

Summary
Inhibition of HIF-1 is an attractive therapeutic strategy to target the tumor microenvironment. However, HIF-1 inhibitors may have limited activity as single agents and combination therapies may be required. We tested the hypothesis that HIF-1 inhibition in a hypoxic stressed tumor microenvironment, which could be generated by administration of antiangiogenic agents, may result in a more pronounced therapeutic effect. The activity of bevacizumab, either alone or in combination with the HIF-1α inhibitor topotecan, was evaluated in U251-HRE xenografts. Tumor tissue was collected at the end of treatment and changes in tumor oxygenation, angiogenesis, proliferation, apoptosis, HIF-1α levels, HIF-1 target genes and DNA damage were evaluated. Bevacizumab decreased microvessel-density and increased intratumor-hypoxia, but did not induce apoptosis. Moreover, bevacizumab alone caused a significant increase of HIF-1-dependent gene expression in tumor tissue. Addition of a low dose of daily topotecan to bevacizumab significantly inhibited tumor growth, relative to mice treated with topotecan or bevacizumab alone (p<0.01). The addition of topotecan to bevacizumab was also associated with profound inhibition of HIF-1 transcriptional activity, significant inhibition of proliferation and induction of apoptosis. Importantly, DNA damage induced by topotecan alone was not augmented by addition of bevacizumab, suggesting that increased cytotoxic activity did not account for the increased anti-tumor effects observed.
These results strongly suggest that combination of anti-VEGF antibodies with HIF-1 inhibitors is an attractive therapeutic strategy targeting in the hypoxic tumor microenvironment.
Keywords: HIF-1, topotecan, bevacizumab, hypoxia, angiogenesis, cancer therapeutics
Introduction
Areas of low oxygen levels (hypoxia) are frequently found in solid tumors due to an imbalance between oxygen delivery and consumption (1). Hypoxia in solid tumors is associated with resistance to radiation therapy and chemotherapy, selection of more invasive and metastatic clones and poor patient prognosis (2, 3). Hypoxia Inducible Factor-1 (HIF-1) is a master regulator of cellular adaptation to oxygen deprivation and may act as a survival factor of hypoxic cancer cells, primarily by activating transcription of genes involved in angiogenesis, glycolytic metabolism, oxygen consumption, migration and invasion (4). Expression of HIF-1α has been demonstrated in many human cancers and is associated with poor prognosis and treatment failure (5-9). Thus, HIF-1 inhibition is an attractive therapeutic strategy to target hypoxic cancer cells (10, 11). However, the potential therapeutic efficacy of HIF-1 inhibitors remains to be established. The heterogeneity of HIF-1α expression in hypoxic areas of solid tumors suggests that combination strategies may be required to achieve a meaningful therapeutic effect in human cancers. We hypothesized that inhibition of HIF-1 may be more effective under conditions of sustained hypoxia (hypoxic stress), where HIF-1-dependent pathways may become essential for the survival of cancer cells.
Bevacizumab, a humanized monoclonal antibody against vascular endothelial growth factor A (VEGF-A) (12), is the first approved anti-angiogenic agent for cancer therapy based on its efficacy, in combination with standard chemotherapy, in patients with metastatic colorectal cancer (CRC), non-squamous non-small cell lung cancer (NSCLC) and metastatic breast cancer (13-15). However, antiangiogenic agents used as single therapy have yielded disappointing results, which may be due, at least in part, to increased intratumor hypoxia and activation of compensatory survival pathways that negatively impact on the therapeutic outcome (16, 17). Indeed, the effects of antiangiogenic agents on the tumor microenvironment are still controversial. Evidence has been provided supporting either a vascular regression, which is presumably associated with increased intratumor hypoxia, (18), or a so-called “normalization” of tumor vasculature, with a consequent decrease in interstitial pressure and better delivery of chemotherapy (19).
Topotecan is a topoisomerase I poison that induces DNA damage and cytotoxicity. However, topotecan also potently inhibits HIF-1α translation by a DNA-damage independent mechanism (20-22). Notably, daily administration of topotecan inhibited HIF-1α expression in U251-HRE xenografts, providing evidence of a biologically relevant inhibition of HIF-1 function (23).
The objective of this study was to test: a) the effects of bevacizumab on intratumor hypoxia and HIF-1 activity; and b) whether blocking HIF-1 expression in a hypoxic tumor microenvironment increased the therapeutic activity of bevacizumab. We found that bevacizumab (5mg/kg, q3d×4) or TPT (0.5mg/kg, qd×10) alone caused a modest growth delay in U251-HRE xenografts. In contrast, concomitant administration of bevacizumab and topotecan caused a pronounced anti-tumor effect, with tumor regression. Bevacizumab alone increased intratumor hypoxia, HIF-1 transcriptional activity, decreased proliferation but did not induce apoptosis of cancer cells. On the contrary, addition of TPT to bevacizumab dramatically reduced HIF-1 activity, tumor cells proliferation and induced apoptosis, yet did not induce higher levels of DNA damage relative to TPT alone.
These results suggest that targeting HIF-1α activity in a hypoxic stressed tumor microenvironment may significantly improve therapeutic outcome, thus maximizing the effect of abrogating compensatory pathways induced by hypoxia.
Material and Methods
Cell lines and reagents
U251-HRE cells, expressing luciferase under control of 3 copies of a Hypoxic Responsive Element (HRE), were routinely maintained as previously described (22). Experiments under hypoxia (1% O2) were performed in hypoxic workstation INVIVO2 400 (Biotrace International, Cincinnati, OH). TPT was obtained from the Drug Synthesis and Chemistry Branch, DTP, NCI. Bevacizumab (Avastin®) was purchased from the NIH Pharmacy.
Animal studies and imaging
Studies were conducted in female athymic nude (NCr/nu) mice obtained from the Animal Production Area (NCI-Frederick) in an AAALAC-accredited facility with an approved animal protocol. Tumors generated with the U251-HRE cell line were obtained by injecting 1 × 107 tumor cells subcutaneously (s.c.) into the flank. Tumor size was monitored by collecting length and width measurements and calculating the tumor weight (mg) as [tumor length × (tumor width)2]/2. For luminescence imaging, mice received 150 mg firefly luciferase (Biosynth AG, Switzerland) per kg body weight given intraperitoneally (i.p.). Following anesthesia with isoflurane gas (Abbott Laboratories, North Chicago, Il), mice were placed into a Xenogen IVIS® imaging station (Xenogen Corp, Alameda, CA) and imaged using Living Image Software® (Xenogen Corp).
Topotecan (solubilized in sterile water) and Bevacizumab (diluted in 0.05% BSA in saline to a final concentration of 0.5mg/ml) were dosed i.p. and 0.05% BSA in saline was used as the vehicle control.
When mice were sacrificed, tumors from each animal were divided in four equal parts to perform protein analysis, RNA analysis and immunohistochemistry.
Immunoblotting analysis
Western blotting analysis from whole cell lysates and from tumor lysates was performed as described previously (23). Monoclonal anti-HIF-1α antibody was purchased from BD-Biosciences (San Diego, CA), p42/44 and γH2AX antibodies were purchased from Cell Signaling Technology (Danvers, MA). β actin antibody was from Millipore (Danver, MA).
Real time PCR analysis
Total RNA from tumors was isolated using the RNA Mini Kit (Qiagen, Inc., Valencia, CA) according to the manufacturer's procedure. RT-PCR and real time PCR to measure hVEGF, hCXCR4, hLOX, hCA9, hPDK1, hHK2, hPFKFB3, hGLUT3 mRNA expression were performed as previously described (22). 18S rRNA, as internal control, was assessed using premixed reagents from Applied Biosystem (Foster City, CA). The sequences of primers and probes used are available upon request.
