

Abstract
An enzymatic assay system was developed to quantify the distribution of recombinant proteins over various cell structures. The system takes advantage of α-complementation of □-galactosidase. The large ω fragment of □–galactosidase is expressed in predefined cell structures with the aid of attached protein localization signals. The resulting reporter cell lines are infected with a second construct expressing a target protein fused with the shorter α fragment of □-galactosidase. The physical proximity of the two recombinant proteins carrying the □-galactosidase fragments results in the reconstitution of an active enzyme, and its activity is measured with a plate reader. The recombinant constructs are based on lentiviral vectors and can be rapidly and efficiently introduced into cells by infection with stocks of lentivirus particles. The efficiency of the system was demonstrated with the FOXO3A transcription factor, which shuttles between the cytoplasm and nucleus in the model colon carcinoma cell line RKO.
Keywords: reporter system, intracellular transport, protein translocation, α-complementation, □-galactosidase, FOXO3A transcription factor
INTRODUCTION
Protein molecules are continuously transferred within the cell as they perform their functions after being synthesized on the ribosome. Protein translocations accompany various processes, such as intracel-lular transport, secretion, and maturation of proteins and protein complexes, and are associated with various regulatory interactions and signal transduction between and within cells. Studies of the character and time course of intracellular protein translocations are essential for a better understanding of the mechanisms of cell processes.
Many methods are available for detecting the translocations of proteins within the cell. One is based on fractionation of sub-cellular components with subsequent identification and quantification of the target protein in the fractions. However, sub-cellular fractionation may be associated with an artificial redistribution of proteins and substantial cross-contamination of sub-cellular fractions. Microscopy is usually employed in detecting the intracellular translocations of proteins in intact cells. The simplest version is based on protein visualization with specific antibodies. Since cell fixation is necessary in this case, a real-time monitoring of protein translocations is unfeasible. A simple method to monitor the time course of protein translocations in the living cell utilizes protein fusions; i.e., the sequence coding for a protein of interest is fused in frame with the gene for a fluorescent protein [1]. Fluorescence resonance energy transfer (FRET) is a variant of this method [2]. Two potentially interacting proteins are labeled with different fluorescent markers and simultaneously expressed in this case. When the proteins are brought together and are less than 20 nm apart, fluorescence of one protein is excited by fluorescence of the other, producing a specific signal [3, 4]. A drawback of this method is that a bulky fluorescent protein involved in the fusion often hinders the functions, including intracellular translocations, of the target protein [5]. When α-complementation of β-galactosidase fragments is employed, the short N-terminal α peptide (56 amino acid residues) is used as a marker and exerts no effect on the target protein function in most cases [6]. A second component involved in complementation is the ω fragment of β-galactosidase. This fragment represents the enzyme with a deletion of residues 11−51 and is active in the presence of the αpeptide, overlapping the deletion [7]. Enzymatic activity is restored when the two fragments are close together, even if contained in different proteins [8, 9]. In fact, the enzyme is active when the two peptides occur in one intracellular compartment. When one of the components (e.g., the ω peptide) is incorporated in a certain cell structure, the appearance of a protein fusion with the α peptide in its vicinity restores β-galactosidase activity. Thus, measuring the enzymatic activity and its time-dependent changes, it is possible to monitor the translocation of the protein fusion into the predetermined cell compartment. When the ω peptide is in an excess, the enzymatic activity is proportional to the amount of the α peptide, allowing quantification of protein translocations. The interaction between the two β-galactosidase fragments is rather weak and exerts almost no effect on the translocations of the two interacting components within the cell [10]. At the same time, the complementation efficiency is extremely high. With such efficiency, it is possible to detect the translocations of proteins between relatively isolated compartments, e.g., between the cytoplasm and nucleus, and rather difficult to detect the translocations within one compartment, e.g., from the plasma membrane to endoplasmic reticulum or from the cytosol into mitochondria. A pair of peptides with lower affinities is more suitable in this case, since the background enzymatic activity, observed in the absence of a direct contact between the peptides, is significantly reduced [10].
Taking advantage of α-complementation, we designed a convenient vector system to study the translocation of recombinant proteins within the cell. Lentiviral vectors were chosen for delivering the constructs into cells. While simple retroviruses efficiently infect only S-phase cells [11-13], lentiviruses are capable of infecting cells in G1 and G2 [14], and the efficiency of infecting a cell population is consequently higher [15]. This is especially important in the case of slowly proliferating cell lines, since the selection time is substantially reduced. Lentiviruses enter the cell nucleus while transcription is active and genome regions are more open; this probably explains the fact that constructs introduced with lentiviral vectors are less prone to subsequent expression silencing, which is characteristic of constructs introduced via DNA transfection [16, 17].
