunostaining of C. elegans embryonic cells at prometaphase (left column) and metaphase (right column) with anti–BUB-1 antibody and anti-tubulin antibody is shown. DNA is also visualized by staining with DAPI. The merged images of BUB-1 (red), DNA (blue), and tubulin (green) are shown in the bottom panel. In prometaphase, BUB-1 localizes along the entire length of each pair of sister chromatids. In metaphase, sister chromatids are highly compacted and congressed at the metaphase plate, where BUB-1 localizes the pole-ward face of each chromosome, thereby forming the classic two-threads shape. The anti-BUB-1 antibody for immunostaining was kindly provided by A. Desai.
3. ARCHITECTURE OF THE C. ELEGANS KINETOCHORE
3.1. Electron microscopic analysis of C. elegans kinetochore structure
The C. elegans kinetochore is one of the best-characterized holocentric kinetochores. Transmission electron microscopy (EM) of C. elegans mitotic chromosomes revealed the presence of a trilaminar kinetochore structure composed of electron-dense inner and outer layers with an electron-lucent middle layer extending along the entire pole-ward face of each chromatid (1). Recently, the ultrastructure of the C. elegans kinetochore was analyzed using high-pressure freezing and freeze substitution (HPF/FS) for better preservation of structure (2). Using this method, Howe et al. (2001) observed a morphology that differed from the trilaminar structure defined by conventional EM. Instead of the trilaminar structure, a clear zone that excluded ribosomes and other cytoplasmic components was found along each pole-ward face of mitotic chromatids. In some sections, a line of lightly stained material was detected between the clear zone and the chromatin. These features are reminiscent of the structure of monocentric chromosomes observed in rat kangaroo cells prepared with by HPF/FS (3), suggesting that despite their diffuse appearance, the structure of kinetochores assembled on holo-centric chromosomes during mitosis is similar to that of kinetochores assembled on monocentric chromosomes. (EM analysis of the kinetochore structure assembled on meiotic chromosomes will be discussed later in section 6.)
3.2. Conserved kinetochore components in C. elegans
The molecular composition between the C. elegans kinetochore and kinetochores in other organisms [reviewed in Refs. (4, 5)] are also substantially similar (Table 1; Figure 2). More than 30 C. elegans kinetochore proteins have been identified to date by diverse approaches. The Roth group first identified C. elegans kinetochore proteins named holo-centric proteins (HCPs) in 1999 (6, 7). They identified and characterized several HCPs (HCP-1 to -4 and HCP-6) via diverse approaches. Therefore, their designation of HCP has a completely cytological basis. HCP-1 was identified by a screen for antibodies that recognize holo-kinetochore proteins (7); HCP-2 to -4 were identified by homology searches using genomic DNA sequence information (6-8); HCP-6 was identified by a genetic screen (9) and it turned out to be a component of mitotic condensin complex. Before these proteins were identified, there was no marker for C. elegans kinetochores. Although MPM-2, a monoclonal antibody to phospho-epitopes localized at kinetochores in mammals, also cross-reacts with the C. elegans holokinetohores (7), those phospho-proteins recognized by MPM-2 have not been characterized. Thus, the identification of HCPs began a new era for the study in C. elegans kinetochores. More specifically, the identification of C. elegans homologs of CENP-A and CENP-C (HCP-3/CeCENP-A and HCP-4/CeCENP-C, respectively) accelerated our understanding of C. elegans kinetochore structure. The field advanced to its next phase when in 2001 Oegema et al. developed a time-lapse imaging system for quantitative functional analysis of the kinetochore proteins in C. elegans early-stage embryos (10). Stably transformed worms that express proteins of interest fused with GFP in embryonic cells can be generated by ballistic bombardment–mediated integrative transformation (11). One of the most useful transgenic strains currently available for functional analysis of chromosome segregation is the one that expresses GFP::tubulin and GFP::histone H2B. Embryos expressing these proteins fused to GFP are dissected and mounted on a thin layer of a 2% agarose pad on a cover glass, and sequential fluorescent images of the transgenic embryos are filmed [described in Gassmann et al (2007) (12)]. Unlike the analysis of fixed cells, the use of the time-lapse imaging system to analyze chromosome and kinetochore movements after nuclear envelope breakdown in transgenic embryos depleted of a specific protein enables the precise characterization of the defects in chromosome dynamics. Oegema et al. defined the phenotype of 1-cell–staged embryos depleted of HCP-3/CeCENP-A as the kinetochore-null (KNL) phenotype, and used it as a reference for genome-wide functional analysis by RNAi to identify key outer kinetochore components named KNL proteins. Identification of KNL proteins has been followed by biochemical purifications of KNL-associating proteins. These studies in combination with comprehensive cross-reference search identified and characterized many of the kinetochore proteins that are conserved between humans and C. elegans. A brief history of those proteins follows. A list of kinetochore associating proteins identified to date is summarized in Table 1. Most of kinetochore proteins have critical roles both in mitosis and meiosis. The localization and functional studies of these proteins during meiosis will be discussed later (Section 6).
Table 1.
Kinetochore associating proteins in C. elegans
| C. elegans protein (gene name) |
Human homolog |
Notable domain | Reference # | |
|---|---|---|---|---|
| Kinetochore specificity |
HCP-3 (F58A4.3) |
CENP-A | Histone H3 variant | (6, 10, 17- 19) |
| HCP-4 (T03F1.9) |
CENP-C | Consensus: VRR[S/T]XR[T/I/V]R[L/V] KPLEYWRGER[I/V/P]XY |
(8, 10, 17) | |
| KNL-2 (K06A5.4) |
c14orf106 | Myb domain | (13) | |
| Microtubule interface at the outer kinetochore |
KNL-1 (C02F5.1) |
BLINKIN | [S/G]ILK motif RRSVF motif MELT repeats Coiled coil |
(17, 27) |
| KNL-3 (T10B5.6) |
(27) | |||
| MIS-12 (Y47G6A.24) |
MIS12 | (27) | ||
| KBP-1 (R13F6.1) |
(27) | |||
| KBP-2 (F26F4.13) |
(27) | |||
| NDC-80 (W01B6.9.1) |
HEC1 | (17, 27) | ||
| HIM-10 (R12B2.4) |
NUF2 | N-terminal conserved domain (Nuf2 family protein) |
(2) | |
| KBP-3 (F26H11.1) |
SPC25 | (27) | ||
| KBP-4 (Y92C3B.1) |
SPC24 | (27) | ||
| Other outer kinetochore components |
KBP-5 (C34B2.2) |
(27) | ||
| HCP-1 (ZK1055.1) |
CENP-F | Coiled coil Repeated motif conserved between HCP-1 and CENP- F (not present in HCP-2) |
(7, 17, 18, 37) |
|
| HCP-2 (T06E4.1) |
CENP-F | Coiled Coil | (7, 17, 18, 37) |
|
| CLS-2 (R107.6) |
CLASP | CLIP associating | (37) | |
| KLP-7 (K11D9.1) |
MCAK | Kinesin moter domain | (10, 18) | |
| LIS-1 (T03F6.5) |
LIS1 | WD40 repeat | (43) | |
| MEL-28 (C38D4.3) |
AHCTF1 | AT-hook-containing | (45, 46) | |
| Spindle assembly Checkpoint |
BUB-1 (R06C7.8) |
BUB1 | Kinase | (10, 17, 18) |
| BUB-3 (Y54G9A.6) |
BUB3 | WD40 repeat | (38, 39) | |
| MDF-1 (C50F4.11) |
MAD1 | Coiled coil | (18, 84) | |
| MDF-2 (Y69A2AR.30) |
MAD2 | HORMA domain | (18, 84) | |
| SAN-1 (ZC328.4) |
BUBR1/Mad3 | (24, 92, 93) | ||
| RZZ complex | CZW-1 (F20D12.4) |
ZW10 | (88, 97) | |
| ROD-1 (F55G1.4) |
ROD | (88, 97) | ||
| ZWL-1 (Y39G10AR.2) |
ZWILCH | |||
| APC actovator |
FZY-1 (ZK177.6) |
CDC20 | WD40 repeats | (114) |
| Condensin II | SMC-4 (F35G12.8) |
SMC4 | SMC domain | (52, 53) |
| HCP-6 (Y110A7A.1) |
CAP-D3 | HEAT motif | (9, 52, 53) | |
| MIX-1 (M106.1) |
SMC2 | SMC domain | (52, 53) |
Figure 2. Kinetochore proteins of C. elegans.
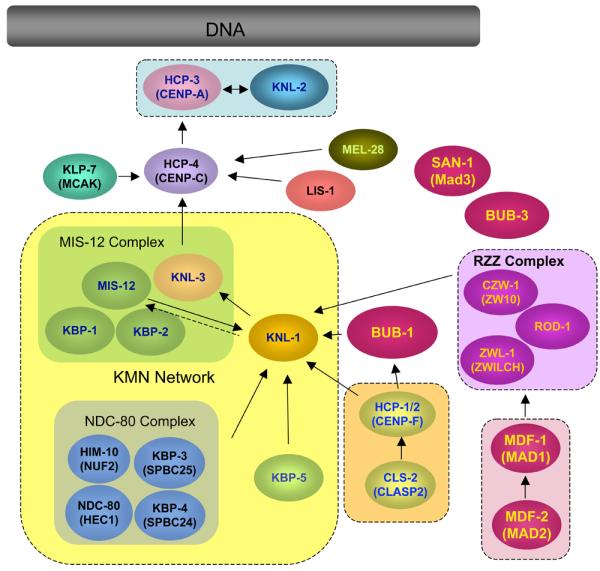
Proteins that localize to C. elegans kinetochores are illustrated with arrows indicating hierarchic molecular dependencies for mitotic kinetochore assembly. Localization dependency was determined by analysis of the effect of RNAi mediated depletion of a protein on kinetochore localization of other protein: For example, if depletion of protein B reduced the amount of protein A at the kinetochore, the dependency is shown as [A → B] i.e. A depends its kinetochore localization on B. Proteins framed in by dashed lines have been biochemically shown to associate each other. See text for detailed information of each protein.
3.2.1. Proteins that establish the foundation for the kinetochore assembly
Kinetochore assembly in C. elegans depends on 2 conserved proteins – a special type of histone H3 variant, HCP-3/CeCENP-A, and its associating protein, HCP-4/CeCENP-C. The HCP-3/CeCENP-A–containing chromatin creates the foundation for kinetochore assembly in the same manner as CENP-A chromatins do in monocentric organisms. HCP-4/CeCENP-C serves as a linker between HCP-3/CeCENP-A chromatins and the highly ordered outer kinetochore complex. To date, no specific sequence requirement for holo-kinetochore assembly in C. elegans has been identified. However, the timing of kinetochore assembly is tightly regulated during the cell cycle. It has recently been found that the protein KNL-2 is required for loading of HCP-3/CeCENP-A onto the chromosomes (13). Of the several known kinetochore-associating proteins, HCP-3/CeCENP-A and KNL-2 are only ones that associate with chromosomes throughout the cell cycle. These proteins play a critical role in establishment of the foundation for kinetochore assembly.
HCP-3/CeCENP-A
HCP-3 was identified by Buchwitz et al. (1999) as a protein that shares sequence similarity in the histone-hold domain with other histone H3 proteins, but its amino-terminal region diverges, as do the centromeric histone H3 variants CENP-A and Cse4 (6, 14-16). Their immunostaining analysis revealed the cell cycle–dependent subcellular localization of HCP-3 (6). During interphase, HCP-3 staining is detected in nuclear foci. In prophase, HCP-3 appears as single lines associated with the condensing chromosomes. At prometaphase, HCP-3 localizes at both sides of condensed chromosomes, along the entire length. At metaphase and anaphase, HCP-3 is detected on the pole-ward face of each set of sister chromatids. Thus, HCP-3 associates with chromosomes throughout the cell cycle and is the first kinetochore component that localizes to C. elegans holo-centric chromosomes.
HCP-3–depleted embryos show varied size and intensity of DAPI-stained nuclei, suggesting that HCP-3 functions in proper chromosome segregation during embryonic cell division cycle (6). The role of this protein in mitotic chromosome segregation has been analyzed further in the living embryos that undergo first mitosis by Oegema et al. (2001) using a time lapse-imaging system. Consistent with the idea that HCP-3 shares the kinetochore function with CENP-A in other organisms, HCP-3 specifies the fundamental kinetochore structure on which localization of all other kinetochore proteins depend (10, 17, 18). Therefore, depletion of HCP-3 causes misassembly of functional kinetochores, leading to complete loss of kinetochore-microtubule attachment. As a consequence, centrosome separation is accelerated, and sister chromatids do not align at a metaphase plate or separate. The phenotype of 1-cell–staged embryos depleted of HCP-3 is defined as the kinetochore-null (KNL) phenotype.
Maddox et al. developed a fluorescent microscopy based method to quantify the extent of chromosome condensation in living embryonic cells expressing GFP::Histone (19). Their analysis using that method revealed that HCP-3 plays a critical role in chromosome condensation during prophase independently of its role in directing kinetochore assembly (19).
HCP-4/CeCENP-C
HCP-4 was identified as a C. elegans protein containing a short sequence similar to that of CENP-C. HCP-4 was found in the cytoplasm of interphase cells but is colocalized with HCP-3 on holo-kinetochores during mitosis (8, 20-23). Kinetochore localization of HCP-4 depends on HCP-3 and is required for the localization of all kinetochore proteins, except HCP-3 (10, 17). The HCP-4–depleted embryos also exhibit the KNL phenotype with normal HCP-3 localization. Thus HCP-4 plays an important role in kinetochore assembly as a downstream of HCP-3. Although depletion of HCP-4 does not interfere with HCP-3 localization to chromosomes, it does hinder sister centromere resolution, which is reflected by a shift in the HCP-3 localization pattern from a single line to paired lines on each chromosome during prophase (24).
Depletion of HCP-4 slows but does not prevent the chromosome condensation during prophase (19), suggesting that HCP-3 containing nucleosomes have two distinctive roles: kinetochore assembly and chromosome condensation, and HCP-4 is specifically involved in kinetochore assembly.
KNL-2
An RNAi-based functional genomic screen of C. elegans chromosomes by using transgenic embryos expressing GFP::histone H2B and GFP::gamma-tubulin or GFP::alpha-tubulin has identified KNL-2 as 1 of 5 kinetochore proteins whose depletion causes the KNL phenotype, including KNL-1, -3, HCP-3/CeCENP-A, and HCP-4/CeCENP-C (13). Unlike other KNL proteins that function downstream of HCP-3/CeCENP-A in kinetochore assembly (see section 3.2.2), KNL-2 is required for loading of HCP-3/CeCENP-A onto chromosomes (13). When interphase nuclei containing GFP::KNL-2 were isolated, shred into chromatin fragments of 500–1500 bp, and immunoprecipitated with the GFP antibody, HCP-3/CeCENP-A but not histone H3 was coenriched with KNL-2 (13), suggesting that KNL-2 and HCP-3/CeCENP-A are in physical proximity on the chromatin. Furthermore, depletion of KNL-2 causes the same extent of chromosome condensation defects as that observed in cells depleted of HCP-3/CeCENP-A (13). These results suggest that KNL-2 plays an important role in chromosome condensation and kinetochore assembly by physically and functionally interacting with HCP-3/CeCENP-A. KNL-2 has a divergent version of the DNA-binding domain, designated as the Myb domain, at its C-terminus (13). Although the human homolog of KNL-2 has a highly diverse sequence, its role in CENP-A chromatin assembly is conserved (13).
3.2.2. Outer kinetochore components that serve as core microtubule biding sites
Although numerous kinetochore proteins have been identified, and great strides have been made toward understanding the kinetochore structure in the last few decades, the identities of the proteins that constitute the core microtubule-binding sites of the kinetochore have remained elusive until recently. An elegant biochemical approach by Cheeseman et al. (2006) to characterize the KNL-1/MIS-12/NDC-80 (KMN) complex network identified two distinct microtubule-binding activities within the network (25). Their data obtained using the reconstituted in vitro system strongly suggests that KMN network constitutes the core microtubule-binding sites of the C. elegans holo-kinetochore. The MIS-12 complex consists of at least four components, MIS-12, KNL-3, KBP-1 and -2, and NDC-80 complex is comprised of NDC-80/CeHEC1, HIM-10/CeMIF2, KBP-3/CeSPBC25 and -4/CeSPBC24. A brief description of each constituent of the complex follows:
KNL-1 protein
KNL-1 was originally identified by an RNAi-based functional genomic screen of C. elegans chromosome III as one of six genes, including smc-4, hcp-3, and cls-2, whose depletion by RNAi causes defects in chromosome segregation based on DIC microscope analysis (26). Further characterization of chromosome segregation defects by 3-dimensional time-lapse analysis using transgenic embryos expressing GFP::histone H2B and GFP::gamma-tubulin revealed that in KNL-1–depleted embryos, two masses of DNA derived from sperm nuclei and oocyte nuclei stay in the middle of two centrosomes without segregating toward each pole, while two centrosomes rapidly and prematurely separate (17). These phenotypes are identical to the KNL phenotype observed in HCP-3/CeCENP-A–depleted embryos. KNL-1 localizes at holo-kinetochores from early prophase until late anaphase (17). This localization requires HCP-3/CeCENP-A, HCP-4/CECENP-C, and KNL-3 (17, 27). KNL-1 has an asymmetric codependency with components of NDC-80 complex, NDC-80, and HIM-10/CeNUF-2 for its kinetochore localization (17). KNL-1 forms a network with KNL-3 and the MIS-12 and NDC-80 complexes (17, 27). KNL-1 is also required to target multiple outer kinetochore components such as HCP-1/CeCENP-F, BUB-1, and CLS-2/CeCLASP2 (17). These proteins have not been identified in KNL-1 immunoprecipitates (17, 27), suggesting that KNL-1 targets these proteins by recruiting additional intermediates and/or directing the assembly of the higher-order structure. Thus, most known outer kinetochore proteins are downstream of KNL-1 in the kinetochore-assembly hierarchy. This finding suggests that the KNL phenotype of KNL-1–depleted embryos arises from the loss of formation of the functional outer kinetochore domain. KNL-1 shares primary sequence features with the yeast kinetochore protein Spc105 and the human Mis12-associating protein AF15q14. All three proteins have the N-terminal [S/G]ILK and RRSVF motifs, the N-terminal MELT repeats, and the C-terminal coiled-coil domain (27). Biochemical characterization of KNL-1 revealed that the protein directly associates with microtubules in the reconstituted KNL-1/MIS-12 complex (25).