Immunohistochemistry
Tumors were fixed in 4% paraformaldehyde and then processed and embedded in paraffin. HIF-1α immunostaining was performed following heat induced antigen retrieval using target retrieval solution (Dako North America, Inc, Carpinteria, CA). Monoclonal anti-HIF-1α antibody (clone H1alpha67) was used at a dilution of 1:2,000 (Novus Biologicals Inc., Littleton, CO). Detection was carried out with CSA II (Dako) on an automated robot (Autostainer) according to the manufacturer's recommendations. Monoclonal anti PECAM-1 antibody was used at a dilution of 1:100 (Santa Cruz Biotechnology, Inc. Santa Cruz, CA). Detection of hypoxic areas was performed using the Hypoxyprobe ™ staining kit obtained from NPI Inc. (Burlington, MA). Antigen retrieval was performed with a citrate solution (BioGenex, San Ramon, CA) in a microwave processor. Hypoxyprobe Mab-1 was used at a dilution of 1:50 for 30 minutes at room temperature. Polyclonal rabbit anti-Ki67 antibody was used at a dilution of 1:10000. TUNEL staining was performed using the ApopTag kit from Millipore (Danver, MA), according to the manufacturer's instructions. Detection was performed by standard ABC methods. Slides were stained for γH2Ax expression using biotinylated MAb JBW301 (Upstate Biotechnology, Lake Placid NY) as the primary antibody, and Streptavidin labeled with Alexafluor 488 (Molecular Probes, Eugene OR) and processed in a Vision Biosystems Autostainer. Slides were coverslipped after embedding in Prolong Gold containing DAPI (Molecular Probes) and allowed to harden at least 1 hour before viewing. Staining specificity controls were mouse testis, a strong positive tissue, and mouse intestine, which is γH2AX negative. Controls were run with every slide set and all controls were stained with the JBW301 MAb.
Images were captured using a Retiga 2000R CCD camera outfitted with a RGB filter (Qimaging) on a Nikon Eclipse 80i upright microscope. All images were captured under consistent illumination and exposure for their respective stains. CD31, Ki67, TUNEL, H&E and Hypoxyprobe slides were scanned in using a Scanscope CS (Aperio, Vista, CA) at 20× and 40× magnification using default system settings. Images were extracted with Imagescope. No image post-processing was done to enhance or alter the original images. Necrotic areas were subtracted from Hypoxyprobe positive values, to evaluate areas of hypoxia only in living cells. For CD31, Ki67 and TUNEL analysis, 10 fields per sample were randomly selected. Image analysis was conducted through Image-Pro Plus v6.1 (MediaCybernetics). Custom made scripts were developed to analyze the respective target signals using various color and morphological segmentation tools. Post-capture image modification was limited to contrast enhancement. Gamma-H2AX response was quantified by counting the number of nuclei in a high-power (200X) field. Two or three fields were selected (according to the size of the specimen) while illuminating in Phase Contrast, such that Phase-dense fields were selected for imaging in blue and green channels, and counts were performed on the merged and color converted photograph. Autofluorescence due to blood cells was determined in the red channel.
Statistical analysis
All data were analyzed using GraphPad Prism 4 (GraphPad Software Inc., San Diego, CA) and the Mann-Whitney non-parametric t-test. The results of the in vivo experiments were tested for outliers using Grubb's test (www.graphpad.com). A p value of less than 0.05 was considered statistically significant.
Results
The combination of bevacizumab with daily topotecan inhibits tumor growth in U251-HRE xenografts
We first tested the effects of the combination of bevacizumab and daily TPT on U251 cells cultured under normoxic or hypoxic conditions in vitro. Bevacizumab, alone or in combination with TPT, did not affect cell proliferation and survival of U251, relative to TPT alone, suggesting that bevacizumab did not have a direct effect on U251 cells (data not shown). U251-HRE cells were then injected subcutaneously into female nude mice and when tumors reached ∼175 mg in size, the mice (n=10/group) were randomized into treatment groups, which included vehicle control, bevacizumab (5mg/kg, q3d×4) or TPT (0.5mg/kg, qd×10, 50% of the dose that inhibited tumor growth in previous experiments) (23), either alone or in combination. Tumors from vehicle treated mice showed a 2.64 fold increase in size over the course of the experiment. Bevacizumab or TPT alone caused a modest reduction in tumor growth (40% and 30%, respectively, relative to vehicle treated mice, Figure 1A). In contrast, the combination of bevacizumab and TPT induced significantly more pronounced tumor inhibition (80% relative to vehicle-treated mice, p=0.0043), and a 47% tumor shrinkage compared to the size at the beginning of treatment.
Figure 1. Daily administration of TPT, in combination with bevacizumab, inhibits tumor growth and luciferase activity in U251-HRE xenografts.
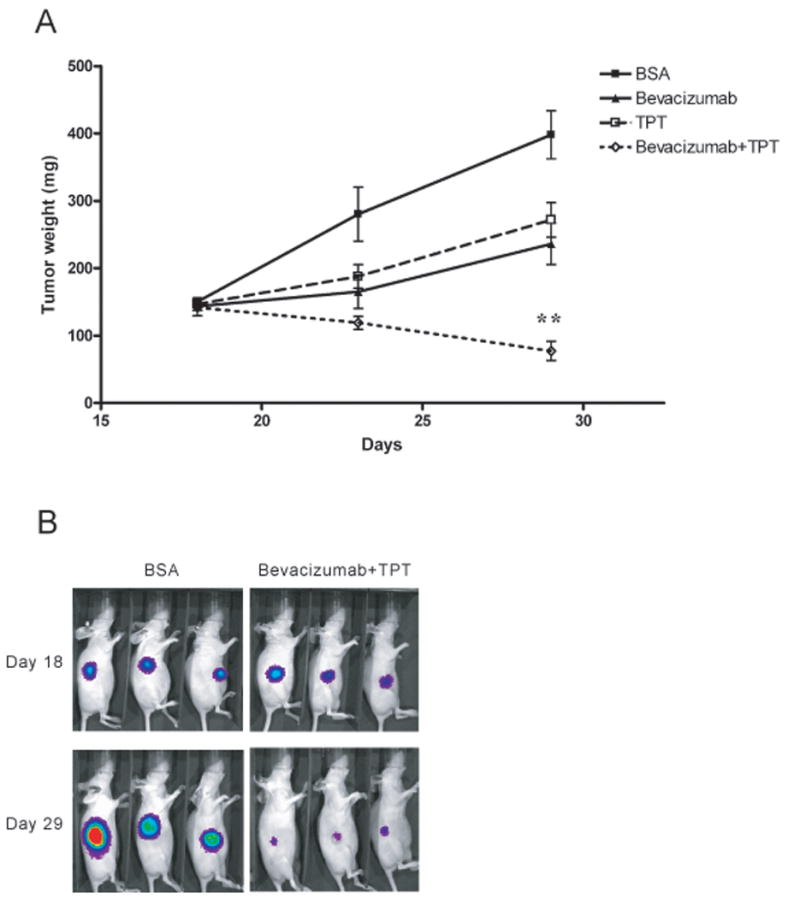
A. U251-HRE cells were implanted into nude mice (n=10 per group) and allowed to grow up to day 18 when treatment was started as indicated. Tumor weight was measured as described in Materials and Methods. On day 29 (end of treatment), there was a statistically significant reduction of tumor weight in mice treated with the combination of TPT and bevacizumab, relative to vehicle-treated controls (Mann-Whitney test, ** indicates p = 0.0043). B. Representative image of luciferase expression from vehicle-treated mice and mice treated with the combination of bevacizumab plus TPT.
U251-HRE cells express the luciferase reporter gene under control of HIF-1 and non-invasive detection of luciferase expression is an indirect measure of HIF-1 transcriptional activity and intratumor hypoxia (23), under conditions in which oxygen availability (a required cofactor for light emission (24)) is not limiting. As shown in Figure 1B, increased expression of luciferase was detected in vehicle-treated mice by the end of treatment (day 10), which reflects the increase of tumor size and presumably indicates an increase in intratumor hypoxia and HIF-1 activity. On the contrary, luciferase activity was decreased in mice treated with TPT or bevacizumab alone, respectively, relative to vehicle-treated controls. While the inhibition of luciferase by TPT is consistent with both an inhibition of tumor size and the previously reported inhibition of HIF-1 activity (23), the inhibition of luciferase expression in mice treated with bevacizumab alone might be due not only to a decrease in tumor size but also to a better tumor oxygenation, with a consequent decrease of intratumor hypoxia or, conversely, to an increase of intratumor hypoxia and a decrease in luciferase activity due to limited availability of oxygen. The combination of TPT with bevacizumab also caused a significant decrease in luciferase emission.
Overall, these data show that the combination of TPT and bevacizumab significantly reduces tumor growth, relative to either agent used alone, and they are suggestive of changes in tumor oxygenation by bevacizumab.
Bevacizumab increases intratumor hypoxia, decreases tumor cells proliferation but does not induce apoptosis in U251-HRE xenografts
The hypothesis underlying this study was that bevacizumab might increase intratumor hypoxia by causing vascular regression. To assess the effects of bevacizumab on the tumor microenvironment, sections from tumors harvested at the end of treatment (day 10) were stained with CD31, an endothelial cell marker, to measure microvessel density. As shown in Figure 2A, tumors from control mice had numerous large vessels, which were decreased in number (34%, data not shown) and size (47%, p=0.0029) in mice treated with bevacizumab. Interestingly, administration of TPT alone slightly increased vessel number and size compared to vehicle control, although these effects were not statistically significant. Notably, the combination of bevacizumab and TPT caused a marked decrease in vessel number (50%, data not shown) and size (80%, p=0.0007), compared to either agent alone and to vehicle-treated mice, demonstrating that the combination of the two agents is more effective in decreasing tumor vasculature.
Figure 2. Bevacizumab increases intratumor hypoxia, decreases tumor cells proliferation but does not induce apoptosis in U251-HRE xenografts.
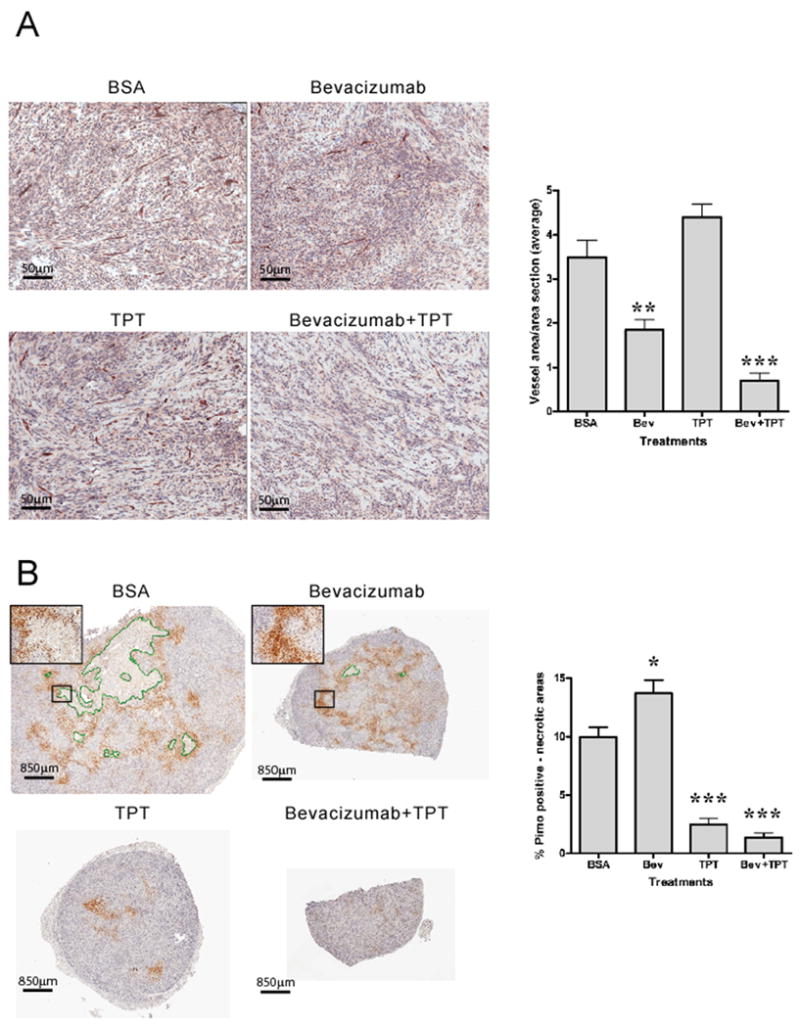
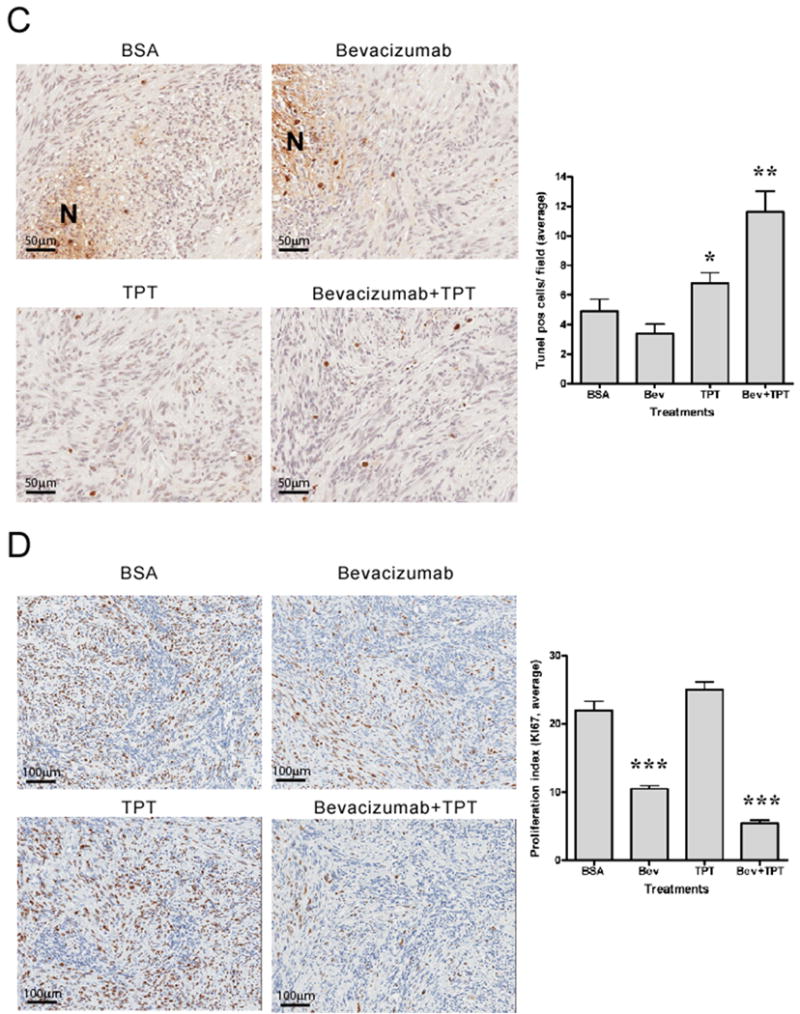
A. CD31 staining (20X) of representative tumor sections from different treatment groups. Graph represents total CD31 positive areas normalized by section areas (mean ± SE, 5 mice/group; (**p=0.029, *** p= 0.0007, relative to vehicle treated mice). B. Pimonidazole staining (1.2X) of representative tumor sections from different treatment groups. Necrotic areas are highlighted in green. Insets are representative fields at higher magnification (10X). Graph represents total pimonidazole positive areas after subtraction of necrotic areas and normalization by section areas (mean ± SE, 5 mice/group, * p=0.0379, *** p= 0.0002). C. TUNEL staining (20X) of regions of representative tumor sections from different treatment groups. “N” indicates necrotic areas. Graph represents total TUNEL positive cells normalized by section area (mean ± SE, 5 mice/group, * p=0.0172, ** p= 0.0027). D. Ki67 staining (10X) of regions of representative tumor sections from different treatment groups. Graph represents total Ki67 positive nuclei normalized by total nuclei number (mean ± SE, 5 mice/group, *** p<0.0001).
To test the effect of bevacizumab on intratumor hypoxia, we measured viable hypoxic areas by staining tumor sections with pimonidazole. As shown in Figure 2B, tumor sections from vehicle-treated mice showed areas of necrosis (highlighted in green) and a classic zonal pattern of pimonidazole positive areas, reflective of hypoxic regions, scattered throughout the section and in viable rims surrounding necrotic areas. Tumors from mice treated with bevacizumab showed minimal areas of necrosis and a 38% increase in pimonidazole positive areas, statistically significant compared to vehicle-treated mice (p=0.0379) and consistent with the decrease in vessel number and size shown in Figure 2A. On the contrary, tumor sections from mice treated with TPT alone showed a marked decrease of intratumor hypoxia (75%, p=0.0002), which was even more pronounced (86%, p=0.0002) in mice treated with TPT plus bevacizumab. The extent of intratumor hypoxia, detected by pimonidazole, may reflect not only changes in tumor vasculature, but also changes in tumor size and oxygen consumption.