MIS-12 protein
MIS-12 was first copurified with KNL-1 and KNL-3 and then named according to its sequence similarity to MIS12 in other organisms (27). Further biochemical work identified the subprotein complex, which consists of three proteins, MIS-12, KBP-1, and KBP-2. Bacterial expression of recombinant KNL-3 protein was significantly stabilized by coexpression of all three components of the MIS-12 complex (25). Kinetochore localization of MIS-12 depends on KNL-3 and partially on KNL-1 (27). The formation of the MIS-12 complex affects the recruitment of KNL-3 into the kinetochore. Depletion of MIS-12, KBP-1 or KBP-2 causes a less severe defect in chromosome segregation in early-stage embryos than the defect observed in embryos depleted of KNL proteins or components of NDC-80 complex (27). In MIS-12–depleted embryos, the timing of chromosome separation was similar to that of wild-type embryos. However, defects in chromosome alignment or segregation were frequently observed. Mitotic chromosomes congressed at the metaphase plate were less compact than those in wild-type embryos. In addition to the chromosome segregation defects, MIS-12–depleted embryos undergoing the first mitosis showed a “spindle-bounce phenotype,” which reflects unique spindle pole–separation kinetics: centrosomes separated rapidly after nuclear envelope break down (NEBD) but then moved back toward the metaphase plates; then they separated again upon sister chromatid separation (27). The spindle-bounce phenotype is a common characteristic of embryos in which components of the MIS-12 complex have been depleted. This phenotype suggests that robust kinetochore-microtubule attachment is initially defective but subsequently corrected in the absence of the MIS-12 complex. Furthermore, depletion of MIS-12 complex components results in delayed and reduced assembly of the outer kinetochore (27). Thus, MIS-12 controls the rate and extent of the outer kinetochore assembly.
KNL-3 protein
KNL-3 was also identified as a kinetochroe protein whose depletion causes the KNL phenotype. Kinetochore localization of KNL-3 coincides with HCP-4/CeCENP-C and KNL-1 (27). KNL-3 is downstream of HCP-3/CeCENP-A and HCP-4/CeCENP-C and is upstream of KNL-1 in a linear-assembly hierarchy (27). KNL-3 and the MIS-12 complex are also codependent for the kinetochore localization (27). KNL-3 copurified with KNL-1, MIS-12 complex, NDC-80 complex, and HCP-4/CeCENP-C (27). This finding suggests that HCP-4/CeCENP-C connects the KMN network to HCP-3/CeCENP-A chromatin by interacting with KNL-3.
KBP-1 and KBP-2
KBP-1 and -2 were identified as KNL-binding proteins by immunoprecipitation/mass spectrometry using specific antibodies to KNL-1 or KNL-3 and a tandem affinity-purification method (27). Both proteins are components of the MIS-12 complex, and embryos depleted of either protein exhibit the same phenotypes as those observed in MIS-12–depleted embryos (27).
NDC-80/CeHEC1
NDC-80 copurified with HIM-10/CeNUF-2 in KNL-1 immunoprecipitates (17). The kinetochore localization of NDC-80 depends on KNL-1 and HIM-10/CeNUF-2 (17). Depletion of NDC-80 slightly reduces kinetochore localization of KNL-1 but does not affect KNL-3 or MIS-12 localization (17) (27). Loss of NDC-80 causes substantial but less severe defects in chromosome segregation than those observed in embryos depleted of KNL protein (17) (27). In embryos depleted of NDC-80, mitotic chromosomes form a disorganized metaphase plate; sister chromatid separation is delayed; chromosomes are often missegregated; and spindle poles are prematurely and rapidly separated (17). NDC-80 forms a heterodimer with HIM-10/CeNUF-2 and interacts with the KBP-3–KBP-4 dimer in the NDC-80 complex (25). Biochemical experiments revealed that the NDC-80–HIM-10 dimer binds to and bundles microtubules but does not bind the MIS-12 complex or KNL-1 in a reconstituted system (25). In this system, the microtubule-binding activity of the NDC-80–HIM-10 dimer is regulated by phosphorylation of NDC-80 by the Aurora B kinase AIR-2 (25).
HIM-10/CeNUF-2
HIM-10 was originally isolated as a mutant strain that shows the high incidence of males (HIM) phenotype that reflects the high frequency of X-chromosome nondisjunction during meiosis (28). Thus, unlike other kinetochore components, HIM-10 was first identified as a protein required for proper chromosome segregation during meiotic division. Further genetic characterization of a him-10–mutant strain demonstrated that the loss of HIM-10 function reduces the transmission frequency of free duplications (inheritable chromosome fragments) during somatic cell division. This finding suggests that HIM-10 has a role in chromosome segregation during mitosis (29). Molecular characterization of the gene product by Howe et al. (2001) revealed that HIM-10 is a C. elegans homolog of NUF2, a conserved kinetochore protein (2, 30). As suggested by its structural similarity to NUF2, HIM-10 plays a role in kinetochore function as a component of the NDC-80 complex that forms a dimer with NDC-80 (25). Depletion of HIM-10 disrupts the ultrastructure of the kinetochore (2) and causes chromosome missegregation and premature spindle pole separation (17, 27).
KBP-3 and KBP-4
KBP-3 and -4 copurified with KNL-1 or with KNL-3 (27). The phenotype of embryos depleted of KBP-3 or KBP-4 is similar to that of embryos depleted of NDC-80. Furthermore, the sequences of KBP-3 and KBP-4 are similar to those of SPBC25 and SPBC24, human kinetochore components that form a complex with HEC1(human homolog of NDC-80) (31, 32). These findings helped characterize these components of the NDC-80 complex. KBP-3 and KBP-4 form a dimer that binds to the NDC-80–HIM-10 dimer in the NDC-80 complex (25). The KBP-3–KBP-4 dimer mediates the interaction of the NDC-80 complex with KNL-1 and the MIS-12 complex in the reconstituted system (25).
3.2.3. Other proteins that localize to kinetochores
Since the sequencing of the entire C. elegans genome was completed, continuous efforts have been made to identify and characterize proteins that are conserved between C. elegans and yeast or humans. Many C. elegans kinetochore proteins have been identified based on the similarity of their amino acid sequence to that of kinetochore components identified in other organisms. Furthermore, parallel biochemical and genetic approaches such as the purification of physical interactors with known kinetochore components or screens for genes required for proper chromosome segregation during early embryogenesis also identified additional kinetochore components. Described below are proteins that localize to holo-kinetochores in C. elegans embryos undergoing mitotic division.
HCP-1/2/CeCENP-F
Similar to human CENP-A, -B, and -C proteins, which were identified as epitopes of anti-centromere antibody (ACA) in patients with CREST (calcinosis, Raynaud's phenomenon, esophageal dysmotility, sclerodactyly, telangiectasia) (15, 33-36), HCP-1 was first identified as an epitope of the mouse monoclonal antibody 6C4, which was isolated in a screen of hybridoma cell lines (7). HCP-1 is a coiled-coil protein with a direct repeat of 132 amino acids and 45% similarity with a direct repeat of 179 amino acids of CENP-F (20-23). Dynamic localization of HCP-1 throughout mitosis was first demonstrated by Moore et al. (1999) by immunostaining with 6C4 (7). Further analysis by other proteins using a specific anti–HCP-1 polyclonal antibody or a transgenic strain expressing GFP::HCP-1 confirmed the earlier finding (17, 18, 37). No association with chromosomes was detectable during interphase; punctate distribution along the entire chromosomes appeared in prophase; and holo-kinetochore localization was observed between late prometaphase to early anaphase.
HCP-2 was identified as a homolog of HCP-1. HCP-2 has a 54% similarity to HCP-1 in the entire amino acid sequence but lacks the direct repeat found in HCP-1 and CENP-F (7). The 6C4 antibody most likely recognizes only HCP-1. Cheeseman et al. (2005) generated an anti–HCP-2 antibody and confirmed that the subcellular localization of this protein was identical to that of HCP-1 (37). Furthermore, they demonstrated coimmunoprecipitation of HCP-1 and HCP-2 (37), suggesting that these proteins function in a complex. Depletion of more than 77% of HCP-1 does not reduce viability or induce defects in chromosome segregation; this finding can be explained by the functional redundancy with HCP-2 (7). Therefore, in most cases, physiologic function of HCP-1 and -2 has been analyzed by characterizing the phenotype caused by their simultaneous depletion, which is often referred to as hcp-1/2(RNAi). Codepletion of HCP-1/2 causes embryonic arrest at a high penetrance (7, 18, 37). In arrested embryos, aneuploid nuclei, multinucleate cells, and anaphase bridges are observed, suggesting that these embryos have defects in chromosome segregation, though there is no sign of defects in chromosome condensation (7). Finer characterization of the hcp-1/2 RNAi phenotype in 1-cell–staged embryos undergoing their first mitosis was made possible by a time lapse imaging system. Encalada et al. (2005) and Cheeseman et al. (2005) defined more precise roles of HCP-1 and -2 in chromosome segregation (18, 37) using this system: In 1-cell–staged embryos depleted of HCP-1/2, mitotic spindles form normally during early mitosis, but the midzone spindle between separating sister chromatids at anaphase is missing (18); centrosome separation is accelerated; centrosome-chromsome distance is decreased; and sister chromatids are segregated to the same pole (37). Together, HCP-1 and -2 are required for sister chromatid biorientation. Depletion of HCP-1/2 bypasses the mitotic delay induced by chemical or mutational disruption of microtubules in early-stage embryos, suggesting that these proteins have a role in the spindle assembly checkpoint pathway (18). Kinetochore localization of HCP-1/2 depends on BUB-1, KLP-7/CeMCAK, HCP-3/CeCENP-A, HCP-4/CeCENP-C, and KNL-1 but does not depend on the NDC-80/CeHEC1 complex (17). HCP-1/2 associates with and targets CLS-2/CeCLASP to kinetochores (27). Because the HCP-1/2 and CLS-2 RNAi phenotypes are indistinguishable, the major function of HCP-1/2 may be to recruit CLS-2 to the kinetochores. Recent RNAi-based synthetic genetic analysis has revealed that depletion of hcp-1 but not hcp-2 is synthetically lethal with deletion of the spindle assembly checkpoint (38, 39), suggesting that HCP-1 and HCP-2 have distinct functions.
CLS-2/CeCLASP2
CLS-2 was identified as a C. elegans homolog of CLASP2 (cytoplasmic linker–associated protein 2), a microtubule-binding protein that acts in microtubule stabilization, mitotic spindle formation, and chromosomal alignment during mitotic progression (20-23). Depletion of CLS-2 causes sister chromatid nondisjunction, presumably due to failure to form bioriented microtubule attachment, accelerated centrosome separation, dramatic oscillation of sister chromatids between centrosomes during prometaphase, and reduction of the interval between centrosome and sister chromatid (37). On the other hand, global microtubule-dependent processes such as pronuclear migration and rotation to align the centrosomes with the long axis of embryos are not affected by the lack of CLS-2 (37). Thus, CLS-2–depleted embryos exhibit mitotic defects similar to those of HCP-1/2/CeCENP-F–depleted embryos.
CLS-2 copurified with HCP-1/2 and requires those proteins for its kinetochore localization. Furthermore, codepletion of CLS-2 and HCP-1/2 does not cause more severe mitotic defects than those caused by depletion of either CLS-1 alone or HCP-1/2 (37). On the basis of these observations, it is thought that the major function of HCP-1/2 is to recruit CLS-2 to the kinetochore. Because chromatid nondisjunction and premature centrosome separation can be suppressed by depletion of GPR-1 and GPR-2, which are required for generating astral pulling forces (37), the observed defects may also require the astral pulling forces.
KLP-7/CeMCAK
klp-7 was identified as a gene encoding a kinesin-like protein related to MKAC (40). After the NEBD during prometaphase, KLP-7 localizes at the outer kinetochores on condensed chromosomes and remains there until telophase (10, 18). Kinetochore localization of KLP-7 depends on HCP-4/CeCENP-C but not on KNL-1, ICP-1 (C. elegans homolog of INCENP, CeINCENP), AIR-2 (C. elegans homolog of Aurora B), SCC-3 or HCP-1/2/CeCENP-F (10, 17, 18, 24). KLP-7 also localizes at centrosomes during mitosis, independently of HCP-3/CeCENP-A and HCP-4/CeCENP-C (10). Embryos depleted of KLP-7 lack the spindle midzone, but the astral microtubule is unaffected, resulting in rapid and premature centrosome separation (10, 18). In KLP-7–depleted embryos, kinetochore localization of HCP-1 and HCP-2 are compromised (18). Depletion of KLP-7 reduces the microtubule growth rate but increases microtubule nucleation and retrograde movement (41).
LIS-1
LIS-1 was identified as a C. elegans homolog of LIS1, a conserved WD40 repeat–containing protein encoded by a human disease gene responsible for lissencephaly, a severe neurological disorder characterized by a lack of proliferation, migration, and survival of cortical neurons during development (42). LIS-1 plays roles in dynein-dependent cellular processes such as pronuclear migration, centrosome separation, spindle assembly, and cell division during embryogenesis most likely via interaction with cytoplasmic dynein heavy chain DHC-1 (43). LIS-1 localizes to the cytoplasm, cell cortex, nuclear periphery microtubule asters, and kinetochores (43). LIS-1 and DHC-1 are codependent for localization to the cell cortex, nuclear periphery, and microtubule asters (43). In contrast, LIS-1 localization to the kinetochore does not depend on DHC-1 or ICP-1/CeINCENP but does depend on HCP-4/CeCENP-C (43). LIS-1 does not require microtubule assembly for its localization to the cell cortex, nuclear periphery, or kinetochores (43). The role of LIS-1 at the kinetochore remains unknown.
KBP-5
KBP-5 copurified with KNL-1, or with KNL-3, as one of KBP proteins (27). KBP-5 is the only nonessential protein among KBP proteins. Kinetochore localization of KBP-5 depends on KNL-1 (27). The molecular function of KBP-5 remains unknown.
MEL-28
MEL-28 is a protein encoded by mel-28, a gene originally isolated as a maternal–lethal effect mutant (44). It has two adenine-thymine (AT) hooks in the C terminus. MEL-28 localizes to the nuclear envelope during interphase and to kinetochores during mitosis (45, 46). Both nuclear envelope localization and kinetochore localization depend on components of the Ran cycle such as RAN-1 (Ran), RAN-2 (RanGAP), NPP-9 (Nup358 or RanGAP cofactor RanBP2), IMB-1 (importin b), and IMA-2 (importin a) (45). Kinetochore localization of MEL-28 also requires HCP-3/CeCENP-A and HCP-4/CeCENP-C (45). Depletion of MEL-28 reduces KNL-3 at the kinetochore, separates centrosomes prematurely, and forms chromosome bridges during anaphase (45). The depletion of MEL-28 also causes a disorganized nuclear envelope (45, 46). KNL-3 depletion does not affect the nuclear envelope structure, and disruption of the nuclear envelope by depletion of NPP-1, a nuclear pore component, does not affect the kinetochore function. Thus, the maintenance of nuclear envelope integrity and kinetochore function are genetically independent cellular processes. MEL-28 is required for both processes, suggesting that this protein functions as a downstream effector of the RAN cycle, which has dual functions in both mitotic spindle assembly and nuclear envelope reassembly.
3.3. Summary and perspectives
C. elegans kinetochores are assembled along the longitudinal axis of mitotic chromosomes, and are therefore termed holo-kinetochores; thus, they are cytologically distinct from kinetochores of other organisms. However, EM analysis of kinetochore architecture reveals that C. elegans holo-kinetochores are structurally similar to those assembled on localized centromeres in monocentric organisms. Identification of proteins that localize to C. elegans kinetochores, isolation of mutants that exhibit defects in chromosome segregation (forward genetics), and RNAi-based functional analysis of proteins homologous to constituents of kinetochores in other organisms (reverse genetics) have identified many C. elegans kinetochore proteins that are structurally and functionally conserved. Thus, with regard to molecular composition, the C. elegans holo-kinetochore shares many common features with monocentric kinetochores in other organisms; however, how the C. elegans holo-kinetochore is assembled on a DNA sequence remains unknown. The key component that specifies the fundamental kinetochore structure is CENP-A.proteins. Before reorganization of the kinetochore structure during prophase, HCP-3/CeCENP-A forms nuclear foci during interphase (G1-S-G2 phase). Chromatin immunoprecipitation (ChIP) is a good technique to identify the chromosomal regions at which HCP-3/CeCENP-A first associates with chromosomes. Characterization of the cis-elements, which mediates HCP-3/CeCENP-A–chromosome interaction, will help us understand how the C. elegans holo-kinetochore is first established before it is eventually assembled independent of any specific DNA sequence. Recently, a Myb DNA-binding domain containing protein, KNL-2 has been identified as a protein required for HCP-3/CENP-A chromatin assembly (13). How the Myb domain of KNL-2 contributes to load HCP-3/CENP-A would be of great interest, too.