To evaluate the effects of changes in tumor vasculature and oxygenation caused by bevacizumab on cancer cells we first examined tissues stained for H&E (Supplementary Figure 1). Sections of tumors from mice treated with bevacizumab alone showed multifocal areas of vacuolated cells with pyknotic nuclei, consistent with induction of cell death, compared to vehicle treated controls. On the contrary, tumors from mice treated with TPT as single agent, showed large multifocal areas of cell death characterized by pyknotic nuclei and cytoplasmic vacuolation or loss. Combination of TPT and bevacizumab showed large areas of multifocal cell death (pyknotic and karyolytic nuclei with disrupted cytoplasm) but also an extensive acellular eosinophilic matrix deposition, suggesting that the combination, but not bevacizumab or TPT alone, had a significant impact on tumor cellularity.
We then assessed tumor cell apoptosis by staining tumor sections for DNA fragmentation (TUNEL). Tumors from vehicle-treated mice showed a low number of TUNEL positive nuclei in areas of viable tissue, while nuclear and stromal stain was observed in areas of necrosis (Figure 2C). Despite the decrease in microvessel density and the increase in intratumor hypoxia, bevacizumab alone did not significantly increase the number of apoptotic cells and showed a pattern of TUNEL stained areas similar to tumors from vehicle treated animals. On the contrary, the combination of TPT with bevacizumab showed a 70% increase in TUNEL positive nuclei (scattered throughout the viable tumor tissue) compared to vehicle treated mice and a 38% increase compared to the levels observed in mice treated with TPT alone (p= 0.0027).
These results suggested that induction of apoptosis by the combination of TPT and bevacizumab, compared to TPT alone, might contribute, at least in part, to the inhibition of tumor growth.
We then tested whether inhibition of tumor cell proliferation might also contribute to the anti-tumor activity of bevacizumab in combination with TPT. As shown in Figure 2D, tumors from vehicle treated mice showed extensive areas of Ki67 positive cells throughout the viable areas of the section, with a proliferation index (Ki67 positive nuclei/total nuclei) of 21.93. Tumors treated with bevacizumab alone showed a 50% decrease in tumor cells proliferation consistent with the decreased tumor growth observed (p<0.0001). On the contrary, TPT alone did not significantly alter tumor cells proliferation compared to vehicle treated mice (proliferation index of 24.98). Interestingly, tumors from mice that received the combination of TPT and bevacizumab showed a 75% decrease in proliferation (proliferation index of 5.44, p<0.0001), with very few cells positive for Ki67. Moreover, the inhibition of proliferation achieved with the combination of bevacizumab and TPT was significantly higher than the decrease achieved with bevacizumab alone, suggesting a synergistic activity of this combination.
In conclusion, these results show that bevacizumab alone decreased microvessel density and proliferation, but did not induce significant apoptosis. In contrast, addition of TPT to bevacizumab markedly decreased tumor cells proliferation and increased cell death, consistent with the increased anti-tumor activity observed.
Bevacizumab and TPT differentially affect HIF-1-dependent gene expression in U251-HRE xenografts
Data presented to this point suggest that bevacizumab increased hypoxia in the tumor microenvironment, which did not appear to be associated with cell death. Since HIF-1α is a key mediator of adaptive responses to hypoxia we tested whether bevacizumab affected the levels of HIF-1α and/or its transcriptional activity. As U251 cells do not express detectable levels of HIF-2α protein in vitro (data not shown), we limited our studies to the HIF-1α subunit. As shown in Figure 3A, western blot analysis of tumor lysates collected at the end of treatment showed heterogeneous levels of HIF-1α expression in vehicle treated mice (high levels of HIF-1α expression in 2 mice out of 4 examined). Tumors from mice treated with bevacizumab alone showed high levels of HIF-1α in all samples examined, suggesting that HIF-1α levels might mirror the increased intratumor hypoxia observed with bevacizumab. As expected, TPT either alone or in combination with bevacizumab decreased HIF-1α protein accumulation.
Figure 3. HIF-1α protein accumulation in tumor tissues.
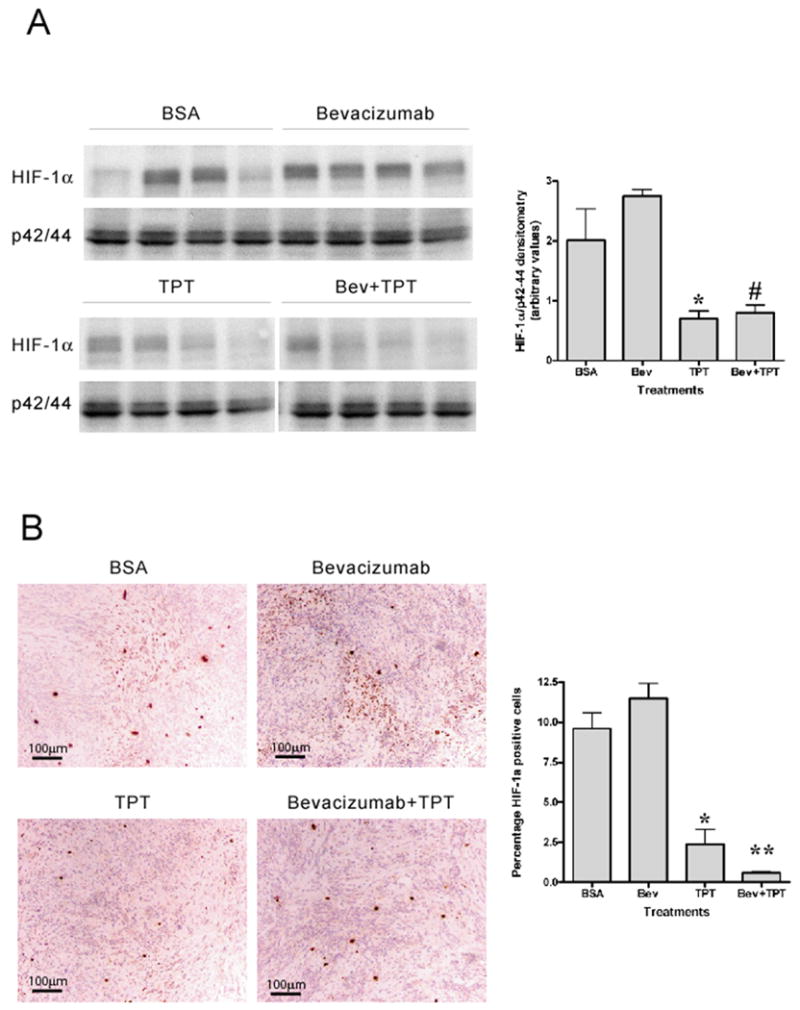
A. Analysis of HIF-1α protein expression by western blotting in tumor lysates harvested on day 10 of treatment from four mice per treatment group. Total p42/44 was used as loading control. Graph represents densitometry analysis of HIF-1α levels normalized by total p42/44 amount (mean ± SE, 4 mice/group, * p=0.0286, between control mice and mice treated with TPT; # p= 0.0286, between mice treated with bevacizumab alone and the combination). B. HIF-1α staining (10X) of representative regions of tumor sections from different treatment groups. Graph represents percentage of HIF-1α positive cells (mean ± SE, 5 mice/group, * p=0.0159, and ** p= 0.0079, relative to control mice).
Similar results were obtained by immunohistochemistry analysis of HIF-1α nuclear staining in tumor sections. As previously shown, U251-HRE xenografts exhibited a zonal pattern of HIF-1α positive cells (Figure 3B), consistent with the presence of intratumor hypoxia. Bevacizumab slightly increased the number of HIF-1α positive cells in tumor sections, while TPT either alone (p=0.0159) or in combination with bevacizumab (p=0.0079) significantly decreased HIF-1α positive cells, compared to vehicle treated mice.
Direct assessment of HIF-1α levels in tumor tissues is informative but might be affected by manipulation and handling of tissues. We therefore assessed HIF-1 transcriptional activity by measuring the mRNA levels of a panel of HIF-1-dependent genes (VEGF, CXCR4, LOX, CA9, GLUT3, PDK1, HK2 and PFKFB3) in tumor lysates. All the genes examined were expressed at higher levels in mice treated with bevacizumab alone compared to vehicle treated mice (Figure 4A). Interestingly, a slight increase was observed at day 5 of treatment (data not shown), while significantly higher levels of expression of HIF-1 target genes were detected at day 10 (Figure 4A), suggesting a time dependent effect. The extent of induction ranged from 2 (PFKFB3) to 4.2 (VEGF) fold and was statistically significant for all genes examined but Glut3. TPT alone significantly inhibited the expression of VEGF and CA9, only marginally affected the levels of GLUT3, PDK1, HK2 and PFKFB3, and did not inhibit CXCR4 and LOX, relative to control animals (Figure 4B), suggesting that HIF-1 independent mechanism might also contribute to the expression of these genes under these conditions. In contrast, TPT, in combination with bevacizumab, decreased the expression of all the genes tested compared to the levels observed in bevacizumab treated mice (Figure 4C). The extent of inhibition ranged from 58% (LOX) to 99% (CA9), and was statistically significant for all genes examined but LOX, strongly suggesting that, under these conditions, inhibition of HIF-1 by TPT profoundly affect hypoxia-induced gene expression.
Figure 4. Bevacizumab increases expression of HIF-1 target genes.
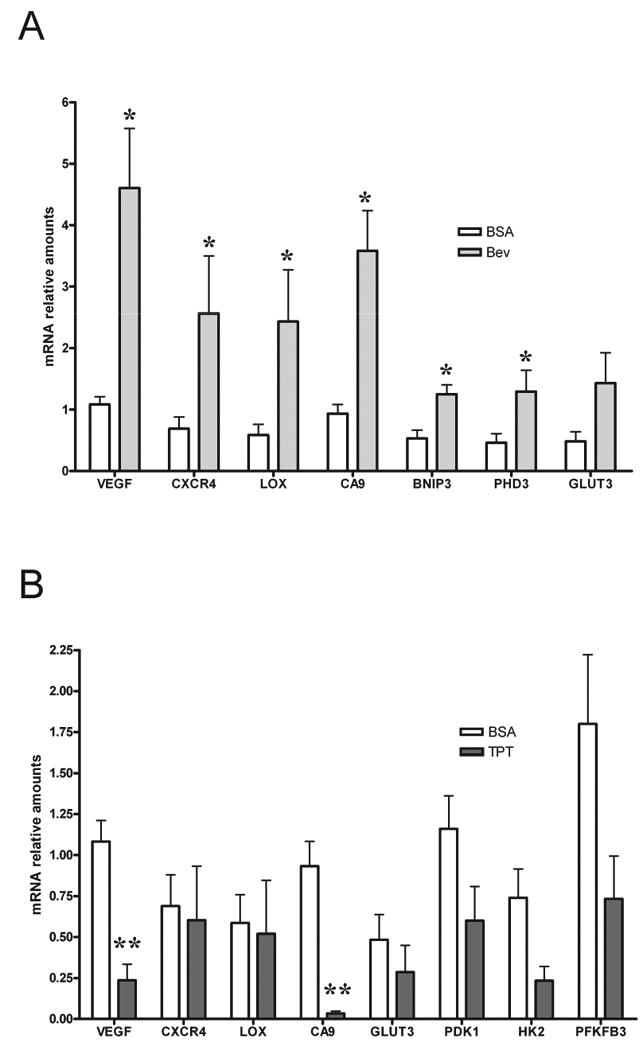
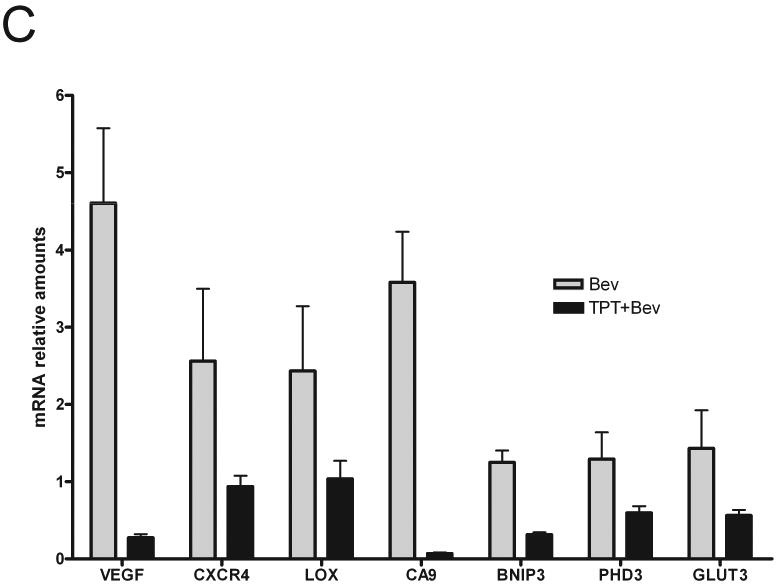
A. Expression of 8 HIF-1 dependent genes in tumor lysates from mice treated with vehicle or bevacizumab measured by Real time PCR. Results are presented as mean ± SE (5 mice/group), * represent a statistical difference between control and treated mice (p<0.05). B. Expression of 8 HIF-1 dependent genes in tumor lysates from mice treated with vehicle or TPT measured by Real time PCR. Results are presented as mean ± SE (5 mice/group), ** represent a statistical difference between control and treated mice (p<0.01). C. Expression of 8 HIF-1 dependent genes in tumor lysates from mice treated with bevacizumab alone or in combination with TPT. Results are presented as mean ± SE (5 mice/group), ** p<0.01, * p<0.05.
In conclusion, these results demonstrate that bevacizumab increases accumulation of transcriptionally active HIF-1α and that addition of TPT abrogates the expression and the function of HIF-1 in the tumor microenvironment.
TPT, alone or in combination with bevacizumab, induces comparable levels of DNA damage in U251-HRE xenografts
TPT is a Topoisomerase I poison known to induce DNA damage (25). To rule out the possibility that the increased inhibition of tumor growth exerted by the combination of TPT plus bevacizumab was due to a better delivery of TPT to the tumor tissue, with a consequent increase in DNA damage and cell death, we measured levels of histone H2AX phosphorylation (γH2AX), a validated biomarker of DNA damage (26), in tumor tissues collected on day 5 or 10 of treatment. Occasional γH2AX positive nuclei were detected in sections from mice treated with vehicle or bevacizumab on day 5 and day 10 (Figure 5A-B). TPT induced, as expected, significantly higher levels of γH2AX positive staining both at day 5 (p=0.0357) and day 10 (p<0.0001), relative to vehicle or bevacizumab-treated mice. Importantly, addition of bevacizumab to TPT did not affect the levels of γH2AX induced by TPT on day 5 (Figure 5A) and actually decreased the number of γH2AX positive nuclei by 40% on day 10 compared to TPT alone (p=0.007) (Figure 5B), suggesting that bevacizumab does not enhance DNA damage induced by TPT and strongly arguing against the possibility that the increased anti-tumor activity might be due to increased cytotoxicity of TPT. Consistent with these results, similar levels of γH2AX, measured by western blot analysis on tumor lysates obtained on day 10, were observed in mice treated with TPT alone or in combination with bevacizumab (data not shown).
Figure 5. Induction of γH2AX by TPT is not affected by addition of bevacizumab.