4. PROTEIN COMPLEXES REQUIRED FOR MAINTENANCE OF MITOTIC CHROMOSOME ORGANIZATION
To achieve proper chromosome segregation, kinetochores must capture bioriented spindle microtubules before sister chromatid separation. In other words, improper kinetochore–microtubule interaction must be prevented or corrected before anaphase onset. The conserved protein complexes condensin and cohesin maintain the integrity of mitotic chromosomes. The chromosome passenger complex detects and corrects aberrant microtubule–kinetochore binding, thereby ensuring proper kinetochore–microtubule attachment and efficient chromosome transmission during cell division. Most constituents in these three complexes are highly conserved between C. elegans and other organisms. This section discusses the current understanding about the function of each complex in chromosome segregation in C. elegans.
4.1. Role of the Condensin complex during chromosome segregation in C. elegans
One key event that occurs at the beginning of mitosis is chromosome condensation. Condensation makes chromosomes short enough to achieve proper sister chromatid separation without the sisters becoming entangled. This condensation depends on the conserved protein complex condensin. Condensin consists of a heterodimer of structural maintenance of chromosomes (SMC) family proteins, SMC2-SMC4, and three non-SMC proteins (reviewed in Refs. (47, 48). In vertebrates, there are two types of condensin complexes, condensin I and condensin II (49). Both complexes contain the SMC2-SMC4 heterodimer as a common constituent, but their other components diverge, making them functionally distinct complexes. The RNAi mediated depletion or immuno-depletion of condensin I- or II-specific subunits in HeLa cells or in Xenopus egg extracts causes distinct phenotypes: chromosomes appear swollen or curly in the absence of condensin I or II, respectively (49). Furthermore, analysis of protein dynamics during the cell cycle progression in living mammalian cells revealed that condensin I associates with mitotic chromosomes after NEBD, and is required for mechanical stabilization of chromosomes rather than chromosome compaction, while condensin II associates with chromosome during prophase and is required for condensation of chromosomes during early prophase (50, 51).
Unlike most organisms that have only one set of SMC2 and SMC4 genes, C. elegans has one SMC2 named MIX-1(mitosis and X-associated)/CeSMC-2 and two SMC4s named DPY-27 and SMC-4. MIX-1/CeSMC-2 forms a heterodimer with DPY-27 in a complex required for dosage compensation of X-linked genes and her-1 gene (referred to as condensin IDC: DC stands for dosage compensation), which contains DPY-26(CAP-H) and DPY-28(CAP-D2) as non-SMC subunits. MIX-1/CeSMC-2 also forms a heterodimer with SMC-4 in the condensin II mitotic complex (52) (Figure 3, Table 2), which contains HCP-6/CeCAP-D3 as a non-SMC subunit (53). A gene encoding HCP-6 was originally isolated as the temperature-sensitive mutant allele mr17 in a screen for chromosome-segregation mutants (9). The mr17-mutant embryos arrest at the nonpermissive temperature, and abnormal chromosome segregation is reflected by unequally distributed nuclei, multinucleate cells, and anaphase bridging (9). The mr17 mutation was mapped in the hcp-6 gene based on the facts that the mr17 phenotype was phenocopied by depletion of the gene by RNAi and is complemented by ectopic expression of the gene. Mitotic chromosomes in embryos depleted of HCP-6 are not condensed, are less rigid, and are more susceptible to twisting (9). Such a loss of chromosome integrity causes merotelic-oriented chromosomes at a high frequency, leading to anaphase bridging or lagging chromosomes. In interphase cells, HCP-6 is found in the nuclear foci, which are distinct from HCP-3/CeCENP-A foci (9). During mitosis, localization of HCP-6 to holo-kinetochores requires HCP-3/CeCENP-A and HCP-4/CeCENP-C (9, 53). HCP-6 coimmunoprecipitated with the C. elegans condensin subunit MIX-1/CeSMC-2 (53). Although HCP-6 is loaded to chromatin independently of MIX-1/CeSMC-2, HCP-6 and MIX-1/CeSMC-2 colocalize to holo-kinetochores (53). Furthermore, embryos depleted of HCP-6 and those depleted of MIX-1/CeSMC-2 exhibit similar chromosome segregation defects (9, 53). Thus, consistent with the role of condensin II in other organisms, HCP-6/CeCAP-D3 is required for maintaining the integrity of mitotic chromosomes, which facilitates proper orientation of sister kinetochores toward each pole, thereby preventing the formation of merotelic-oriented chromosomes, suggesting that HCP-6 functions in mitotic chromosome segregation as a constituent of condensin II.
Figure 3. Condensin complex and cohesin complex in C. elegans.
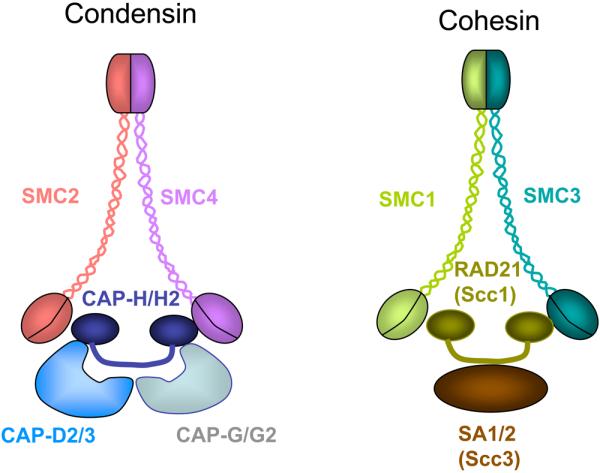
SMC-protein complexes, condensin, and cohesin, are conserved in eukaryotes. Their common structures are shown. An SMC2-SMC4 heterodimer functions as the core of condensin I and II. Condensin I contains the non-SMC subunits CAP-D2, CAP-H, and CAP-G; Condensin II contains CAP-D3, CAP-H2, and CAP-G2. An SMC1-SMC3 heterodimer functions as the core of the cohesin complex, which contains two non-SMC subunits, Rad21/Scc1 and SA proteins/Scc3. Redrawn with permission from Fig. 1 in (45).
Table 2.
Components of condensins and cohesins in C. elegans
| Subunit | Condensin I like Dosage compensation (Condensin IDC) |
Condensin II | Condensin IM | Mitotic Cohesin |
Meiotic Cohesin |
Unkown | |
|---|---|---|---|---|---|---|---|
| Condensin I&II | SMC2 | MIX-1/SMC-2 | MIX-1/SMC-2 | MIX-1/SMC-2 | |||
| SMC4 | DPY-27 | SMC-4 | SMC-4 | ||||
| CAP-D2 | DPY-28 | DPY-28 | |||||
| Condensin I subunit | CAP-H | DPY-26 | DPY-26 | ||||
| CAP-G | N/D | N/D | |||||
| CAP-D3 | HCP-6 | ||||||
| Condensin II subunit | CAP-H2 | (C29E4.2) | |||||
| CAP-G2 | (F55C5.4) | ||||||
|
| |||||||
| Cohesin | SMC1 | HIM-1/SMC-1 | HIM-1/SMC-1 | ||||
| SMC3 | SMC-3 | SMC-3 | |||||
| RAD21(Scc1) | SCC-1/COH-2 | REC-8 | COH-1, COH-3 | ||||
| SA1,SA2(Scc3) | SCC-3 | SCC-3 | |||||
In the early days, C. elegans was thought to use only its condensin II complex for mitosis (9), while its condensing I-like complex, condensin IDC functioned instead in gene regulation during dosage compensation (47, 54). However, the Hagstrom group recently found that MIX-1–SMC-4 dimer also interacts with non-SMC subunits of condensin IDC (DPY-26 and DPY-28) to form a distinct complex (referred to as condensin IM :M stands for mitotic) which fulfills the mitotic function of condensin I (Hagstrom, unpublished data). Although condensins of other organisms associate with the central chromosome axis, C. elegans condensin localizes to holokinetochores during mitosis (52). Unlike HCP-6/CeCAP-D3 whose association with chromatin depends on HCP-3/CeCENP-A, the loading of SMC-4 and MIX-1/SMC-2 onto chromosomes does not require the kinetochore structure assembled on the centromere region specified by HCP-3/CeCENP-A. However, their properly restricted distribution does depend on HCP-3/CeCENP-A (52). Likewise, HCP-3/CeCENP-A loads onto mitotic chromosomes independently of condensin, but this loading requires the mitotic chromosome organization promoted by condensin to maintain its normal orientation toward the mitotic spindle (52). AIR-2 is required for the loading of condensin onto mitotic chromosomes (52), and the loss of condensin by depletion of SMC-4 or MIX-1/SMC-2 causes an aberrant wispy morphology of prometaphase chromosomes but does not affect the degree of compaction at metaphase (52). Condensin-free sister chromatids fail to resolve their connections and remain attached by chromosome bridges during anaphase (52). Despite the sister chromatid–separation defect, cytokinesis proceeds normally (52).
Moore et al. (2001) demonstrated that condensin II is required for sister centromere resolution when the microtubule-kinetochore attachment is abrogated. In the absence of HCP-4/CeCENP-C, kinetochore localization of HCP-6/CeCAP-D3 and centromere resolution during prophase is restored by depletion of cohesin components or cohesin establishment factors (24). This finding suggests that the role of HCP-4/CeCENP-C in centromere resolution is to remove cohesin from kinetochores to enable the recruitment of condensin II to kinetochores.
4.2. Role of the cohesin complex during chromosome segregation in C. elegans
Upon completion of DNA replication, duplicated chromatids are connected until anaphase via sister chromatid cohesion, which consists of the protein complex cohesin. Establishment and maintenance of cohesin are critical factors for proper chromosome segregation. Loss of functional cohesin often causes nondisjunction of paired chromatids. The formation of the cohesin complex is required for generating appropriate tension at kinetochores, which captures bioriented microtubules. Cohesin complex consists of a heterodimer of SMC proteins, SMC1-SMC3, and non-SMC subunits, RAD21/SCC1 and SCC3 (55-59). At the metaphase-anaphase transition, RAD21/SCC1 is cleaved by the endopeptidase separase (60, 61). This cleavage causes the dissociation of cohesin complex from chromosomes, leading to sister chromatid separation. Separase is bound and inactivated by the anaphase inhibitor securin until the anaphase-promoting complex also known as cyclosome (APC/C) targets securin for degradation (62). Thus, cohesin components play crucial roles in determining the timing of anaphase onset.
C. elegans has single homologs of SMC1, SMC3, and SCC3 and four homologs of RAD21/SCC1 (COH-1, COH-2, COH-3, and REC-8) (63) (Figure 3, Table 2). C. elegans SMC1 was originally identified as a mutant exhibiting the HIM phenotype and was named him-1 (28). Chan et al. (2003) identified him-1 mutations in a coding region for a protein homologous to SMC1 protein family members and confirmed that those mutations were responsible for the mutant phenotypes by complementation assay (64). Phenotypic and immunolocalization analyses of each RAD21/SCC1 homolog revealed that their functions are distinct from each other (63, 65). REC-8 expression is restricted in meiotic cells, and depletion of REC-8 causes substantial meiotic defects, suggesting that REC-8 is a meiosis-specific cohesin component (63) (described in more detail in Section 6.3). Because COH-2 copurified with HIM-1/SMC-1, SMC-3, and SCC-3 from embryonic lysate (64) and depletion of COH-2 causes chromosome-segregation defects in embryos that were similar to those observed in embryos depleted of other cohesin components (65), COH-2 was considered an ortholog of RAD21/SCC1 and was renamed SCC-1. Careful examination of the pattern of protein expression during development demonstrated by Mito et al. (2003) revealed that SCC-1/COH-2 is expressed in the nuclei of every dividing cell throughout development. They also demonstrated that the subnuclear localization of SCC-1/COH-2 changes in a cell cycle–dependent manner: SCC-1/COH-2 associates with chromosomes during interphase, dissociates from prophase to anaphase, and then reassociates at telophase (65). In contrast, COH-1 was not detected in early embryos but becomes detectable in the nuclei of all embryonic cells after about the 100-cell stage (65), when the level of protein expression in embryos elevates toward the morphogenesis stage.
During larval development, COH-1 is expressed in every somatic cell but not in germ cells (65). Moreover, COH-1 associates with chromosomes, regardless of the stage of cell cycle (65). These observations suggest that COH-1 regulates general gene expression by affecting the chromatin organization instead of functioning in chromosomal cohesion. As for COH-3, RNAi-mediated depletion of COH-3 does not cause any significant phenotype, making determination of its function difficult (63).
Embryos depleted of either cohesin component exhibit a similar chromosome missegregation phenotype called the cut-like phenotype. In these embryos, a mass of chromosomes stays in the middle of the bipolar spindle and is often divided by a cleavage furrow; multiple micronuclei of variable size are formed in daughter cells (63, 65, 66). Despite this severe chromosome-segregation defect, the loss of cohesin by depletion of any cohesin component did not affect the timing of cytokinesis, the condensation and decondensation of chromosomes, or the formation of the nuclear envelope (65). Chan et al. (2003) identified an additional component of the cohesin complex, TIM-1, which is a C. elegans homolog of the Drosophila Timeless, which regulates a circadian rhythm. TIM-1 coimmunoprecipitated with HIM-1/SMC-1 (64). Embryos depleted of TIM-1 are arrested with aneuploid nuclei or with nuclei containing prematurely separated sister chromatids. Depletion of TIM-1 causes the mislocalization of the non-SMC cohesin subunit but does not affect the localization of SMC subunit to chromosomes in meiotic prophase (64).
4.3. Chromosomal passenger proteins in chromosome segregation
The conserved chromosomal passenger complex plays a crucial role in accurate chromosome segregation by correcting the aberrant microtubule-kinetochore attachment (reviewed in Refs. (67, 68).
C. elegans chromosomal passenger complex (or ABI complex) is composed of AIR-2 and three nonenzymatic proteins, BIR-1/Survivin, ICP-1/INCENP, and CSC-1 (chromosome segregation and cytokinesis defective-1) (69, 70). Although the structure of first three subunits are highly conserved among eukaryotes, CSC-1 does not share common structural similarity to Borealin/Dasra-B, the fourth subunit of chromosomal passenger complex in other organisms (67). Depletion of any one of the four proteins causes an identical defect in mitotic chromosome segregation: chromosomes are condensed but not well congressed to the metaphase plate. Subsequently, the chromatin is stretched between two poles (69-71). Although the depletion of chromosomal passenger proteins causes misloading of condensin to chromosomes during metaphase, the depletion of condensin causes a defect in chromosome condensation only during prophase but does not affect metaphase plate formation. This finding suggests that mitotic defects caused by the loss of the chromosomal passenger complex is not merely due to condensin missing from the chromosomes (71).
AIR-2 kinase depends on nonenzymatic subunits for its dynamic localization during mitosis (71). CSC-1 and BIR-1 form a subcomplex, which can associate with ICP-1 but not with AIR-2 (71). AIR-2 kinase activity is stimulated by ICP-1 but not by the CSC-1–BIR-1 complex (71). Nevertheless, depletion of CSC-1 or BIR-1 causes a defect in mitotic chromosome segregation that is identical to that caused by depletion of AIR-2, suggesting that the function of the CSC-1–BIR-1 complex is to target subunits for the AIR-2–ICP-1 subcomplex. The yeast Aurora kinase Ipl1 phosphorylates NDC-80 and reduces the microtubule-binding activity of NDC-80 in vitro (25). This finding suggests that AIR-2 activity regulates NDC-80's microtubule-binding status, which in turn regulates the chromosomal passenger complex–dependent elimination of incorrect kinetochore-microtubule attachment.
4.4. Summary and perspectives
For proper sister chromatid segregation, sister kinetochores must capture microtubules emanating from opposite poles. This bioriented microtubule binding is ensured in part by proper mitotic chromosome organization, which is maintained by orchestrated functioning of the cohesin and condensin complexes. In addition, aberrant microtubule–kinetochore attachment, such as merotelic microtubule attachment, is corrected by the chromosome passengers complex before sister chromatid separation.
The basic function and molecular composition of condensins are conserved between C. elegans and other organisms. However, C. elegans condensin has some unique properties. For example, C. elegans condensin II localizes to holo-kinetochores, which may reflect the holocentric chromosome–specific function of condensin II. In C. elegans, only condensin II has been characterized as the condensin with a mitotic function. Although C. elegans has a condensin I–like protein complex, condensin IDC, the complex has a dosage-compensation function and not a mitotic function. The Hagstrom group has recently identified an additional condensin complex named condensin IM, which consists of SMC subunits of condensin II and non-SMC subunits of condensin IDC (Hagstrom, unpublished data). Further characterization of this complex will resolve the paradox that C. elegans appears to lack the complex with conserved mitotic functions.
During mitosis of mammalian cells, cohesin localized along the chromosome arms is released during prophase and prometaphase via a pathway dependent on phosphorylation of the cohesin subunit SA2, most likely by Plk/Polo kinase (72). On the other hand, cohesin at the centromere is retained because it is protected by shugoshin (73-77) and released at the metaphase–anaphase transition by cleavage of the RAD21 subunit by separase (78, 79). The C. elegans cohesin appears to be loaded onto chromosomes during interphase, most likely at late telophase in the previous round of the cell cycle, and dissociates from chromosomes during prophase (65). Regulation of cohesin association with and dissociation from holo-centric chromosomes needs further study.
In contrast to the structure of three of four subunits of the chromosomal passenger complex, which are highly conserved among eukaryotes, the fourth subunit, CSC-1, the C. elegans ortholog of Borealin/Dasra-B, is structurally highly diverse. Borealin/Dasra-B forms a complex with survivin and recruits the Aurora B kinase-INCENP complex to the centromeric region of chromosomes. Because CSC-1 and Borealin/Dasra-B have similar molecular functions, their structural diversity may reflect the difference between the architectures of C. elegans holokinetochores and monocentric chromosomes in other organisms.