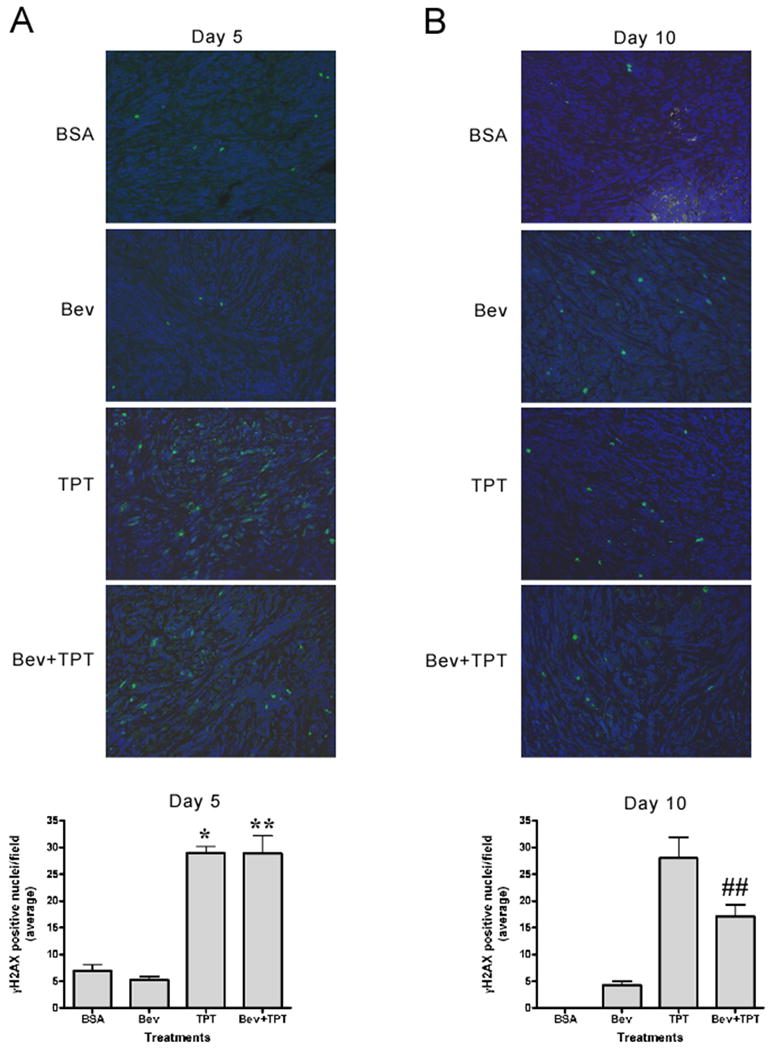
A-B. γH2AX staining (200X) of representative regions of tumor sections from different treatment groups at day 5 (A) and day 10. (B). Graphs represent total γH2AX positive cells from tumors at day 5 (left) and 10 (right) normalized by section area. Tissues from vehicle treated mice on day 10 (panel B) showed a large number of degenerated cells that precluded an accurate count of positive nuclei for this group. Statistical analysis for day 10 samples was done using the bevacizumab samples as reference. (mean ± SE, 5 mice/group, *** p<0.001 compared to bevacizumab, ## p<0.01 compared to TPT).
In conclusion, these results are consistent with the conclusion that the combination of TPT and bevacizumab does not increase the levels of DNA damage induced by TPT alone, and they suggest that abrogation of HIF-1-dependent responses, rather than increased cytotoxic activity, may be responsible for the increased antitumor effects of bevacizumab in combination with TPT.
Discussion
The pharmacological validation of HIF-1 as a target has been hampered by the lack of selective inhibitors and reliable biomarkers predictive of therapeutic activity. Indeed, the potential outcome of HIF-1 inhibition in cancer therapy remains poorly understood, both in preclinical models and in the clinical setting. Genetic approaches targeting HIF-1α have overwhelmingly demonstrated that HIF-1 inhibition translates into impaired tumor growth (27, 28). However, genetic approaches suffer from intrinsic limitations, including but not limited to abrogation of HIF-1 activity before tumor implantation and inhibition which is only limited to tumor cells, thus missing a potential effect on the tumor microenvironment. Approaches using inducible expression of shRNA targeting HIF-1α at different stages of tumor progression have indeed demonstrated that early, but not late, inhibition of HIF-1α may affect tumor growth (29). These somewhat conflicting results may be reconciled in light of the heterogeneous expression of HIF-1α in solid tumors, which may result in an unpredictable contribution to the biological behavior of cancer cells. Indeed, unlike human cancers in which genetic alterations, e.g. VHL loss of function, drive normoxic expression of HIF-α subunits, the majority of solid tumors express HIF-1α in an oxygen dependent fashion. It is then plausible that HIF-1 inhibitors may have a limited clinical activity when used as single agents and that they might be more effective in situations in which survival of the majority of tumor cells depends on HIF-1. Hence, combination therapies may be required to fully exploit the therapeutic potential of HIF-1 inhibition.
We hypothesized that a hypoxic stressed tumor microenvironment, which might be generated by administration of antiangiogenic agents, would be an ideal scenario in which HIF-1 inhibition might be associated with increased therapeutic effects. However, the physiological and molecular consequences of anti-angiogenic therapies still remain poorly understood. On the one hand, anti-angiogenic therapies may lead to vascular regression, which would increase intratumor hypoxia (18). On the other hand, inhibition of VEGF signaling may be associated with a transient “normalization window”, which would improve tumor oxygenation and delivery of chemotherapy (19). Preclinical models have yielded controversial results showing vasculature normalization in some models (30) and an increase in intratumor hypoxia in others (31).
One of the objectives of this study was to test whether bevacizumab, an approved antiangiogenic agent, increased intratumor hypoxia and induced HIF-1-transcriptional activity. If so, we hypothesized, addition of a HIF-1 inhibitor might provide a significant therapeutic benefit. Despite the known limitations of using bevacizumab in mouse models, i.e., bevacizumab only neutralizes human VEGF, we found that bevacizumab (5mg/kg, q3d×4) decreased tumor growth by 40% in U251 xenografts, a result in agreement with other reports showing activity of bevacizumab in tumor xenografts (32). To better understand the effects of bevacizumab on the tumor microenvironment we investigated a number of biomarkers, including HRE-dependent bioluminescence, intratumor hypoxia, microvessel density, HIF-1α protein levels and expression of HIF-1-target genes, which reflect changes in tumor vasculature and oxygenation and that might be predictive of biological responses to bevacizumab. Expression of luciferase in U251-HRE cells reflects HIF-1 transcriptional activity (23) under conditions in which oxygen, a required cofactor for luciferase activity, is not limiting (24). In this study we found that bioluminescence was decreased by bevacizumab. Albeit luciferase expression is affected by tumor size, the extent of intratumor hypoxia, measured by pimonidazole, was increased, suggesting that an increase in intratumor hypoxia might limit oxygen availability and further decrease luciferase activity. Indeed, treatment with bevacizumab resulted in decreased microvessel density and increased HIF-1α protein levels and transcriptional activity. Interestingly, bevacizumab also caused a 50% decrease in tumor cell proliferation (measured by Ki67 labeling) possibly associated with the increase in intratumor hypoxia and G1 arrest of cancer cells (33). It is then conceivable that the inhibition of tumor growth observed in mice treated with bevacizumab alone might be due primarily to a decrease in cell proliferation. Interestingly, bevacizumab did not induce significant levels of apoptosis, possibly because of the activation of HIF-1-dependent compensatory responses, which might also explain, at least in part, the overall lack of single agent activity of anti-VEGF therapies.
The effects of bevacizumab on the tumor microenvironment suggest that combination with a HIF-1 inhibitor might be a rational therapeutic strategy. However, the lack of specific small molecule inhibitors of HIF-1 raises the legitimate question as to the best approach to down-regulate HIF-1α in tumor tissue. We have previously shown that TPT, a topoisomerase I poison that causes DNA damage, inhibited HIF-1α protein accumulation and angiogenesis in U251-HRE xenografts (20, 22, 23). Notably, we also demonstrated that inhibition of HIF-1α protein was independent of DNA damage, suggesting that different schedules of administration of TPT may be used to maximize DNA damage or HIF-1 inhibition, respectively. In this study, a lower daily dose of TPT (0.5 mg/kg), compared with previous studies (23), was able to inhibit HIF-1α protein accumulation, but only marginally decreased the majority of HIF-1-dependent genes analyzed and tumor growth, but slightly increased vessel density, suggesting that under these conditions HIF-1 independent pathways may also be relevant for the induction of angiogenesis and that inhibition of HIF-1 alone was not sufficient to generate a meaningful therapeutic response. In a previous study (23) TPT, used at 1mg/kg., inhibited HIF-1a expression and MVD. In this study TPT was used at a lower dose (0.5mg/kg), which achieved partial inhibition of HIF-1 transcriptional activity, (Figure 4B), but did not decrease MVD. Higher doses of topotecan might have a direct effect on endothelial cells (34). Conversely, this data suggest that HIF-1 inhibition alone does not necessarily correlate with decrease of MVD (35) due to HIF-1-independent pathways that affect endothelial cell survival. These findings emphasize that the context in which HIF-1 inhibition occurs is a critical parameter that may determines the therapeutic outcome. Indeed, TPT induced γH2AX but did not affect tumor cells proliferation, suggesting that inhibition of tumor growth caused by TPT alone was mainly due to its cytotoxic effects.
What is the underlying mechanism of the interaction between bevacizumab and TPT? At least two possibilities, which are not mutually exclusive, should be considered. The first is that TPT in combination with bevacizumab exerted more pronounced cytotoxic effects. It has been recently shown that bevacizumab induced vascular normalization and improved tumor perfusion in a neuroblastoma xenograft model and that addition of a single dose of TPT significantly decreased tumor growth (30). However, this approach was aimed at emphasizing the ability of TPT to induce DNA damage and cytotoxicity, effects minimized under our experimental conditions by the low dose of TPT and protracted schedule of administration. Indeed, we found that bevacizumab did not increase the ability of TPT to induce γH2AX, a hallmark of TPT-induced DNA damage (26) and a potential biomarker to detect DNA damage in patients (36). Actually, addition of bevacizumab slightly decreased the levels of γH2AX at the end of treatment (day 10), relative to TPT alone, possibly due to inhibition of tumor cells proliferation caused by bevacizumab, arguing against increased cytotoxic effects of TPT when combined with bevacizumab.
The alternative possibility to explain the increased inhibition of tumor growth exerted by the combination of bevacizumab and TPT is that inhibition of HIF-1 activity in a hypoxic stressed tumor microenvironment was sufficient to both thwart proliferation and induce cell death. Indeed, we observed that addition of TPT caused a significant decrease in the expression of HIF-1-dependent genes, which were induced by bevacizumab alone. A number of HIF-1 inducible genes have been implicated in the switch to glycolitic metabolism and inhibition of mitochondrial respiration (37), and their inhibition might have contributed to inhibition of proliferation and induction of cell death. Notably, we also observed a marked decrease in tumor cellularity and an increase matrix deposition, but only a mild induction of apoptosis (measured by TUNEL) which, in light of the antitumor activity observed, suggest that induction of apoptosis or cell death might have occurred early during treatment with the combination.
The combination of bevacizumab and TPT also markedly reduced angiogenesis relative to either agent alone. This finding may result from either a converging inhibition of these two agents on the VEGF pathway or from the inhibition of VEGF-independent angiogenic pathways controlled by HIF-1 transcriptional activity. A further implication of this possibility is the inhibition of circulating endothelial progenitor cells (CEPs) recruitment to the tumor site, which is dependent upon the production of angiogenic factors, including but not limited to VEGF, by the hypoxic tumor (38).
Combination of HIF-1 inhibitors with other therapeutic strategies has been recently explored. Direct cellular effects of HIF-1 inhibitors, e.g., sensitization to chemotherapeutic agents (39), or indirect effects on tumor metabolism, e.g., increased oxygen consumption (40), have been implicated as potential mechanisms for improving therapeutic efficacy. Here we propose that the combination with antiangiogenic agents may synergize by inducing a hypoxic stress in the tumor microenvironment. However, it is conceivable that anti-angiogenic therapies may yield heterogeneous responses in different tumor types, which may not always result in increased tumor hypoxia and HIF-1 activation. Identification of biomarkers predictive of therapeutic outcome is then pivotal to select patients that might respond to this combination. Hypoxic tumors or tumors that depend on VEGF for sustained angiogenesis might be more sensitive to the combination of anti-VEGF and HIF-1 inhibitors. It will be essential to further investigate this therapeutic strategy in early clinical trials, not only by assessing therapeutic activity but also by developing biomarkers that validate its mechanism of action. This study provides both a rationale for further clinical development of this combination and preliminary evidence of potential biomarkers that can be used in early clinical trials to validate the activity, or lack thereof, of this therapeutic strategy.
Supplementary Material
H&E staining (10X and 40X) of regions of representative tumor sections from different treatment groups. “m” indicates acellular eosinophilic matrix, arrown indicate areas of apoptosis/necrosis (nuclear pyknosis or karyolysis and disrupted or vacuolated cytoplasm), “V” indicates viable tumor cells.
Acknowledgments
The authors would like to thank Keith Rogers and Scott M. Lawrence for help with the CD31, Pimonidazole, Ki67 and TUNEL staining. This project has been funded in whole or in part with Federal funds from the National Cancer Institute, National Institutes of Health, under Contract No. N01-CO-12400. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government. This research was supported [in part] by the Developmental Therapeutics Program, DCTD, of the National Cancer Institute, NIH. NCI-Frederick is accredited by AAALACi and follows the Public Health Service Policy on the Care and Use of Laboratory Animals. Animal care was provided in accordance with the procedures outlined in the Guide for Care and Use of Laboratory Animals (NIH publication No. 86-23, 1985).
Reference List
- 1.Brown JM, Wilson WR. Exploiting tumour hypoxia in cancer treatment. Nat Rev Cancer. 2004;4:437–47. doi: 10.1038/nrc1367. [DOI] [PubMed] [Google Scholar]
- 2.Harris AL. Hypoxia--a key regulatory factor in tumour growth. Nat Rev Cancer. 2002;2:38–47. doi: 10.1038/nrc704. [DOI] [PubMed] [Google Scholar]
- 3.Hockel M, Vaupel P. Tumor hypoxia: definitions and current clinical, biologic, and molecular aspects. J Natl Cancer Inst. 2001;93:266–76. doi: 10.1093/jnci/93.4.266. [DOI] [PubMed] [Google Scholar]
- 4.Semenza GL. Hypoxia and cancer. Cancer Metastasis Rev. 2007;26:223–4. doi: 10.1007/s10555-007-9058-y. [DOI] [PubMed] [Google Scholar]
- 5.Koukourakis MI, Bentzen SM, Giatromanolaki A, et al. Endogenous markers of two separate hypoxia response pathways (hypoxia inducible factor 2 alpha and carbonic anhydrase 9) are associated with radiotherapy failure in head and neck cancer patients recruited in the CHART randomized trial. J Clin Oncol. 2006;24:727–35. doi: 10.1200/JCO.2005.02.7474. [DOI] [PubMed] [Google Scholar]
- 6.Aebersold DM, Burri P, Beer KT, et al. Expression of hypoxia-inducible factor-1alpha: a novel predictive and prognostic parameter in the radiotherapy of oropharyngeal cancer. Cancer Res. 2001;61:2911–6. [PubMed] [Google Scholar]
- 7.Birner P, Schindl M, Obermair A, et al. Overexpression of hypoxia-inducible factor 1alpha is a marker for an unfavorable prognosis in early-stage invasive cervical cancer. Cancer Res. 2000;60:4693–6. [PubMed] [Google Scholar]
- 8.Birner P, Schindl M, Obermair A, Breitenecker G, Oberhuber G. Expression of hypoxia-inducible factor 1alpha in epithelial ovarian tumors: its impact on prognosis and on response to chemotherapy. Clin Cancer Res. 2001;7:1661–8. [PubMed] [Google Scholar]