5. SPINDLE ASSEMBLY CHECKPOINT IN C. ELEGANS
To ensure faithful chromosome segregation, the spindle assembly checkpoint monitors the status of microtubule-kinetochore binding and inhibits the activity of the APC/C, thereby delaying anaphase onset until all of the kinetochores are properly attached to the spindles [reviewed in Musacchio and Salmon (2007), and May and Hardwick (2006) (80, 81)]. Every eukaryotic cell relies on the spindle assembly checkpoint to coordinate the cell division cycle with sister chromatid separation, and C. elegans is no exception. Nevertheless, until recently it was believed that spindle assembly checkpoint did not function in early-stage embryos due to the observation that treatment with the microtubule-depolymerizing drug nocodazole did not cause metaphase arrest of blastomeres in early-stage C. elegans embryos (82, 83). However, the molecular identification of spindle assembly checkpoint components and careful cytologic examination of the cell cycle progression revealed the conserved checkpoint function in response to disruption of mitotic spindle microtubules (18).
5.1. Spindle assembly checkpoint in embryonic cells
In C. elegans, most of the known spindle assembly checkpoint components are conserved. A cell cycle delay in mitosis induced treatment with nocodazole was detected in fixed premeiotic germ cells as an increase in the number of nuclei that react with phosphorylated histone H3 antibody, demonstrating the presence of the spindle assembly checkpoint in proliferating germ cells (84). Meanwhile, due to the lack of an apparent mitotic-arrest phenotype (82, 83), the presence of the spindle assembly checkpoint function in C. elegans embryos has been uncertain. However, a recent advance in the fluorescent microscope imaging technology made possible time-lapse experiments in which the mitotic duration was precisely measured. These studies revealed that chemical or mutational disruption of microtubules induces the spindle assembly checkpoint–dependent extension of the interval between the NEBD to anaphase onset or that between NEBD to nuclear envelope reformation (NER) as much as two fold (18). Thus, spindle assembly checkpoint activation in C. elegans embryos causes only a mild extension in the duration of mitotic progression but does not cause extensive mitotic arrest. Nevertheless, the mild extension of the mitotic duration is crucial for maintaining the viability of individual worms.
The modest mitotic delay may be sufficient for holo-centric kinetochores to capture bioriented microtubules. Because the timing of division of every cell during embryogenesis is tightly regulated for proper positioning of descendants, an excess mitotic delay in any blastomere must be fatal. Therefore, C. elegans may have evolved into an organism with limited mitotic duration, even in the presence of mitotic spindle defects. Thus, cytologically distinct mitotic events such as chromosome condensation, NEBD, anaphase onset, or NER are useful parameters for identifying proteins required for activation of the spindle assembly checkpoint, because the depletion of such proteins should affect the intervals between these events.
5.2. Spindle assembly checkpoint components
The spindle assembly checkpoint components, MAD1, MAD2, MAD3, BUB1, and BUB3, are conserved in C. elegans (85, 86). All of these proteins are required for microtubule defect–induced mitotic delay in early-stage embryos (our unpublished data), suggesting the conservation of molecular functions. The C. elegans homolog of MPS1 has not been identified, presumably because of its highly diverse sequence (87). Higher eukaryote–specific spindle assembly checkpoint proteins RZZ (ROD-ZW10-ZWILCH) are also conserved (88-91).
5.2.1. MDF-1/CeMAD-1 and MDF-2/CeMAD-2
Identification of MDF-1 and MDF-2 facilitated our understanding of the spindle assembly checkpoint in C. elegans. MDF-2 is a C. elegans homolog of Mad2 that shares 40% sequence identity with budding yeast Mad2. Ectopically overexpressed MDF-2 complements the benomyl-sensitive phenotype of the mad2-deletion yeast strain, thereby confirming the functional conservation of the protein (84). MDF-1 was identified as an MDF-2–binding protein (84). Although MDF-1 has relatively diverse amino acid sequence, its predicted coiled-coil structure and the similarity of the short amino acid sequence in the C-terminal domain to those of human MAD1 led us to categorize the protein as a member of the Mad1 protein family. MDF-1 and MDF-2 accumulated at the unattached kinetochore. Immunofluorescent microscopic analysis with anti–MDF-1 antibody revealed that MDF-1 localizes to the nuclear periphery during interphase. The protein accumulates in the nuclear matrix late in prophase, but its association with chromosomes is hardly detectable throughout the progression of the normal cell cycle during embryogenesis. However, it accumulates at holo-kinetochores in embryos treated with a microtubule-depolymerizing drug such as nocodazole during prometaphase and metaphase (Figure 4).
Figure 4. Subcellular localization of MDF-1/CeMAD-1.
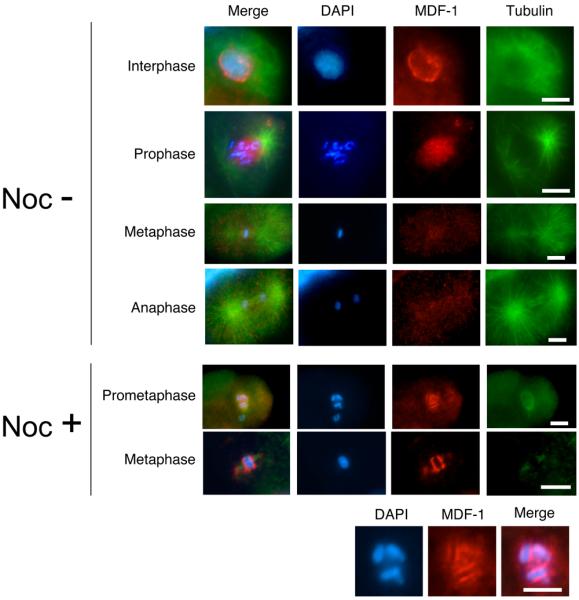
Fluorescence micrographs of early-stage wild-type embryos undergoing mitosis are shown. At this stage, MDF-1/CeMAD-1 localizes to kinetochore regions only in the presence of spindle damage. Wild-type embryos were dissected from adult hermaphrodites and either incubated for 15 min in 30 μg/ml nocodazole (Noc+) to induce kinetochore localization of MDF-1 or not incubated (Noc−). The embryos were then fixed and stained with DAPI (blue), anti–MDF-1 antibody (red), and anti-tubulin antibody (green). Images of chromosomes in the Noc+ cells in prometaphase are shown at the higher magnification in the bottom row. Scale bars: 10 μm.
The mdf-1–deletion strain exhibited various developmental defects (e.g., embryonic arrest, larval arrest, abnormal vulval development, and sterility due to abnormal germ cell development) (84) (Figure 5). These defects were comparable to phenotypes observed in worms in which either MDF-1 or MDF-2 expression was suppressed by RNAi. Genome instability was reflected by the high incidence of males (i.e., increased frequency of males in self-cross progeny of a hermaphrodite worm indicates the increased frequency of X-chromosome missegregation during meiosis). The presence of aneuploid oocytes also suggested the spindle assembly checkpoint function of MDF-1. More direct evidence for MDF-1 having a role in the spindle assembly checkpoint in C. elegans was that MDF-1 is required for nocodazole-induced mitotic arrest in germ cells. Furthermore, Encalada et al. (18) recently demonstrated that in embryonic cells, MDF-1 and MDF-2 are required for mitotic delays induced by chemical or mutational disruption of the microtubule's cytoskeleton. Thus, MDF-1 and MDF-2 play a role in the spindle assembly checkpoint activity in both embryonic cells and germ cells. Unlike mammalian MAD2, whose depletion shortens the duration of normal mitosis and results in premature sister chromatid separation, the depletion of MDF-1 or MDF-2 does not affect the duration of mitosis in embryonic cells during normal cell cycle progression. Nevertheless, these proteins are required for the normal development and maintenance of viability of individual animals, suggesting the prominent requirement of MDF-1 and MDF-2 during postembryonic development.
Figure 5. Genome instability and developmental defects in the mdf-1/CeMAD-1–deletion strain. A.

Germline defects in gonads dissected from mdf-1–homozygous (Δmdf-1) hermaphrodites. Single gonad arms of N2 (wild-type) adult hermaphrodites (top panel) and sterile Δmdf-1 adult hermaphrodites (bottom panel) were dissected, fixed, and stained with DAPI. The distal end of each gonad is indicated by a white arrow. N2 germ cells are arrayed from distal to proximal (right to left): premeiotic proliferation, transition zone, and meiotic prophase (pachytene followed by diplotene/diakinesis). Sterile Δmdf-1 adult hermaphrodites exhibit variable defective phenotypes such as endomitotic germ cells (Emo) or tumor-like gonad (Tum). In the Tum gonad, a whole gonad arm is filled with premeiotically proliferating cells. In the Emo gonad, normal meiotic prophase progression occurs until cells enter diakinesis; chromosomes decondense (D) before entering meiosis I division; and polyploidal nuclei (P) are observed in the region of spermatheca. B. Chromosome instability in Δmdf-1 embryonic cells. In the left panels, the nuclei of early-stage N2 embryos carrying a pair of GFP-tagged chromosomes [256 repeats of lacO sequence and hsp-16 promoter::GFP::lacI fusion gene were integrated into a single locus of a chromosome by gamma irradiation induced-chromosomal recombination: the integrated line was isolated in our laboratory from the transgenic strain provided by Barbara Meyer (132)] are shown at various cell cycle stages. Each nucleus contains two GFP dots. In the right panel, the nuclei of three Δmdf-1 embryonic cells are shown. More than three GFP dots were detected in the nuclei, indicating either premature sister chromatid separation or aneuploidy. C. Defective gonad development in Δmdf-1 hermaphrodites. An N2 adult hermaphrodite (upper panels) and a sterile Δmdf-1 adult hermaphrodite (lower panels) were fixed and stained with DAPI. The body (blue) and gonad (red) are outlined in the accompanying illustrations. In the N2 adult hermaphrodite, two symmetrically developed gonad arms are detected. In the Δmdf-1 adult hermaphrodite, a single, poorly developed gonad arm is seen.
5.2.2. SAN-1/CeMAD-3
SAN-1 (for suspended animation-1), a MAD3 homolog, was first identified by an RNAi-based genome-wide screen for genes required for survival during anoxia (<0.001% oxygen) (92). Depletion of SAN-1 substantially reduces the viability of embryos exposed to 24 h of anoxia. Phenotypic analysis revealed that SAN-1 is required for the oxygen-deprivation–induced metaphase arrest in blastomeres (92). MDF-2/CeMAD-2 is also required for this anoxia-induced suspended animation, suggesting that the spindle assembly checkpoint is activated by anoxia. SAN-1 localizes to kinetochores during prophase and metaphase or to kinetochores in embryos treated with nocodazole or anoxia (24, 92, 93). Despite its essential role in the maintenance of viability under the stressed condition, SAN-1 is not required for normal cell cycle progression during development (94). Unlike the mdf-1–deletion homozygotes, san-1–deletion homozygotes are viable and exhibit only a mild reduction of brood size under normal experimental conditions (94).
5.2.3. BUB-1
BUB-1 was identified as a C. elegans homolog of BUB1 kinase. BUB-1 localizes to kinetochores in prophase prior to NEBD and remains associated with kinetochores until midanaphase (10, 18). Kinetochore localization of BUB-1 depends on HCP-3/CeCENP-A, HCP-4/CeCENP-C, and KNL-1 (10, 17, 18). Depletion of BUB-1 causes embryonic arrest at high penetrance most likely due to defects in mitotic progression (95). In BUB-1–depleted embryos, lagging chromosomes often arise during anaphase (10, 18). Nevertheless, BUB-1 depletion does not cause any gross defects in spindle microtubule formation (10, 18): The spindle midzone is formed normally; the kinetic properties of centrosome separation are indistinguishable from those of untreated embryos.
Although BUB-1 is required for kinetochore localization of HCP-1/2/CeCENP-F, the mitotic defects exhibited by BUB-1–depleted embryos are distinct from those observed in HCP-1/2/CeCENP-F–depleted embryos (18). This finding suggests that either HCP-1/2/CeCENP-F proteins, which do not localize to the kinetochore, still play a role in mitotic progression, or that HCP-1/2/CeCENP-F still localizes to kinetochores at an undetectable level but enough to exert its mitotic function in BUB-1–depleted embryos. BUB-1 is required for mitotic delays induced by chemical or mutational disruption of the microtubule cytoskeleton (18). However, BUB-1 depletion extends the duration of mitosis from NEDB to anaphase by 1.5 fold, compared with that seen in untreated embryos, rather than causing precocious anaphase onset, which is a hallmark of spindle assembly checkpoint defects. Thus, BUB-1 appears to have dual functions: one in mitotic progression and another in the spindle assembly checkpoint.
5.2.4. BUB-3
BUB-3 was identified as a protein with five repeats of the WD-40 domains related to human BUB3. BUB-3–depleted embryos do not exhibit any significant defects in growth or fertility. However, depletion of BUB-3 is synthetically lethal with deletion of MDF-1, MDF-2 or SAN-1 (38, 39). High-throughput yeast two-hybrid analysis identified an interaction between BUB-3 and BUB-1 (96). Furthermore, BUB3 is required for nocodazole-induced mitotic delay (our unpublished data). These data suggest the functional conservation of BUB-3 and BUB3 in other organisms.
5.2.5. RZZ (ROD-ZW10-ZWILCH) complex
ROD-1 (CeROD), CZW-1(CeZW10), and ZWL-1(CeZWILCH) have been identified as components of the C. elegans RZZ complex (88, 97). Depletion of any of these three proteins causes the embryonic-lethal phenotype at high penetrance (95). Chromosome bridges arise at a high frequency during anaphase in embryos depleted of CZW-1 or ROD-1 (88, 97).
5.3. Mutations that activate the spindle assembly checkpoint
In C. elegans, the spindle assembly checkpoint–dependent extension of mitotic duration is induced by mutational disruption of the mitotic spindle, as well as microtubule depolymerization caused by treatment with nocodazole. Encalada et al. (2005) demonstrated that mitotic delay was induced in embryos carrying a partial loss-of-function mutation of dnc-1, a C. elegans homolog of the p150Glued dynactin subunit that is required for proper assembly and positioning of the mitotic spindle in early embryonic cells (18). In dnc-1–mutant embryos, the duration from chromosome condensation to NER was increased by 1.3 fold compared with that seen in wild-type embryos (18). The interval between NEBD and anaphase onset was also increased almost 2 fold (18). Extended mitosis can be also observed in embryos depleted of ZYG-9, a XMAP215 homolog required for microtubule stability during mitosis (98), or those depleted of DHC-1, the heavy chain of cytoplasmic dynein, a microtubule minus-end–directed motor that, like dynactin, is required for mitotic spindle assembly (99-101). Although its molecular identity has not yet been determined, the apo-5 (anaphase spindle position-defective-5) mutant that causes abnormal spindle positioning with short microtubules and disorganized cytoplasmic tubulin fragments also induces the extension of mitotic duration to 1.86 fold of that of wild-type embryos (18). Extension of mitotic duration by depletion of these proteins depends on the spindle assembly checkpoint proteins MDF-1/CeMAD-1, MDF-2/CeMAD-2, and BUB-1 (18). Similarly, the depletion of LIS-1 induces mitotic arrest followed by apoptotic cell death in premeiotic germ cells (102). This mitotic arrest is MDF-1/CeMAD-1–dependent, suggesting that the loss of LIS-1 activates the spindle assembly checkpoint in proliferating germ cells. Thus, mutational destruction of functional mitotic spindle formation induces moderate but certain mitotic delay in a spindle assembly checkpoint–dependent manner. However, the extension of mitotic duration is not a common phenotype for all spindle-defective mutants. For example, the depletion of AIR-1, an aurora-A kinase required for centrosome maturation, does not affect the mitotic duration despite the poor spindle formation of AIR-1–depleted embryos (103, 104). Further characterization of spindle-defective phenotypes caused by depletion of different proteins should provide further insight into the mechanism of activation of the spindle assembly checkpoint in C. elegans.
In mammalian or yeast cells, not only spindle defects but also kinetochore or cohesion defects activate the spindle assembly checkpoint (55, 56, 105-108). On the other hand, in C. elegans, depletion of cohesion components in early-stage embryos does not induce mitotic delay (65)(our unpublished data). This unresponsiveness of the spindle assembly checkpoint to the cohesion defect suggests that only the unattached kinetochore and not the tension-free kinetochore is recognized as an emergency signal for the spindle assembly checkpoint in C. elegans. Furthermore, mitotic duration of early-stage embryos is not affected by depletion of outer kinetochore components such as components of KMN network (17, 27). Depletion of kinetochore components disables the function of the spindle assembly checkpoint rather than activating it. Depletion of the inner kinetochore component HCP-4/CeCENP-C results in the KNL phenotype and sister chromatids never separate, bypassing the microtubule disruption– induced extension of the duration of mitosis, defined as the interval between NEBD and NER (18). Depletion of HCP-4/CeCENP-C causes misassembly of many outer kinetochore components (10, 17, 18). These observations suggest that intact kinetochore assembly is crucial for a functional spindle assembly checkpoint; that is, the C. elegans spindle assembly checkpoint is activated only when an intact kinetochore is present and has a microtubule-free status. Encalada et al. (2005) also reported that depletion of HCP-1/2/CeCENP-F or KLP-7/CeMKAC bypasses mitotic delay induced by chemical or mutational disruption of mitotic spindles. In one-cell embryos depleted of HCP-1/2, anaphase-like separation and movements of chromosomal masses to opposite poles are observed after pronuclear envelope break down (PNEBD). Encalada et al. considered this chromosome movement anaphase onset and determined the mitotic duration, defined as the interval between PNEBD and anaphase onset. According to them, depletion of HCP-1/2 appeared to cause precocious anaphase onset in embryonic cells similar to the phenotype observed in mammalian cells depleted of spindle assembly checkpoint components. Furthermore, depletion of HCP-1/2 bypassed apo-5–induced mitotic delay. However, Cheeseman et al. demonstrated in GFP::MIS-12 expressing embryos that sister chromatids did not separate but migrated to the same pole in the absence of HCP-1/2 (37). Therefore, separation of chromosomal masses observed in HCP-1/2–depleted embryos is not sister chromatid separation. Furthermore, the mitotic duration, defined as the interval between PNEBD and NER, is not affected by depletion of HCP-1/2. Although Encalada et al. demonstrated that nocodazole-induced extension of the interval between NEDB and NER in blastomeres of two-cell stage embryos is suppressed by depletion of HCP-1/2, still supporting their argument that HCP-1/2 is required for activating the spindle assembly checkpoint, they may need to reconsider their definition of mitotic duration. To measure the activity of the spindle assembly checkpoint, the system to quantify the ubiquitin E3 ligase activity of APC/C or stability of APC/C targets in a single cell needs to be established as an alternative way of measuring mitotic duration.