- 9.Bos R, van der GP, Greijer AE, et al. Levels of hypoxia-inducible factor-1alpha independently predict prognosis in patients with lymph node negative breast carcinoma. Cancer. 2003;97:1573–81. doi: 10.1002/cncr.11246. [DOI] [PubMed] [Google Scholar]
- 10.Melillo G. Targeting hypoxia cell signaling for cancer therapy. Cancer Metastasis Rev. 2007;26:341–52. doi: 10.1007/s10555-007-9059-x. [DOI] [PubMed] [Google Scholar]
- 11.Powis G, Kirkpatrick L. Hypoxia inducible factor-1alpha as a cancer drug target. Mol Cancer Ther. 2004;3:647–54. [PubMed] [Google Scholar]
- 12.Ferrara N, Hillan KJ, Novotny W. Bevacizumab (Avastin), a humanized anti-VEGF monoclonal antibody for cancer therapy. Biochem Biophys Res Commun. 2005;333:328–35. doi: 10.1016/j.bbrc.2005.05.132. [DOI] [PubMed] [Google Scholar]
- 13.Hurwitz H, Fehrenbacher L, Novotny W, et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med. 2004;350:2335–42. doi: 10.1056/NEJMoa032691. [DOI] [PubMed] [Google Scholar]
- 14.Sandler A, Gray R, Perry MC, et al. Paclitaxel-carboplatin alone or with bevacizumab for non-small-cell lung cancer. N Engl J Med. 2006;355:2542–50. doi: 10.1056/NEJMoa061884. [DOI] [PubMed] [Google Scholar]
- 15.Miller K, Wang M, Gralow J, et al. Paclitaxel plus bevacizumab versus paclitaxel alone for metastatic breast cancer. N Engl J Med. 2007;357:2666–76. doi: 10.1056/NEJMoa072113. [DOI] [PubMed] [Google Scholar]
- 16.Bottaro DP, Liotta LA. Cancer: Out of air is not out of action. Nature. 2003;423:593–5. doi: 10.1038/423593a. [DOI] [PubMed] [Google Scholar]
- 17.Blagosklonny MV. Antiangiogenic therapy and tumor progression. Cancer Cell. 2004;5:13–7. doi: 10.1016/s1535-6108(03)00336-2. [DOI] [PubMed] [Google Scholar]
- 18.Kerbel R, Folkman J. Clinical translation of angiogenesis inhibitors. Nat Rev Cancer. 2002;2:727–39. doi: 10.1038/nrc905. [DOI] [PubMed] [Google Scholar]
- 19.Jain RK. Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science. 2005;307:58–62. doi: 10.1126/science.1104819. [DOI] [PubMed] [Google Scholar]
- 20.Rapisarda A, Uranchimeg B, Sordet O, et al. Topoisomerase I-mediated inhibition of hypoxia-inducible factor 1: mechanism and therapeutic implications. Cancer Res. 2004;64:1475–82. doi: 10.1158/0008-5472.can-03-3139. [DOI] [PubMed] [Google Scholar]
- 21.Rapisarda A, Shoemaker RH, Melillo G. Targeting topoisomerase I to inhibit hypoxia inducible factor 1. Cell Cycle. 2004;3:172–5. [PubMed] [Google Scholar]
- 22.Rapisarda A, Uranchimeg B, Scudiero DA, et al. Identification of small molecule inhibitors of hypoxia-inducible factor 1 transcriptional activation pathway. Cancer Res. 2002;62:4316–24. [PubMed] [Google Scholar]
- 23.Rapisarda A, Zalek J, Hollingshead M, et al. Schedule-dependent inhibition of hypoxia-inducible factor-1alpha protein accumulation, angiogenesis, and tumor growth by topotecan in U251-HRE glioblastoma xenografts. Cancer Res. 2004;64:6845–8. doi: 10.1158/0008-5472.CAN-04-2116. [DOI] [PubMed] [Google Scholar]
- 24.Cecic I, Chan DA, Sutphin PD, et al. Oxygen sensitivity of reporter genes: implications for preclinical imaging of tumor hypoxia. Mol Imaging. 2007;6:219–28. [PubMed] [Google Scholar]
- 25.Pommier Y. Topoisomerase I inhibitors: camptothecins and beyond. Nat Rev Cancer. 2006;6:789–802. doi: 10.1038/nrc1977. [DOI] [PubMed] [Google Scholar]
- 26.Furuta T, Takemura H, Liao ZY, et al. Phosphorylation of histone H2AX and activation of Mre11, Rad50, and Nbs1 in response to replication-dependent DNA double-strand breaks induced by mammalian DNA topoisomerase I cleavage complexes. J Biol Chem. 2003;278:20303–12. doi: 10.1074/jbc.M300198200. [DOI] [PubMed] [Google Scholar]
- 27.Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer. 2003;3:721–32. doi: 10.1038/nrc1187. [DOI] [PubMed] [Google Scholar]
- 28.Melillo G. Inhibiting hypoxia-inducible factor 1 for cancer therapy. Mol Cancer Res. 2006;4:601–5. doi: 10.1158/1541-7786.MCR-06-0235. [DOI] [PubMed] [Google Scholar]
- 29.Li L, Lin X, Staver M, et al. Evaluating hypoxia-inducible factor-1alpha as a cancer therapeutic target via inducible RNA interference in vivo. Cancer Res. 2005;65:7249–58. doi: 10.1158/0008-5472.CAN-04-4426. [DOI] [PubMed] [Google Scholar]
- 30.Dickson PV, Hamner JB, Sims TL, et al. Bevacizumab-induced transient remodeling of the vasculature in neuroblastoma xenografts results in improved delivery and efficacy of systemically administered chemotherapy. Clin Cancer Res. 2007;13:3942–50. doi: 10.1158/1078-0432.CCR-07-0278. [DOI] [PubMed] [Google Scholar]
- 31.Franco M, Man S, Chen L, et al. Targeted anti-vascular endothelial growth factor receptor-2 therapy leads to short-term and long-term impairment of vascular function and increase in tumor hypoxia. Cancer Res. 2006;66:3639–48. doi: 10.1158/0008-5472.CAN-05-3295. [DOI] [PubMed] [Google Scholar]
- 32.Ferrara N. VEGF as a therapeutic target in cancer. Oncology. 2005;69 3:11–6. doi: 10.1159/000088479. [DOI] [PubMed] [Google Scholar]
- 33.Brown JM, Giaccia AJ. The unique physiology of solid tumors: opportunities (and problems) for cancer therapy. Cancer Res. 1998;58:1408–16. [PubMed] [Google Scholar]
- 34.Petrangolini G, Pratesi G, De CM, et al. Antiangiogenic effects of the novel camptothecin ST1481 (gimatecan) in human tumor xenografts. Mol Cancer Res. 2003;1:863–70. [PubMed] [Google Scholar]
- 35.Ryan HE, Poloni M, McNulty W, et al. Hypoxia-inducible factor-1alpha is a positive factor in solid tumor growth. Cancer Res. 2000;60:4010–5. [PubMed] [Google Scholar]
- 36.Rao VA, Agama K, Holbeck S, Pommier Y. Batracylin (NSC 320846), a dual inhibitor of DNA topoisomerases I and II induces histone gamma-H2AX as a biomarker of DNA damage. Cancer Res. 2007;67:9971–9. doi: 10.1158/0008-5472.CAN-07-0804. [DOI] [PubMed] [Google Scholar]
- 37.Semenza GL. Oxygen-dependent regulation of mitochondrial respiration by hypoxia-inducible factor 1. Biochem J. 2007;405:1–9. doi: 10.1042/BJ20070389. [DOI] [PubMed] [Google Scholar]
- 38.Shaked Y, Ciarrocchi A, Franco M, et al. Therapy-induced acute recruitment of circulating endothelial progenitor cells to tumors. Science. 2006;313:1785–7. doi: 10.1126/science.1127592. [DOI] [PubMed] [Google Scholar]
- 39.Li L, Lin X, Shoemaker AR, et al. Hypoxia-inducible factor-1 inhibition in combination with temozolomide treatment exhibits robust antitumor efficacy in vivo. Clin Cancer Res. 2006;12:4747–54. doi: 10.1158/1078-0432.CCR-05-2842. [DOI] [PubMed] [Google Scholar]
- 40.Cairns RA, Papandreou I, Sutphin PD, Denko NC. Metabolic targeting of hypoxia and HIF1 in solid tumors can enhance cytotoxic chemotherapy. Proc Natl Acad Sci U S A. 2007;104:9445–50. doi: 10.1073/pnas.0611662104. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
H&E staining (10X and 40X) of regions of representative tumor sections from different treatment groups. “m” indicates acellular eosinophilic matrix, arrown indicate areas of apoptosis/necrosis (nuclear pyknosis or karyolysis and disrupted or vacuolated cytoplasm), “V” indicates viable tumor cells.