5.4. Balance between the activity of the APC/C and that of the spindle assembly checkpoint
Traditional forward-genetics approaches identified an intriguing interaction between spindle assembly checkpoint components and the APC/C components. APC/C is a large ubiquitin ligase E3 protein complex, which consists of at least 11 subunits (109). The APC/C targets and ubiquitinates the anaphase inhibitor Securin and cyclin B for ubiquitin-mediated protein degradation, thereby promoting sister chromatid separation and exit from mitosis. Upon receiving the emergency signal generated from an unattached or tension-free kinetochore, the spindle assembly checkpoint arrests cells before anaphase by inhibiting the APC/C activity.
In C. elegans, 10 orthologs of APC/C subunits including, one paralog of APC5, have been identified to date (110-112). Depletion of any of the seven subunits by RNAi results in a strong maternal-effect embryonic arrest at the 1-cell stage due to a defect in the metaphase-toanaphase transition during meiosis I. Phenotypic analysis using temperature-sensitive allele of the APC subunits revealed that APC/C activity is also required for metaphase-to-anaphase transition during mitosis in embryonic cells and in premeiotically dividing germ cells.
If the observed fatal developmental defects of the mdf-1/CeMAD-1–deletion strain are caused by the loss of a functional spindle assembly checkpoint that results in the deregulation of APC activity, then mutations that reduce the APC/C activity or increase the stability of the APC/C substrates should suppress the lethality of the mdf-1–deletion strain. Consistent with this hypothesis, developmental defects and infertility of mdf-1/CeMAD-1–deletion homozygotes are suppressed by the reduction in APC/C activity, which is caused by hypomorphic mutations in the APC/C component emb-30 (C. elegans homolog of APC4) or in the APC activator fzy-1 (C. elegans homolog of CDC20) (113, 114). Furthermore, Tarailo et al. (2007) isolated and characterized additional mdf-1–suppressor mutants and mapped one suppressor mutation in a gene encoding a newly identified APC5-like protein (115). They also demonstrated that most isolated suppressor mutants exhibit the extended mitotic duration in early-stage embryos with increased level of IFY-1(C. elegans ortholog of Securin), suggesting that the APC/C activity is down-regulated in those suppressor mutants. These results suggest that MDF-1's essential function is to regulate the appropriate APC/C activity for proper development.
Reciprocally, Stein et al. (2007) demonstrated that the 1-cell–arrest phenotype of mat-3(or180), a temperature-sensitive allele of the APC/C component mat-3/CeAPC-8 was suppressed by hypomorphic mutants of spindle assembly checkpoint genes (94, 115). They performed a genetic screen for mutants that suppress the lethality of mat-3(or180) homozygotes and isolated loss-of-function allele of mdf-2/CeMAD-2 and san-1/CeMAD-3 (94). They also isolated an mdf-1/CeMAD-1 allele, mdf-1(av19), which specifically lacks a spindle assembly checkpoint function but retains the other essential function of MDF-1/CeMAD-1 (94). Spindle assembly checkpoint mutants isolated as mat-3(or180) suppressors can also suppress the 1-cell embryonic-arrest phenotype of other APC mutants (94), suggesting that these mutants modify the overall activity of APC/C rather than interact with the specific APC/C subunits.
Together, the tight genetic relations between the spindle assembly checkpoint and APC/C lead to a model in which a certain threshold of APC/C activity is required for progression through the cell cycle. This threshold appears to be higher during meiosis than during mitosis, because a very small reduction in APC/C activity causes defects in meiotic progression but not in mitotic progression. Suppressor mutations may weaken the inhibitory interaction of the spindle assembly checkpoint components with the APC/C, thereby increasing the APC activity so that it is above the threshold limit. On the basis of this hypothesis, the activity of the spindle assembly checkpoint may control the appropriate level of APC/C activity through animal development.
5.5. Summary and perspectives
Although the spindle assembly checkpoint in mammalian or yeast cells causes extensive mitotic arrest in response to a defect in kinetochore–microtubule attachment, the C. elegans spindle assembly checkpoint causes only a mild extension in the duration of mitotic progression. This slight mitotic delay may be sufficient for this holo-centric organism to maintain viability with chromosome transmission fidelity. The cytologically detectable level of mitotic delay in C.elegans embryos is induced only by a microtubule defect and not by depletion of cohesion components, suggesting that the spindle assembly checkpoint is activated by unattached kinetochores but not by tension-free kinetochores. Depletion of core kinetochore components such as HCP-4/CeCENP-C bypasses the microtubule defect–induced mitotic delay, suggesting that the spindle assembly checkpoint is activated only when the intact kinetochore is assembled and present in a microtubule-free status. Depletion of any one of conserved spindle assembly checkpoint components extends rather than shortens the duration of mitosis in embryonic cells undergoing normal cell division cycle. Thus, determining the timing of anaphase onset during normal cell cycle progression does not seem to be a common function for spindle assembly checkpoint components in C. elegans embryonic cells. Nevertheless, the functions of the spindle assembly checkpoint components, such as MDF-1 in regulation of the APC/C activity, are indispensable for proper development. Studying of the role of MDF-1 in postembryonic lineage can help determine the function of spindle assembly checkpoint in multicellular organisms.
6. MEIOTIC CHROMOSOME SEGREGATION IN C. ELEGANS
Meiosis is a specialized cell cycle event that occurs only in the reproductive system to generate haploid gametes. It is initiated by the crossover-mediated pairing (or homologous recombination) of homologous chromosomes. Paired homologous chromosomes are then separated and segregated into daughter cells during meiosis I division (or reductional division). During meiosis II division, sister chromatids, which constitute a single chromosome, are separated. Therefore, sister kinetochores that should be bioriented during mitosis and meiosis II must syntenically capture microtubules emanating from the same pole during meiosis I. To accomplish the sequential chromosome transmission events successfully, C. elegans has localized microtubulekinetochore attachment sites on meiotic chromosomes.
6.1. Architecture of the meiotic kinetochore in C. elegans
Meiotic division can be observed in C. elegans spermatocytes and oocytes. The meiotic spindles formed during spermatogenesis are similar to mitotic spindles assembled in blastomeres during embryogenesis (116). In contrast, during oogenesis, the acentriolar and barrel-shaped spindles are formed in parallel to the surface of the fertilized oocytes (116). Despite the cytologically distinct spindle shapes, meiotic chromosomes in spermatocytes and oocytes have similar architectures. During diakinesis of meiosis I, each pair of homologous chromosomes forms a highly condensed, array-shaped bivalent (116). In situ hybridization analysis with a probe for the ribosomal DNA mapped to the one end of a linkage group revealed that the homologous chromosomes in each bivalent were associated end-to-end, and outer ends of the bivalent facing to the pole attach to microtubules (116). Orientation of each bivalent is random, i.e., the ends of the chromosomes randomly attach to the microtubule. Conventional EM analysis of meiotic chromosomes did not distinguish the trilaminar kinetochore structures during either meiosis I or II divisions. Instead, microtubules appeared to project into the chromatin (116). However, the recent analysis of ultrastructure of meiotic chromosomes using HPF/FS demonstrated by Howe et al. (2001) revealed the presence of a ribosome-free zone, the presumptive meiotic kinetochores surrounding each bivalent (2). Nevertheless, there is no evidence that microtubules attach all along the meiotic chromosomes.
6.2. Localization of kinetochore components on bivalents
Consistent with the EM observation that the ribosome-free zone, which is presumably the meiotic kinetochore structure, surrounds the bivalents, conserved mitotic outer-kinetochore components, HCP-1/CeCENP-F, HIM-10/CeNUF-2, KNL-1, KNL-3, NDC-80, and MIS-12, localized as a halo around the meiotic chromosomes during both meiosis I and II divisions (2, 117) (Figure 6). Particularly, Howe et al. (2001) demonstrated that HIM-10/CeNUF-2 surrounds not only chromosomes but also extrachromosomal arrays that consist of prokaryotic DNA (2), reinforcing the lack of the specificity of DNA sequence for meiotic kinetochore assembly.
Figure 6. Localization of kinetochore components on meiotic chromosomes.

(a) Schematic showing the meiotic chromosomes during meisis I and II division. Each bivalent, from diakinesis to metaphase of meiosis I, consists of a pair of homologous chromosomes comprising a pair of sister chromatids. Homologous chromosomes separate during meiosis I division, and sister chromatids separate during meiosis II division (b – f). The localization pattern of indicated proteins on bivalents during meiosis I and meiosis II divisions are illustrated in red. The meiotic chromosomes (a pair of homologous chromosomes during meiosis I and a pair of sister chromatids during meiosis II) are shown in blue. (b) Outer kinetochore components localize to a cup-like structure (red) surrounding the bivalent. (c) In contrast, inner kinetochore components and condensin components localize through the chromosomal DNA. HCP-3/CeCENP-A is removed from chromosomes prior to meiosis II division. (d) The cohesin complex (red), which localizes to cohesion between homologous chromosomes (blue) and between sister chromatids (blue), forms a cruciform shape. (e) The chromosomal passenger complex (red) colocalizes with cohesin only to be resolved. (f) BUB-1 (red) colocalizes with outer kinetochore components (shown in red in b) and chromosomal passenger proteins (shown in red in e). Redrawn with permission from Fig. 3 in (117).
The inner kinetochore components HCP-3/CeCENP-A and HCP-4/CeCENP-C also localize to meiotic chromosomes, but their localization coincides with that of chromosomal DNA (117) (Figure 6). Both HCP-3/CeCENP-A and HCP-4/CeCENP-C are loaded onto meiotic chromosomes during late prophase of meiosis I (117): HCP-3/CeCENP-A appears around diplotene, and then HCP-4/CeCENP-C appears around diakinesis. HCP-4/CeCENP-C is retained on chromosomes until late meiosis II (117), but HCP-3/CeCENP-A is dramatically reduced on chromosomes at the transition from meiosis I to meiosis II (117). KNL-1 also localizes throughout the chromatin during late prophase of meiosis I. It then relocalizes to a cup-like structure formed on the surface of the bivalents just prior to fertilization (117). In oocytes depleted of HCP-3/CeCENP-A, both HCP-4/CeCENP-C and KNL-1 fail to localize to chromosomes during meiotic prophase (117), suggesting HCP-3/CeCENP-A recruits HCP-4/CeCENP-C and KNL-1 to meiotic chromosomes. In contrast, in fertilized, HCP-3/CeCENPA–depleted oocytes, which undergo meiosis I division or meiosis II division, only HCP-4/CeCENP-C delocalized; KNL-1 localized normally to the surface of the chromosomes (117). Thus, conserved outer kinetochore components localize to the surface of the bivalents during both stages of meiosis independently of HCP-3/CeCENP-A and HCP-4/CeCENP-C.
Despite their localization to meiotic chromosomes, HCP-3/CeCENP-A and HCP-4/CECENP-C depletion does not affect the meiotic cell cycle progression (117). On the other hand, as previously described, a mutant allele of him-10/CeNUF-2 exhibits the Him phenotype reflecting the high frequency of X-chromosome nondisjunction and suggesting HIM-10/CeNUF-2's role in regulation of meiotic chromosome segregation (2). HIM-10/CeNUF-2 coincides with other outer kinetochore components on the surface of meiotic chromosomes; thus, HIM-10/CeNUF-2 and other conserved outer kinetochore components may constitute the meiotic kinetochores that assemble on the surface of meiotic chromosomes independently of HCP-3/CeCENP-A and HCP-4/CeCENP-C, thereby mediating proper microtubule-chromosome attachment during both stages of meiosis. Whether the molecular composition of the meiotic kinetochore is identical to that of the mitotic kinetochore or whether some other meiosis-specific components are required for meiotic kinetochore assembly remains unknown.
6.3. Role of cohesin and condensin in meiosis
Consistent with the fact that the core cohesin subunit HIM-1/SMC-1 was originally identified as a mutant exhibiting the Him phenotype, C. elegans cohesin components play a crucial role in meiotic cohesin. Although mitotic cohesin consists of HIM-1/SMC-1, SMC-3, SCC-3, and SCC-1/COH-2, in meiotic cells, SCC-1/COH-2 is replaced with REC-8, a meiosis-specific non-SMC subunit (63). Meiotic cohesin is loaded onto chromosomes prior to meiotic prophase. In pachytene nuclei, meiotic cohesin localizes along the longitudinal axes of synapsed chromosomes (63, 64, 66). Although this localization pattern coincides with the synaptonemal complex (SC), the localization of cohesin components is not dependent on homolog pairing and SC formation (64). In diakinesis, cohesin components localize between homologous chromosomes and between sister chromatids, thereby distributing in a cruciform pattern on each bivalent (64). SCC-3 localizes throughout the chromatin during diakinesis (64), suggesting that this protein has an additional as yet unknown function. Consistent with the localization pattern, the depletion of meiotic cohesion components disrupts the formation of the synapsis and prematurely separates homologous chromosomes and sister chromatids (63, 64, 66). Meiotic cohesin also plays a role in homologous chromosome pairing before synapsis. Depletion of meiotic cohesin induces chromosome fragmentation at diakinesis (63), suggesting that meiotic cohesin plays a role in repairing double-strand breaks introduced into meiotic chromosomes for initiation of homologous recombination. Another cohesin component, TIM-1, is extensively expressed in premeiotic nuclei, but its expression is dramatically reduced upon entry to meiotic prophase (64), suggesting that TIM-1 establishes or stabilizes meiotic cohesin rather than serving as a constituent of the cohesin structure.
During diakinesis to meiosis I metaphase, the chromosomal passenger complex colocalizes to cohesin to be resolved for separation of homologous chromosomes (71), suggesting that AIR-2/CeAurora B kinase activity contributes to the dissociation of cohesin from homologous chromosomes during meiosis I division. Actually, Rogers et al (2002) have demonstrated that REC-8 is phosphorylated by AIR-2 in vitro. They also showed that depletion of AIR-2 by RNAi stabilized REC-8 (118). Furthermore, their data that depletion of two PP1 phosphatases, CeGLC-7α/β, which antagonize the meiotic activity of AIR-2/aurora kinase (119) by RNAi destabilized REC-8 also suggests that the kinase activity of AIR-2 regulates the degradation of REC-8 thereby controlling the dissociation of cohesin during meiosis. Embryos depleted of chromosomal passenger complex demonstrate normal formation of the metaphase plate with six well-ordered bivalents; however, during anaphase, the chromatin fails to separate and is stretched along the spindle axis (71). The stretched but unseparated chromatids are recondensed and aligned at the metaphase plate. Then they are stretched again during anaphase of meiosis II (71). Thus, the chromosomal passenger complex is specifically required for meiotic chromosome segregation but dispensable for meiotic cell cycle progression.
C. elegans condensin II is first enriched on meiotic chromosomes during the diplotene stage, when each pair of homologous chromosomes starts compacting to form a highly condensed bivalent. At diakinesis, condensin II localizes to individual sister chromatids, forming four foci in each bivalent (53). Although condensin II components HCP-6/CeCAP-D3 and MIX-1/SMC-2 independently associate with mitotic chromosomes, localization of MIX-1 to diplotene and diakinesis chromosomes depends on HCP-6/CeCAP-D3 (53). Furthermore, condensin II does not require HCP-3/CeCENP-A for its association with meiotic chromosomes (53). Consistent with the localization pattern, depletion of condensin II components does not affect the pachytene chromosome organization but does induce defects in chromosome compaction and organization during diplotene and diakinesis (53). As a consequence of aberrant chromosome organization, condensin II-free meiotic chromosomes fail to segregate properly during both stages of meiosis. However, the defect in homologous chromosome segregation (meiosis I) is less severe than that in sister chromatid separation (meiosis II) (52, 53), implying that the sensitivity of condensin II components to reduction differs.
6.4. Role of the spindle assembly checkpoint in meiosis
The mdf-1/CeMAD-1–deletion strain exhibits a defect in germ line development and a weak Him phenotype (84), suggesting the meiotic function of MDF-1/CeMAD-1. Furthermore, the deletion or hypomorphic mutants of mdf-1/CeMAD-1, mdf-2/CeMAD-2, and san-1/CeMAD-3 suppresses the meiotic defect of APC-mutant strains (94), which also suggests that the spindle assembly checkpoint regulates APC activity during meiotic division. Immunofluorescent microscopic analysis of MDF-2/CeMAD-2 in fixed embryos revealed the colocalization of MDF-2/CeMAD-2 with meiotic spindles (84). However, no association with meiotic chromosomes was detected (84). In contrast, BUB-1 localizes to the surface of bivalents during both stages of meiosis (117). In addition, BUB-1 is present between homologous chromosomes during meiosis I division, and between sister chromatids during meiosis II division (117). Thus, BUB-1 localization coincides with cohesion, which will be dissolved during anaphase of each meiotic division. This observation is consistent with the report that BUB1 kinase is required for localization of Shugoshin protein to centromere region, thereby protecting the centromeric cohesion until anaphase (73, 120, 121). Whether BUB-1's spindle assembly checkpoint function is required for proper meiotic chromosome segregation is unknown.
6.5. Summary and perspectives
C. elegans meiotic divisions observed during spermatogenesis or oogenesis are cytologically distinct from mitotic divisions during embryogenesis. To sequentially accomplish two different types of chromosome transmission events - meiosis I division (homologous chromosome separation or reductional division) and meiosis II division (sister chromatid separation or equational division) - C. elegans meiotic chromosomes must have localized at microtubule– kinetochore attachment sites. Components of the mitotic kinetochore, condensin, cohesin, and the chromosomal passenger complex, whose mitotic function have been well characterized, are also required for meiotic division. However, molecular compositions of meiotic and mitotic kinetochores are distinct. Although meiotic and mitotic kinetochores have many common kinetochore proteins, assembly of the former is not dependent on HCP-3/CeCENP-A or HCP-4/CeCENP-C. How the location of the kinetochores on meiotic chromosomes is specified and how meiotic kinetochores are assembled remains to be studied. Phenotypic analysis of the mdf-1/CeMAD-1 deletion strain has revealed a defect in meiotic chromosome segregation, reflected by the Him phenotype. However, because loss of mdf-1 causes severe developmental defects in germ cells that are premeiotically proliferating or arrested in meiotic prophase, the precise role of the spindle assembly checkpoint in meiotic division remains to be elucidated. Isolation of temperature-sensitive alleles or meiotic function–specific alleles of spindle assembly checkpoint genes may facilitate the understanding of the mechanism of maintenance of chromosome transmission fidelity during meiosis.
7. OUTSTANDING ISSUES AND FUTURE DIRECTIONS
In the last decade, significant strides have been made toward understanding the molecular characterization of C. elegans holo-kinetochore. Studies have revealed a striking similarity between holo-kinetochores and monocentric kinetochores in other eukaryotic model organisms. They have also shown that C. elegans mitotic chromosome organization is maintained by structurally and functionally conserved protein complexes, cohesin, condensin, and the chromosome passenger complex. Although spindle assembly checkpoint components are also well conserved in C. elegans, and the molecular mechanism by which spindle assembly checkpoint inhibits the APC/C appears to be similar to that in other organisms, there are some differences between C. elegans and other organisms in how spindle assembly checkpoint functions. C. elegans spindle assembly checkpoint does not cause mitotic delay in response to kinetochore defects or cohesion defects but does cause a delay in response to some kinds of microtubule defects. This responsiveness, which is restricted to only microtubule defects, may be caused by the unique kinetochore structure. Determination of spatiotemporal organization of the kinetochore at the molecular level, including the distribution of spindle assembly checkpoint components, will be required for further understanding of the mechanism that regulates proper chromosome segregation by spindle assembly checkpoint in C. elegans. Several biologically interesting issues arise from the comparative study of spindle assembly checkpoints in different organisms, some of which should be addressed by considering the unique characteristics of C. elegans.
7.1. Spindle assembly checkpoint activation in embryonic cells
The functional analysis using RNAi techniques and mutant strains revealed that most kinetochore proteins (all proteins identified to date, except for KBP-5) are indispensable for proper chromosome segregation in every round of the cell cycle. The loss of any one of kinetochore components causes more or less chromosome missegregation during embryogenesis without affecting the timing of cytokinesis. As a consequence, embryos depleted of kinetochore proteins die with multiple aneuploid blastomeres. Thus, disruption of fully functional kinetochore structure does not induce mitotic delay. Chemical or mutational disruption of the mitotic spindle induces the spindle assembly checkpoint–dependent extension of mitosis up to 2 fold of normal mitosis. Furthermore, spindle defect–induced mitotic delay can be bypassed in embryos depleted of conserved kinetochore components. These observations suggest that spindle assembly checkpoint is activated only when the intact kinetochore is present with microtubule-free status. The spindle defects–induced mitotic delay depends on MDF-1/CeMAD-1, whose depletion barely affects the kinetochore-microtubule attachment. MDF-1/CeMAD-1 localizes to unattached kinetochores (Figure 4), suggesting that despite its holo-centric structure, the C. elegans spindle assembly checkpoint recognizes the microtubule attachment status of kinetochore, just like other monocentric organisms. MDF-1/CeMAD-1 is thought to recruit MDF-2/CeMAD-2, thereby facilitating efficient binding of MDF-2/CeMAD-2 to FZY-1/CeCDC-20 for inhibition of the APC/C activity to cause the mitotic delay. One possibility is that MDF-1/CeMAD-1 depends on the intact kinetochore structure for its association with kinetochores. Exploring what is a minimum requirement for kinetochore localization of MDF-1/CeMAD-1 may help us understand the molecular mechanisms by which the spindle assembly checkpoint is activated. Identification of the kinetochore receptor of MDF-1/CeMAD-1 will shed light on this biologically important topic.
7.2. Spindle assembly checkpoint function during postembryonic development
C. elegans is conceived as a single-cell zygote that undergoes a nearly invariant developmental process requiring genetically programmed cell divisions. From the first mitotic cell division following the pronuclear fusion, every round of the cell cycle of each blastomere is temporary and spatially controlled, and the cell division cycle of each blastomere occurs asynchronously (122, 123). Asynchronous cell division in early-stage embryos, in part, depends on conserved DNA replication checkpoint components, which regulate the duration of the S-phase (124). In contrast to the variability of the duration of the S-phase, the duration of mitosis during early embryogenesis is invariable (18). Therefore, it is unlikely that spindle assembly checkpoint, which regulates the mitotic progression, contributes to asynchronous cell division during early embryogenesis. Moreover, unlike mammalian MAD proteins, which act as pacemakers for mitotic cell cycle progression (125, 126), loss of MDF-1/CeMAD-1, MDF-2/CeMAD-2, or SAN-1/CeMAD-3 does not affect the duration of mitosis in early-stage embryos in C. elegans (18). Particularly, SAN-1/CeMAD-3 is not essential for animal viability, and the deletion of san-1/CeMAD-3 does not cause any severe developmental defects (94). On the other hand, despite the dispensability during early embryogenesis, C. elegans strains carrying the deletion mutations in mdf-1/CeMAD-1 or mdf-2/CeMAD-2 exhibit severe defects in larval development and germ cell development (84) (Figure 5). These results strongly suggest that MDF-1/CeMAD-1 and MDF-2/CeMAD-2 play some essential roles during late embryogenesis or postembryonic development. Because the lethality of the mdf-1/CeMAD-1–deletion strain is suppressed by reduction of the APC/C activity, the essential function of MDF-1/CeMAD-1 may be to regulate the appropriate level of APC/C activity during development.
The spindle assembly checkpoint may also contribute to cell cycle arrest of cells in some lineages when animals are exposed to undesirable environmental conditions. During postembryonic development, unfavorable environmental conditions cause global interruption of the reproducible pattern of cell division. For example, in response to nutrient deprivation, many somatic precursors in hatchlings are arrested in the G0/G1 stage in a cyclin-dependent kinase inhibitor, CKI-1(C. elegans KIP)–dependent manner (127). Thus, in both genetically programmed and environmentally regulated cell cycle progression, the timing of cell division of each somatic cell is controlled mainly during the G1 or S phase. An exception is the primordial germ cells that are genetically programmed to arrest in early mitosis during embryogenesis, and this mitotic arrest is sustained when animals are starved during postembryonic development (128). Recently, Watanabe et al. demonstrated that MDF-1/CeMAD-1 is required for this starvation–induced mitotic arrest of germ cell precursors (129). Hemizygosity of mdf-1 caused inappropriate germ cell proliferation in larvae exposed to the starved condition (129). Molecular biology studies revealed that MDF-1/CeMAD-1 functions as one of the downstream targets of the PI3K-AKT kinase signaling pathway in response to nutritional status (129), exemplifying that the spindle assembly checkpoint functions as a cell cycle regulator during postembryonic development by cross-talking with other signaling pathways. Future studies should focus on identifying additional roles of the spindle assembly checkpoint during postembryonic development and elucidating the molecular mechanisms by which the spindle assembly checkpoint is activated in response to environmental cues instead of spindle damage.
7.3. A reverse-genetics approach to identify synthetic genetic interactors with the spindle assembly checkpoint components
san-1/CeMAD-3 is dispensable for animal viability but essential for proper response to emergencies during chromosome segregation, making san-1 an ideal query gene for studying synthetic genetic interactions. The synthetic-lethal screen identifies genes whose deletion activates the spindle assembly checkpoint. Characterizing the function of synthetic-lethal genes will increase our knowledge about the molecular mechanism of activation of spindle assembly checkpoint. Systematic comprehensive analyses by using a collection of deletion alleles of nonessential genes – synthetic genetic array (SGA) or synthetic lethal analysis by microarray (SLAM) – have previously identified synthetic lethality in combination with deletion mutants of SAC components in budding yeast. Consistent with the prediction that simultaneous defects in kinetochore–microtubule attachment and SAC function cause fatal chromosomal missegregation, these analyses have identified many genes that play a role in chromosome transmission or mitotic spindle function (130, 131). Some of the genetic interactions identified in yeast have also been demonstrated in C. elegans, using the mdf-1/MAD1 deletion allele (38). Thus, screening for synthetic genetic interactors with SAC components is an effective approach to identify genes whose depletion activates the SAC pathway. Furthermore, although yeast SGA analyses have not identified synthetic genetic interactions between any two SAC components assayed, the synthetic genetic interaction among mdf-1/MAD1, san-1/MAD3, and bub-3 has been demonstrated in C. elegans (38, 39). These results strongly suggest that the screen for san-1 synthetic genetic interactors will identify additional genes whose products are required to activate the SAC pathway, particularly signaling pathways that are mediated by MDF-1/MAD1 or BUB-3. In C. elegans embryonic cells, SAC activation causes only a moderate extension in the duration of mitotic progression and not extensive mitotic arrest. The lack of an apparent mitotic-arrest phenotype has hindered the identification of factors required for SAC activation in C. elegans. RNAi-mediated genomewide screens for san-1 synthetic genetic interactors can overcome this problem and identify additional proteins required to activate the spindle assembly checkpoint in C. elegans.
7.4. Forward genetics approaches to studying the spindle assembly checkpoint in C. elegans
Tarailo et al. (2007) isolated mutants that suppress the lethality of the strain homozygous for the mdf-1 deletion. Consistent with the previous finding that hypomorphic mutants of emb-30/CeAPC-4 and fzy-1/CeCDC-20 suppress the lethality of the mdf-1/CeMAD-1–deletion strain (113, 114), many of the mutants compensate for the loss of the spindle assembly checkpoint function, presumably by reducing the APC/C activity, thereby extending the mitotic duration. Interestingly, two of the uncharacterized mutants suppress their lethality without affecting the mitotic duration (115). Molecular characterization of these mutants will provide new insight into the essential function of spindle assembly checkpoint components in multicellular organisms.
A genetic screen for suppressors of the meiotic defect of the APC mutant mat-3/CeAPC-8 identified many loss-of-function mutants of spindle assembly checkpoint genes (94). The mat-3/CeAPC-8–suppressor screen also isolated some uncharacterized mutations mapped to different chromosomal loci from known spindle assembly checkpoint genes (94). Determination of molecular identity of these suppressors will help us find additional spindle assembly checkpoint components. One mat-3/CeAPC-8-–suppressor mutant, mdf-1(av19), is defective only in the meiotic role of mdf-1/CeMAD-1 but is capable of robust mitotic function, a so-called separation-of-function allele (94). The isolation and characterization of the av19 allele has led to several models for differential activity of MDF-1/CeMAD-1. First, MDF-1/CeMAD-1 may play dual roles: one domain may provide spindle assembly checkpoint function, and the other domain may regulate an as yet unknown function required for embryogenesis. Second, MDF-1/CeMAD-1 may interact with different APC/C substrates during meiosis and mitosis, and the mdf-1(av19) allele shows defective interaction with meiosis-specific APC/C substrates but retains binding to mitosis-specific substrates. Third, a higher level of MDF-1/CeMAD-1 activity may be required for meiosis rather than mitosis, and mdf-1(av19) is simply a reduced-function mutant: the activity of mdf-1(av19) product is not sufficient for meiotic function but is sufficient for mitotic requirement. Further characterization of the mdf-1(av19)–mutant will elucidate the discrete functions of MDF-1/CeMAD-1 in mitosis and meiosis. Thus, the traditional genetic screen allows us to obtain a separation-of-function allele, which would not have been identified by the analysis of a mere loss-of-function phenotype generated by RNAi experiments. Isolation of additional alleles for each spindle assembly checkpoint gene will expand our knowledge about the function of the spindle assembly checkpoint components.
8. ACKNOWLEDGEMENTS
I thank the members of St. Jude Mitosis Club for their discussion and helpful comments. I also thank Kirsten Hagstrom and Frontiers in Bioscience reviewers for critical reading of this manuscript and useful suggestions. This work was supported by the Cancer Center Support Grant CA021765 from the National Cancer Institute and by the American Lebanese Syrian Associated Charities (ALSAC).
9. REFERENCES
- 1.Albertson DG, Thomson JN. The kinetochores of Caenorhabditis elegans. Chromosoma. 1982;86(3):409–28. doi: 10.1007/BF00292267. [DOI] [PubMed] [Google Scholar]
- 2.Howe M, McDonald KL, Albertson DG, Meyer BJ. HIM-10 is required for kinetochore structure and function on Caenorhabditis elegans holocentric chromosomes. J Cell Biol. 2001;153(6):1227–38. doi: 10.1083/jcb.153.6.1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McEwen BF, Hsieh CE, Mattheyses AL, Rieder CL. A new look at kinetochore structure in vertebrate somatic cells using high-pressure freezing and freeze substitution. Chromosoma. 1998;107(6-7):366–75. doi: 10.1007/s004120050320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Maddox PS, Oegema K, Desai A, Cheeseman IM. “Holo”er than thou: chromosome segregation and kinetochore function in C. elegans. Chromosome Res. 2004;12(6):641–53. doi: 10.1023/B:CHRO.0000036588.42225.2f. [DOI] [PubMed] [Google Scholar]
- 5.Oegema K, Hyman AA. WormBook. 2006. Cell division; pp. 1–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Buchwitz BJ, Ahmad K, Moore LL, Roth MB, Henikoff S. A histone-H3-like protein in C. elegans. Nature. 1999;401(6753):547–8. doi: 10.1038/44062. [DOI] [PubMed] [Google Scholar]
- 7.Moore LL, Morrison M, Roth MB. HCP-1, a protein involved in chromosome segregation, is localized to the centromere of mitotic chromosomes in Caenorhabditis elegans. J Cell Biol. 1999;147(3):471–80. doi: 10.1083/jcb.147.3.471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Moore LL, Roth MB. HCP-4, a CENP-C-like protein in Caenorhabditis elegans, is required for resolution of sister centromeres. J Cell Biol. 2001;153(6):1199–208. doi: 10.1083/jcb.153.6.1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stear JH, Roth MB. Characterization of HCP-6, a C. elegans protein required to prevent chromosome twisting and merotelic attachment. Genes Dev. 2002;16(12):1498–508. doi: 10.1101/gad.989102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Oegema K, Desai A, Rybina S, Kirkham M, Hyman AA. Functional analysis of kinetochore assembly in Caenorhabditis elegans. J Cell Biol. 2001;153(6):1209–26. doi: 10.1083/jcb.153.6.1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Praitis V, Casey E, Collar D, Austin J. Creation of low-copy integrated transgenic lines in Caenorhabditis elegans. Genetics. 2001;157(3):1217–26. doi: 10.1093/genetics/157.3.1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gassmann R, Kline SL, Carvalho A, Desai A. Analysis of kinetochore assembly and function in Caenorhabditis elegans embryos and human cells. Methods. 2007;41(2):177–89. doi: 10.1016/j.ymeth.2006.07.027. [DOI] [PubMed] [Google Scholar]
- 13.Maddox PS, Hyndman F, Monen J, Oegema K, Desai A. Functional genomics identifies a Myb domain-containing protein family required for assembly of CENP-A chromatin. J Cell Biol. 2007;176(6):757–63. doi: 10.1083/jcb.200701065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stoler S, Keith KC, Curnick KE, Fitzgerald-Hayes M. A mutation in CSE4, an essential gene encoding a novel chromatin-associated protein in yeast, causes chromosome nondisjunction and cell cycle arrest at mitosis. Genes Dev. 1995;9(5):573–86. doi: 10.1101/gad.9.5.573. [DOI] [PubMed] [Google Scholar]
- 15.Sullivan KF, Hechenberger M, Masri K. Human CENP-A contains a histone H3 related histone fold domain that is required for targeting to the centromere. J Cell Biol. 1994;127(3):581–92. doi: 10.1083/jcb.127.3.581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Meluh PB, Yang P, Glowczewski L, Koshland D, Smith MM. Cse4p is a component of the core centromere of Saccharomyces cerevisiae. Cell. 1998;94(5):607–13. doi: 10.1016/s0092-8674(00)81602-5. [DOI] [PubMed] [Google Scholar]
- 17.Desai A, Rybina S, Muller-Reichert T, Shevchenko A, Shevchenko A, Hyman A, Oegema K. KNL-1 directs assembly of the microtubule-binding interface of the kinetochore in C. elegans. Genes Dev. 2003;17(19):2421–35. doi: 10.1101/gad.1126303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Encalada SE, Willis J, Lyczak R, Bowerman B. A spindle checkpoint functions during mitosis in the early Caenorhabditis elegans embryo. Mol Biol Cell. 2005;16(3):1056–70. doi: 10.1091/mbc.E04-08-0712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maddox PS, Portier N, Desai A, Oegema K. Molecular analysis of mitotic chromosome condensation using a quantitative time-resolved fluorescence microscopy assay. Proc Natl Acad Sci U S A. 2006;103(41):15097–102. doi: 10.1073/pnas.0606993103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Akhmanova A, Hoogenraad CC, Drabek K, Stepanova T, Dortland B, Verkerk T, Vermeulen W, Burgering BM, De Zeeuw CI, Grosveld F, Galjart N. Clasps are CLIP-115 and −170 associating proteins involved in the regional regulation of microtubule dynamics in motile fibroblasts. Cell. 2001;104(6):923–35. doi: 10.1016/s0092-8674(01)00288-4. [DOI] [PubMed] [Google Scholar]
- 21.Mimori-Kiyosue Y, Grigoriev I, Lansbergen G, Sasaki H, Matsui C, Severin F, Galjart N, Grosveld F, Vorobjev I, Tsukita S, Akhmanova A. CLASP1 and CLASP2 bind to EB1 and regulate microtubule plus-end dynamics at the cell cortex. J Cell Biol. 2005;168(1):141–53. doi: 10.1083/jcb.200405094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lansbergen G, Grigoriev I, Mimori-Kiyosue Y, Ohtsuka T, Higa S, Kitajima I, Demmers J, Galjart N, Houtsmuller AB, Grosveld F, Akhmanova A. CLASPs attach microtubule plus ends to the cell cortex through a complex with LL5beta. Dev Cell. 2006;11(1):21–32. doi: 10.1016/j.devcel.2006.05.012. [DOI] [PubMed] [Google Scholar]
- 23.Mimori-Kiyosue Y, Grigoriev I, Sasaki H, Matsui C, Akhmanova A, Tsukita S, Vorobjev I. Mammalian CLASPs are required for mitotic spindle organization and kinetochore alignment. Genes Cells. 2006;11(8):845–57. doi: 10.1111/j.1365-2443.2006.00990.x. [DOI] [PubMed] [Google Scholar]
- 24.Moore LL, Stanvitch G, Roth MB, Rosen D. HCP-4/CENP-C promotes the prophase timing of centromere resolution by enabling the centromere association of HCP-6 in Caenorhabditis elegans. Mol Cell Biol. 2005;25(7):2583–92. doi: 10.1128/MCB.25.7.2583-2592.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cheeseman IM, Chappie JS, Wilson-Kubalek EM, Desai A. The conserved KMN network constitutes the core microtubule-binding site of the kinetochore. Cell. 2006;127(5):983–97. doi: 10.1016/j.cell.2006.09.039. [DOI] [PubMed] [Google Scholar]
- 26.Gonczy P, Echeverri C, Oegema K, Coulson A, Jones SJ, Copley RR, Duperon J, Oegema J, Brehm M, Cassin E, Hannak E, Kirkham M, Pichler S, Flohrs K, Goessen A, Leidel S, Alleaume AM, Martin C, Ozlu N, Bork P, Hyman AA. Functional genomic analysis of cell division in C. elegans using RNAi of genes on chromosome III. Nature. 2000;408(6810):331–6. doi: 10.1038/35042526. [DOI] [PubMed] [Google Scholar]
- 27.Cheeseman IM, Niessen S, Anderson S, Hyndman F, Yates JR, 3rd, Oegema K, Desai A. A conserved protein network controls assembly of the outer kinetochore and its ability to sustain tension. Genes Dev. 2004;18(18):2255–68. doi: 10.1101/gad.1234104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hodgkin J, Horvitz HR, Brenner S. Nondisjunction Mutants of the Nematode CAENORHABDITIS ELEGANS. Genetics. 1979;91(1):67–94. doi: 10.1093/genetics/91.1.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hedgecock EM, Herman RK. The ncl-1 gene and genetic mosaics of Caenorhabditis elegans. Genetics. 1995;141(3):989–1006. doi: 10.1093/genetics/141.3.989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nabetani A, Koujin T, Tsutsumi C, Haraguchi T, Hiraoka Y. A conserved protein, Nuf2, is implicated in connecting the centromere to the spindle during chromosome segregation: a link between the kinetochore function and the spindle checkpoint. Chromosoma. 2001;110(5):322–34. doi: 10.1007/s004120100153. [DOI] [PubMed] [Google Scholar]
- 31.Bharadwaj R, Qi W, Yu H. Identification of two novel components of the human NDC80 kinetochore complex. J Biol Chem. 2004;279(13):13076–85. doi: 10.1074/jbc.M310224200. [DOI] [PubMed] [Google Scholar]
- 32.McCleland ML, Kallio MJ, Barrett-Wilt GA, Kestner CA, Shabanowitz J, Hunt DF, Gorbsky GJ, Stukenberg PT. The vertebrate Ndc80 complex contains Spc24 and Spc25 homologs, which are required to establish and maintain kinetochoremicrotubule attachment. Curr Biol. 2004;14(2):131–7. doi: 10.1016/j.cub.2003.12.058. [DOI] [PubMed] [Google Scholar]
- 33.Saitoh H, Tomkiel J, Cooke CA, Ratrie H, 3rd, Maurer M, Rothfield NF, Earnshaw WC. CENP-C, an autoantigen in scleroderma, is a component of the human inner kinetochore plate. Cell. 1992;70(1):115–25. doi: 10.1016/0092-8674(92)90538-n. [DOI] [PubMed] [Google Scholar]
- 34.Palmer DK, O'Day K, Wener MH, Andrews BS, Margolis RL. A 17-kD centromere protein (CENP-A) copurifies with nucleosome core particles and with histones. J Cell Biol. 1987;104(4):805–15. doi: 10.1083/jcb.104.4.805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Earnshaw WC, Sullivan KF, Machlin PS, Cooke CA, Kaiser DA, Pollard TD, Rothfield NF, Cleveland DW. Molecular cloning of cDNA for CENP-B, the major human centromere autoantigen. J Cell Biol. 1987;104(4):817–29. doi: 10.1083/jcb.104.4.817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Earnshaw WC, Rothfield N. Identification of a family of human centromere proteins using autoimmune sera from patients with scleroderma. Chromosoma. 1985;91(3-4):313–21. doi: 10.1007/BF00328227. [DOI] [PubMed] [Google Scholar]
- 37.Cheeseman IM, MacLeod I, Yates JR, 3rd, Oegema K, Desai A. The CENP-F-like proteins HCP-1 and HCP-2 target CLASP to kinetochores to mediate chromosome segregation. Curr Biol. 2005;15(8):771–7. doi: 10.1016/j.cub.2005.03.018. [DOI] [PubMed] [Google Scholar]
- 38.Tarailo M, Tarailo S, Rose AM. Synthetic lethal interactions identify phenotypic “interologs” of the spindle assembly checkpoint components. Genetics. 2007;177(4):2525–30. doi: 10.1534/genetics.107.080408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hajeri VA, Stewart AM, Moore LL, Padilla PA. Genetic analysis of the spindle checkpoint genes san-1, mdf-2, bub-3 and the CENP-F like homologues hcp-1 and hcp-2 in Caenorhabditis elegans. Cell Div. 2008;3(1):6. doi: 10.1186/1747-1028-3-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Siddiqui SS. Metazoan motor models: kinesin superfamily in C. elegans. Traffic. 2002;3(1):20–8. doi: 10.1034/j.1600-0854.2002.30104.x. [DOI] [PubMed] [Google Scholar]
- 41.Srayko M, Kaya A, Stamford J, Hyman AA. Identification and characterization of factors required for microtubule growth and nucleation in the early C. elegans embryo. Dev Cell. 2005;9(2):223–36. doi: 10.1016/j.devcel.2005.07.003. [DOI] [PubMed] [Google Scholar]
- 42.Shu T, Ayala R, Nguyen MD, Xie Z, Gleeson JG, Tsai LH. Ndel1 operates in a common pathway with LIS1 and cytoplasmic dynein to regulate cortical neuronal positioning. Neuron. 2004;44(2):263–77. doi: 10.1016/j.neuron.2004.09.030. [DOI] [PubMed] [Google Scholar]
- 43.Cockell MM, Baumer K, Gonczy P. lis-1 is required for dynein-dependent cell division processes in C. elegans embryos. J Cell Sci. 2004;117(Pt 19):4571–82. doi: 10.1242/jcs.01344. [DOI] [PubMed] [Google Scholar]
- 44.Gonczy P, Schnabel H, Kaletta T, Amores AD, Hyman T, Schnabel R. Dissection of cell division processes in the one cell stage Caenorhabditis elegans embryo by mutational analysis. J Cell Biol. 1999;144(5):927–46. doi: 10.1083/jcb.144.5.927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fernandez AG, Piano F. MEL-28 is downstream of the Ran cycle and is required for nuclear-envelope function and chromatin maintenance. Curr Biol. 2006;16(17):1757–63. doi: 10.1016/j.cub.2006.07.071. [DOI] [PubMed] [Google Scholar]
- 46.Galy V, Askjaer P, Franz C, Lopez-Iglesias C, Mattaj IW. MEL-28, a novel nuclear-envelope and kinetochore protein essential for zygotic nuclear-envelope assembly in C. elegans. Curr Biol. 2006;16(17):1748–56. doi: 10.1016/j.cub.2006.06.067. [DOI] [PubMed] [Google Scholar]
- 47.Hagstrom KA, Meyer BJ. Condensin and cohesin: more than chromosome compactor and glue. Nat Rev Genet. 2003;4(7):520–34. doi: 10.1038/nrg1110. [DOI] [PubMed] [Google Scholar]
- 48.Hirano T. At the heart of the chromosome: SMC proteins in action. Nat Rev Mol Cell Biol. 2006;7(5):311–22. doi: 10.1038/nrm1909. [DOI] [PubMed] [Google Scholar]
- 49.Ono T, Losada A, Hirano M, Myers MP, Neuwald AF, Hirano T. Differential contributions of condensin I and condensin II to mitotic chromosome architecture in vertebrate cells. Cell. 2003;115(1):109–21. doi: 10.1016/s0092-8674(03)00724-4. [DOI] [PubMed] [Google Scholar]
- 50.Gerlich D, Hirota T, Koch B, Peters JM, Ellenberg J. Condensin I stabilizes chromosomes mechanically through a dynamic interaction in live cells. Curr Biol. 2006;16(4):333–44. doi: 10.1016/j.cub.2005.12.040. [DOI] [PubMed] [Google Scholar]
- 51.Hirota T, Gerlich D, Koch B, Ellenberg J, Peters JM. Distinct functions of condensin I and II in mitotic chromosome assembly. J Cell Sci. 2004;117(Pt 26):6435–45. doi: 10.1242/jcs.01604. [DOI] [PubMed] [Google Scholar]
- 52.Hagstrom KA, Holmes VF, Cozzarelli NR, Meyer BJ. C. elegans condensin promotes mitotic chromosome architecture, centromere organization, and sister chromatid segregation during mitosis and meiosis. Genes Dev. 2002;16(6):729–42. doi: 10.1101/gad.968302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chan RC, Severson AF, Meyer BJ. Condensin restructures chromosomes in preparation for meiotic divisions. J Cell Biol. 2004;167(4):613–25. doi: 10.1083/jcb.200408061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lieb JD, Albrecht MR, Chuang PT, Meyer BJ. MIX-1: an essential component of the C. elegans mitotic machinery executes X chromosome dosage compensation. Cell. 1998;92(2):265–77. doi: 10.1016/s0092-8674(00)80920-4. [DOI] [PubMed] [Google Scholar]
- 55.Guacci V, Koshland D, Strunnikov A. A direct link between sister chromatid cohesion and chromosome condensation revealed through the analysis of MCD1 in S. cerevisiae. Cell. 1997;91(1):47–57. doi: 10.1016/s0092-8674(01)80008-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Michaelis C, Ciosk R, Nasmyth K. Cohesins: chromosomal proteins that prevent premature separation of sister chromatids. Cell. 1997;91(1):35–45. doi: 10.1016/s0092-8674(01)80007-6. [DOI] [PubMed] [Google Scholar]
- 57.Toth A, Ciosk R, Uhlmann F, Galova M, Schleiffer A, Nasmyth K. Yeast cohesin complex requires a conserved protein, Eco1p(Ctf7), to establish cohesion between sister chromatids during DNA replication. Genes Dev. 1999;13(3):320–33. doi: 10.1101/gad.13.3.320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Losada A, Hirano M, Hirano T. Identification of Xenopus SMC protein complexes required for sister chromatid cohesion. Genes Dev. 1998;12(13):1986–97. doi: 10.1101/gad.12.13.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Losada A, Yokochi T, Kobayashi R, Hirano T. Identification and characterization of SA/Scc3p subunits in the Xenopus and human cohesin complexes. J Cell Biol. 2000;150(3):405–16. doi: 10.1083/jcb.150.3.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ciosk R, Zachariae W, Michaelis C, Shevchenko A, Mann M, Nasmyth K. An ESP1/PDS1 complex regulates loss of sister chromatid cohesion at the metaphase to anaphase transition in yeast. Cell. 1998;93(6):1067–76. doi: 10.1016/s0092-8674(00)81211-8. [DOI] [PubMed] [Google Scholar]
- 61.Uhlmann F, Lottspeich F, Nasmyth K. Sister-chromatid separation at anaphase onset is promoted by cleavage of the cohesin subunit Scc1. Nature. 1999;400(6739):37–42. doi: 10.1038/21831. [DOI] [PubMed] [Google Scholar]
- 62.Cohen-Fix O, Peters JM, Kirschner MW, Koshland D. Anaphase initiation in Saccharomyces cerevisiae is controlled by the APC-dependent degradation of the anaphase inhibitor Pds1p. Genes Dev. 1996;10(24):3081–93. doi: 10.1101/gad.10.24.3081. [DOI] [PubMed] [Google Scholar]
- 63.Pasierbek P, Jantsch M, Melcher M, Schleiffer A, Schweizer D, Loidl J. A Caenorhabditis elegans cohesion protein with functions in meiotic chromosome pairing and disjunction. Genes Dev. 2001;15(11):1349–60. doi: 10.1101/gad.192701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chan RC, Chan A, Jeon M, Wu TF, Pasqualone D, Rougvie AE, Meyer BJ. Chromosome cohesion is regulated by a clock gene paralogue TIM-1. Nature. 2003;423(6943):1002–9. doi: 10.1038/nature01697. [DOI] [PubMed] [Google Scholar]
- 65.Mito Y, Sugimoto A, Yamamoto M. Distinct developmental function of two Caenorhabditis elegans homologs of the cohesin subunit Scc1/Rad21. Mol Biol Cell. 2003;14(6):2399–409. doi: 10.1091/mbc.E02-09-0603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pasierbek P, Fodermayr M, Jantsch V, Jantsch M, Schweizer D, Loidl J. The Caenorhabditis elegans SCC-3 homologue is required for meiotic synapsis and for proper chromosome disjunction in mitosis and meiosis. Exp Cell Res. 2003;289(2):245–55. doi: 10.1016/s0014-4827(03)00266-0. [DOI] [PubMed] [Google Scholar]
- 67.Ruchaud S, Carmena M, Earnshaw WC. Chromosomal passengers: conducting cell division. Nat Rev Mol Cell Biol. 2007;8(10):798–812. doi: 10.1038/nrm2257. [DOI] [PubMed] [Google Scholar]
- 68.Pinsky BA, Biggins S. The spindle checkpoint: tension versus attachment. Trends Cell Biol. 2005;15(9):486–93. doi: 10.1016/j.tcb.2005.07.005. [DOI] [PubMed] [Google Scholar]
- 69.Romano A, Guse A, Krascenicova I, Schnabel H, Schnabel R, Glotzer M. CSC-1: a subunit of the Aurora B kinase complex that binds to the survivin-like protein BIR-1 and the incenp-like protein ICP-1. J Cell Biol. 2003;161(2):229–36. doi: 10.1083/jcb.200207117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kaitna S, Mendoza M, Jantsch-Plunger V, Glotzer M. Incenp and an aurora-like kinase form a complex essential for chromosome segregation and efficient completion of cytokinesis. Curr Biol. 2000;10(19):1172–81. doi: 10.1016/s0960-9822(00)00721-1. [DOI] [PubMed] [Google Scholar]
- 71.Kaitna S, Pasierbek P, Jantsch M, Loidl J, Glotzer M. The aurora B kinase AIR-2 regulates kinetochores during mitosis and is required for separation of homologous Chromosomes during meiosis. Curr Biol. 2002;12(10):798–812. doi: 10.1016/s0960-9822(02)00820-5. [DOI] [PubMed] [Google Scholar]
- 72.Hauf S, Roitinger E, Koch B, Dittrich CM, Mechtler K, Peters JM. Dissociation of cohesin from chromosome arms and loss of arm cohesion during early mitosis depends on phosphorylation of SA2. PLoS Biol. 2005;3(3):e69. doi: 10.1371/journal.pbio.0030069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kitajima TS, Kawashima SA, Watanabe Y. The conserved kinetochore protein shugoshin protects centromeric cohesion during meiosis. Nature. 2004;427(6974):510–7. doi: 10.1038/nature02312. [DOI] [PubMed] [Google Scholar]
- 74.McGuinness BE, Hirota T, Kudo NR, Peters JM, Nasmyth K. Shugoshin prevents dissociation of cohesin from centromeres during mitosis in vertebrate cells. PLoS Biol. 2005;3(3):e86. doi: 10.1371/journal.pbio.0030086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Watanabe Y. Shugoshin: guardian spirit at the centromere. Curr Opin Cell Biol. 2005;17(6):590–5. doi: 10.1016/j.ceb.2005.10.003. [DOI] [PubMed] [Google Scholar]
- 76.Watanabe Y. Sister chromatid cohesion along arms and at centromeres. Trends Genet. 2005;21(7):405–12. doi: 10.1016/j.tig.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 77.Yu H, Tang Z. Bub1 multitasking in mitosis. Cell Cycle. 2005;4(2):262–5. [PubMed] [Google Scholar]
- 78.Hauf S, Waizenegger IC, Peters JM. Cohesin cleavage by separase required for anaphase and cytokinesis in human cells. Science. 2001;293(5533):1320–3. doi: 10.1126/science.1061376. [DOI] [PubMed] [Google Scholar]
- 79.Waizenegger IC, Hauf S, Meinke A, Peters JM. Two distinct pathways remove mammalian cohesin from chromosome arms in prophase and from centromeres in anaphase. Cell. 2000;103(3):399–410. doi: 10.1016/s0092-8674(00)00132-x. [DOI] [PubMed] [Google Scholar]
- 80.Musacchio A, Salmon ED. The spindle-assembly checkpoint in space and time. Nat Rev Mol Cell Biol. 2007;8(5):379–93. doi: 10.1038/nrm2163. [DOI] [PubMed] [Google Scholar]
- 81.May KM, Hardwick KG. The spindle checkpoint. J Cell Sci. 2006;119(Pt 20):4139–42. doi: 10.1242/jcs.03165. [DOI] [PubMed] [Google Scholar]
- 82.Hyman AA, White JG. Determination of cell division axes in the early embryogenesis of Caenorhabditis elegans. J Cell Biol. 1987;105(5):2123–35. doi: 10.1083/jcb.105.5.2123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Strome S, Wood WB. Generation of asymmetry and segregation of germ-line granules in early C. elegans embryos. Cell. 1983;35(1):15–25. doi: 10.1016/0092-8674(83)90203-9. [DOI] [PubMed] [Google Scholar]
- 84.Kitagawa R, Rose AM. Components of the spindle-assembly checkpoint are essential in Caenorhabditis elegans. Nat Cell Biol. 1999;1(8):514–21. doi: 10.1038/70309. [DOI] [PubMed] [Google Scholar]
- 85.Hoyt MA, Totis L, Roberts BT. S. cerevisiae genes required for cell cycle arrest in response to loss of microtubule function. Cell. 1991;66(3):507–17. doi: 10.1016/0092-8674(81)90014-3. [DOI] [PubMed] [Google Scholar]
- 86.Li R, Murray AW. Feedback control of mitosis in budding yeast. Cell. 1991;66(3):519–31. doi: 10.1016/0092-8674(81)90015-5. [DOI] [PubMed] [Google Scholar]
- 87.Weiss E, Winey M. The Saccharomyces cerevisiae spindle pole body duplication gene MPS1 is part of a mitotic checkpoint. J Cell Biol. 1996;132(1-2):111–23. doi: 10.1083/jcb.132.1.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Starr DA, Williams BC, Li Z, Etemad-Moghadam B, Dawe RK, Goldberg ML. Conservation of the centromere/kinetochore protein ZW10. J Cell Biol. 1997;138(6):1289–301. doi: 10.1083/jcb.138.6.1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Chan GK, Jablonski SA, Starr DA, Goldberg ML, Yen TJ. Human Zw10 and ROD are mitotic checkpoint proteins that bind to kinetochores. Nat Cell Biol. 2000;2(12):944–7. doi: 10.1038/35046598. [DOI] [PubMed] [Google Scholar]
- 90.Williams BC, Li Z, Liu S, Williams EV, Leung G, Yen TJ, Goldberg ML. Zwilch, a new component of the ZW10/ROD complex required for kinetochore functions. Mol Biol Cell. 2003;14(4):1379–91. doi: 10.1091/mbc.E02-09-0624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Karess R. Rod-Zw10-Zwilch: a key player in the spindle checkpoint. Trends Cell Biol. 2005;15(7):386–92. doi: 10.1016/j.tcb.2005.05.003. [DOI] [PubMed] [Google Scholar]
- 92.Nystul TG, Goldmark JP, Padilla PA, Roth MB. Suspended animation in C. elegans requires the spindle checkpoint. Science. 2003;302(5647):1038–41. doi: 10.1126/science.1089705. [DOI] [PubMed] [Google Scholar]
- 93.Hajeri VA, Trejo J, Padilla PA. Characterization of sub-nuclear changes in Caenorhabditis elegans embryos exposed to brief, intermediate and long-term anoxia to analyze anoxia-induced cell cycle arrest. BMC Cell Biol. 2005;6:47. doi: 10.1186/1471-2121-6-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Stein KK, Davis ES, Hays T, Golden A. Components of the spindle assembly checkpoint regulate the anaphase-promoting complex during meiosis in Caenorhabditis elegans. Genetics. 2007;175(1):107–23. doi: 10.1534/genetics.106.059105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Sonnichsen B, Koski LB, Walsh A, Marschall P, Neumann B, Brehm M, Alleaume AM, Artelt J, Bettencourt P, Cassin E, Hewitson M, Holz C, Khan M, Lazik S, Martin C, Nitzsche B, Ruer M, Stamford J, Winzi M, Heinkel R, Roder M, Finell J, Hantsch H, Jones SJ, Jones M, Piano F, Gunsalus KC, Oegema K, Gonczy P, Coulson A, Hyman AA, Echeverri CJ. Full-genome RNAi profiling of early embryogenesis in Caenorhabditis elegans. Nature. 2005;434(7032):462–9. doi: 10.1038/nature03353. [DOI] [PubMed] [Google Scholar]
- 96.Li S, Armstrong CM, Bertin N, Ge H, Milstein S, Boxem M, Vidalain PO, Han JD, Chesneau A, Hao T, Goldberg DS, Li N, Martinez M, Rual JF, Lamesch P, Xu L, Tewari M, Wong SL, Zhang LV, Berriz GF, Jacotot L, Vaglio P, Reboul J, Hirozane-Kishikawa T, Li Q, Gabel HW, Elewa A, Baumgartner B, Rose DJ, Yu H, Bosak S, Sequerra R, Fraser A, Mango SE, Saxton WM, Strome S, Van Den Heuvel S, Piano F, Vandenhaute J, Sardet C, Gerstein M, Doucette-Stamm L, Gunsalus KC, Harper JW, Cusick ME, Roth FP, Hill DE, Vidal M. A map of the interactome network of the metazoan C. elegans. Science. 2004;303(5657):540–3. doi: 10.1126/science.1091403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Scaerou F, Starr DA, Piano F, Papoulas O, Karess RE, Goldberg ML. The ZW10 and Rough Deal checkpoint proteins function together in a large, evolutionarily conserved complex targeted to the kinetochore. J Cell Sci. 2001;114(Pt 17):3103–14. doi: 10.1242/jcs.114.17.3103. [DOI] [PubMed] [Google Scholar]
- 98.Matthews LR, Carter P, Thierry-Mieg D, Kemphues K. ZYG-9, a Caenorhabditis elegans protein required for microtubule organization and function, is a component of meiotic and mitotic spindle poles. J Cell Biol. 1998;141(5):1159–68. doi: 10.1083/jcb.141.5.1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Sweeney HL, Holzbaur EL. Mutational analysis of motor proteins. Annu Rev Physiol. 1996;58:751–92. doi: 10.1146/annurev.ph.58.030196.003535. [DOI] [PubMed] [Google Scholar]
- 100.Skop AR, White JG. The dynactin complex is required for cleavage plane specification in early Caenorhabditis elegans embryos. Curr Biol. 1998;8(20):1110–6. doi: 10.1016/s0960-9822(98)70465-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Gonczy P, Pichler S, Kirkham M, Hyman AA. Cytoplasmic dynein is required for distinct aspects of MTOC positioning, including centrosome separation, in the one cell stage Caenorhabditis elegans embryo. J Cell Biol. 1999;147(1):135–50. doi: 10.1083/jcb.147.1.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Buttner EA, Gil-Krzewska AJ, Rajpurohit AK, Hunter CP. Progression from mitotic catastrophe to germ cell death in Caenorhabditis elegans lis-1 mutants requires the spindle checkpoint. Dev Biol. 2007;305(2):397–410. doi: 10.1016/j.ydbio.2007.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Schumacher JM, Ashcroft N, Donovan PJ, Golden A. A highly conserved centrosomal kinase, AIR-1, is required for accurate cell cycle progression and segregation of developmental factors in Caenorhabditis elegans embryos. Development. 1998;125(22):4391–402. doi: 10.1242/dev.125.22.4391. [DOI] [PubMed] [Google Scholar]
- 104.Hannak E, Kirkham M, Hyman AA, Oegema K. Aurora-A kinase is required for centrosome maturation in Caenorhabditis elegans. J Cell Biol. 2001;155(7):1109–16. doi: 10.1083/jcb.200108051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Sonoda E, Matsusaka T, Morrison C, Vagnarelli P, Hoshi O, Ushiki T, Nojima K, Fukagawa T, Waizenegger IC, Peters JM, Earnshaw WC, Takeda S. Scc1/Rad21/Mcd1 is required for sister chromatid cohesion and kinetochore function in vertebrate cells. Dev Cell. 2001;1(6):759–70. doi: 10.1016/s1534-5807(01)00088-0. [DOI] [PubMed] [Google Scholar]
- 106.Tomonaga T, Nagao K, Kawasaki Y, Furuya K, Murakami A, Morishita J, Yuasa T, Sutani T, Kearsey SE, Uhlmann F, Nasmyth K, Yanagida M. Characterization of fission yeast cohesin: essential anaphase proteolysis of Rad21 phosphorylated in the S phase. Genes Dev. 2000;14(21):2757–70. doi: 10.1101/gad.832000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Pangilinan F, Spencer F. Abnormal kinetochore structure activates the spindle assembly checkpoint in budding yeast. Mol Biol Cell. 1996;7(8):1195–208. doi: 10.1091/mbc.7.8.1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Hoque MT, Ishikawa F. Cohesin defects lead to premature sister chromatid separation, kinetochore dysfunction, and spindle-assembly checkpoint activation. J Biol Chem. 2002;277(44):42306–14. doi: 10.1074/jbc.M206836200. [DOI] [PubMed] [Google Scholar]
- 109.Harper JW, Burton JL, Solomon MJ. The anaphase-promoting complex: it's not just for mitosis any more. Genes Dev. 2002;16(17):2179–206. doi: 10.1101/gad.1013102. [DOI] [PubMed] [Google Scholar]
- 110.Golden A, Sadler PL, Wallenfang MR, Schumacher JM, Hamill DR, Bates G, Bowerman B, Seydoux G, Shakes DC. Metaphase to anaphase (mat) transition-defective mutants in Caenorhabditis elegans. J Cell Biol. 2000;151(7):1469–82. doi: 10.1083/jcb.151.7.1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Davis ES, Wille L, Chestnut BA, Sadler PL, Shakes DC, Golden A. Multiple subunits of the Caenorhabditis elegans anaphase-promoting complex are required for chromosome segregation during meiosis I. Genetics. 2002;160(2):805–13. doi: 10.1093/genetics/160.2.805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Shakes DC, Sadler PL, Schumacher JM, Abdolrasulnia M, Golden A. Developmental defects observed in hypomorphic anaphase-promoting complex mutants are linked to cell cycle abnormalities. Development. 2003;130(8):1605–20. doi: 10.1242/dev.00385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Furuta T, Tuck S, Kirchner J, Koch B, Auty R, Kitagawa R, Rose AM, Greenstein D. EMB-30: an APC4 homologue required for metaphase-to-anaphase transitions during meiosis and mitosis in Caenorhabditis elegans. Mol Biol Cell. 2000;11(4):1401–19. doi: 10.1091/mbc.11.4.1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kitagawa R, Law E, Tang L, Rose AM. The Cdc20 homolog, FZY-1, and its interacting protein, IFY-1, are required for proper chromosome segregation in Caenorhabditis elegans. Curr Biol. 2002;12(24):2118–23. doi: 10.1016/s0960-9822(02)01392-1. [DOI] [PubMed] [Google Scholar]
- 115.Tarailo M, Kitagawa R, Rose AM. Suppressors of spindle checkpoint defect (such) mutants identify new mdf-1/MAD1 interactors in Caenorhabditis elegans. Genetics. 2007;175(4):1665–79. doi: 10.1534/genetics.106.067918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Albertson DG, Thomson JN. Segregation of holocentric chromosomes at meiosis in the nematode, Caenorhabditis elegans. Chromosome Res. 1993;1(1):15–26. doi: 10.1007/BF00710603. [DOI] [PubMed] [Google Scholar]
- 117.Monen J, Maddox PS, Hyndman F, Oegema K, Desai A. Differential role of CENP-A in the segregation of holocentric C. elegans chromosomes during meiosis and mitosis. Nat Cell Biol. 2005;7(12):1248–55. doi: 10.1038/ncb1331. [DOI] [PubMed] [Google Scholar]
- 118.Rogers E, Bishop JD, Waddle JA, Schumacher JM, Lin R. The aurora kinase AIR-2 functions in the release of chromosome cohesion in Caenorhabditis elegans meiosis. J Cell Biol. 2002;157(2):219–29. doi: 10.1083/jcb.200110045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Hsu JY, Sun ZW, Li X, Reuben M, Tatchell K, Bishop DK, Grushcow JM, Brame CJ, Caldwell JA, Hunt DF, Lin R, Smith MM, Allis CD. Mitotic phosphorylation of histone H3 is governed by Ipl1/aurora kinase and Glc7/PP1 phosphatase in budding yeast and nematodes. Cell. 2000;102(3):279–91. doi: 10.1016/s0092-8674(00)00034-9. [DOI] [PubMed] [Google Scholar]
- 120.Tang Z, Sun Y, Harley SE, Zou H, Yu H. Human Bub1 protects centromeric sister-chromatid cohesion through Shugoshin during mitosis. Proc Natl Acad Sci U S A. 2004;101(52):18012–7. doi: 10.1073/pnas.0408600102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Kitajima TS, Hauf S, Ohsugi M, Yamamoto T, Watanabe Y. Human Bub1 defines the persistent cohesion site along the mitotic chromosome by affecting Shugoshin localization. Curr Biol. 2005;15(4):353–9. doi: 10.1016/j.cub.2004.12.044. [DOI] [PubMed] [Google Scholar]
- 122.Sulston JE, Horvitz HR. Post-embryonic cell lineages of the nematode, Caenorhabditis elegans. Dev Biol. 1977;56(1):110–56. doi: 10.1016/0012-1606(77)90158-0. [DOI] [PubMed] [Google Scholar]
- 123.Sulston JE, Schierenberg E, White JG, Thomson JN. The embryonic cell lineage of the nematode Caenorhabditis elegans. Dev Biol. 1983;100(1):64–119. doi: 10.1016/0012-1606(83)90201-4. [DOI] [PubMed] [Google Scholar]
- 124.Brauchle M, Baumer K, Gonczy P. Differential activation of the DNA replication checkpoint contributes to asynchrony of cell division in C. elegans embryos. Curr Biol. 2003;13(10):819–27. doi: 10.1016/s0960-9822(03)00295-1. [DOI] [PubMed] [Google Scholar]
- 125.Gorbsky GJ, Chen RH, Murray AW. Microinjection of antibody to Mad2 protein into mammalian cells in mitosis induces premature anaphase. J Cell Biol. 1998;141(5):1193–205. doi: 10.1083/jcb.141.5.1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Taylor SS, McKeon F. Kinetochore localization of murine Bub1 is required for normal mitotic timing and checkpoint response to spindle damage. Cell. 1997;89(5):727–35. doi: 10.1016/s0092-8674(00)80255-x. [DOI] [PubMed] [Google Scholar]
- 127.Fukuyama M, Gendreau SB, Derry WB, Rothman JH. Essential embryonic roles of the CKI-1 cyclin-dependent kinase inhibitor in cell-cycle exit and morphogenesis in C elegans. Dev Biol. 2003;260(1):273–86. doi: 10.1016/s0012-1606(03)00239-2. [DOI] [PubMed] [Google Scholar]
- 128.Fukuyama M, Rougvie AE, Rothman JH. C. elegans DAF-18/PTEN mediates nutrient-dependent arrest of cell cycle and growth in the germline. Curr Biol. 2006;16(8):773–9. doi: 10.1016/j.cub.2006.02.073. [DOI] [PubMed] [Google Scholar]
- 129.Watanabe S, Yamamoto TG, Kitagawa R. Spindle assembly checkpoint gene mdf-1 regulates germ cell proliferation in response to nutrition signals in C. elegans. Embo J. 2008 doi: 10.1038/emboj.2008.32. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Daniel JA, Keyes BE, Ng YP, Freeman CO, Burke DJ. Diverse functions of spindle assembly checkpoint genes in Saccharomyces cerevisiae. Genetics. 2006;172(1):53–65. doi: 10.1534/genetics.105.046441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Lee MS, Spencer FA. Bipolar orientation of chromosomes in Saccharomyces cerevisiae is monitored by Mad1 and Mad2, but not by Mad3. Proc Natl Acad Sci U S A. 2004;101(29):10655–60. doi: 10.1073/pnas.0404102101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Dawes HE, Berlin DS, Lapidus DM, Nusbaum C, Davis TL, Meyer BJ. Dosage compensation proteins targeted to X chromosomes by a determinant of hermaphrodite fate. Science. 1999;284(5421):1800–4. doi: 10.1126/science.284.5421.1800. [DOI] [PubMed] [Google Scholar]