Using a lentiviral vector, we expressed the ω peptide of β-galactosidase in the cell nucleus of several human cell cultures, which can be used as test systems to analyze the redistribution of target proteins between the nucleus and cytoplasm. Such model reporter cell lines are suitable for studying the mechanisms of protein translocation and provide a convenient tool for screening chemical or genetic libraries for modulators of certain cell functions associated with protein translocations.
The potential of our localization reporter system was demonstrated with the FOXO3A transcription factor, which is involved in the signaling pathway triggered by insulin-like growth factor 1 (IGF1) [18] and belongs to the Fork head family [19]. Proteins of the FOXO subfamily are known to shuttle between the nucleus and cytoplasm, and their activity is thereby regulated [20-22]. Entering the cell nucleus, the FOXO proteins interact with specific sites in the target genes and activate their transcription [20, 22-24]. The FOXO proteins have both the nuclear localization and nuclear export signals [21], and their translocations between the nucleus and cytoplasm seems to be an equilibrium process but depends on several accessory proteins. Nuclear import of FOXO is promoted by importins, while its export from the nucleus is regulated by exportin Crm1 [20, 21]. A key role in the balance between the nuclear import and export of the FOXO proteins is played by serine/threonine protein kinase B (PKB, or Akt) [20-22]. The protein kinase function of Akt is activated by its binding with phosphatidyl inositol, which is phosphorylated by phosphatidyl inositol 3-kinase (PI3K). Activated Akt phosphorylates Thr32 in nuclear FOXO3A [20-24]; as a result, the FOXO-Crm1 complex interacts with the 14−3−3 protein, the nuclear localization signal is suppressed, and the FOXO protein is predominantly redistributed into the cytoplasm [25, 26]. Affecting these regulatory processes, it is possible to change the intracellular localization of FOXO3A, which provides a convenient model for studying the efficiency of our localization reporter system.
EXPERIMENTAL
Lentiviral constructs
Our genetic constructs were based on the lentiviral vector pLCMV, which was supplemented with expression cassettes containing bleo (phleomycin resistance) and neo (G418 resistance). The ωfragment of β-galactosidase was obtained by deleting the region coding for amino acid residues 11−51. For this purpose, PCR was carried out with a plasmid containing the full-length β-galactosidase cDNA and oligonucleotide primers containing the XbaI and BamHI sites. The N-terminal primer was complementary to the region coding for the first 11 residues and residues 51−58. The C-terminal primer covered the stop codon. To obtain the constructs expressing the ω-fragment in certain cell structures, we used the mitochondrial localization signal corresponding to the 29 N-terminal residues of human cytochrome c oxygenase subunit VIII [27], the endoplasmic reticulum localization signal corresponding to the 20 N-terminal residues of calreticulin [28], the plasma membrane localization signal corresponding to the 20 N-terminal residues of neuromodulin [29], and the nuclear localization signal located at the C end and corresponding to four copies of the nuclear localization signal of the SV40 T antigen [30]. To obtain the constructs expressing the α peptide of β-galactosidase, the peptide was placed at the C end of the fusion upstream of the c-Myc antigenic epitope. Some constructs had another order of the elements: the c-Myc epitope was at the N end and was followed by the α peptide and target protein. The above signals were used to direct the fusion proteins to particular compartments.
Lentivirus virions, infection, and selection of target cells
Lentiviral constructs were introduced in 293T cells along with packaging plasmids as described previously [31]. Infected cells were transferred into a selective medium containing 500 μg/ml G418 (14 days) or 25 μg/ml phleomycin (20 days) 48 h after infection. Reporter lines were cloned in 96-well plates by limiting dilution.
β-Galactosidase activity
was measured with a fluorescent substrate, using a GalScreen kit (Perkin-Elmer).
Western blot analysis
of recombinant FOXO3A was carried out according to a standard protocol, using antibody 9E10 against the c-Myc epitope (Abcam) and a ECL-Plus development kit (Amersham).
RESULTS AND DISCUSSION
Lentiviral Reporter Vector Constructs
Our reporter system for studying the intracellular localization of proteins includes constructs of two types.
One construct is introduced in any model cell culture to obtain the standard reporter cell line. The construct serves to continuously produce the ω-fragment of β-galactosidase and to ensure its incorporation in certain cell structures owing to a specific intracellular localization signal. The construct additionally expresses bleo, conferring phleomycin resistance on infected cells and allowing their selection with phleomycin. We obtained several such constructs, directing the protein to the plasma membrane, mitochondria, membranes of the endoplasmic reticulum, or the nucleus. A scheme of pLCMV-ω-lacZ-nuc-bleo, ensuring the nuclear localization of the mutant β-galactosidase, is shown in Fig. 1a. A construct expressing the ωfragment is introduced in cultured cells to obtain a stable cell line expressing the inactive pro-enzyme in a certain cell structure.
Fig. 1.
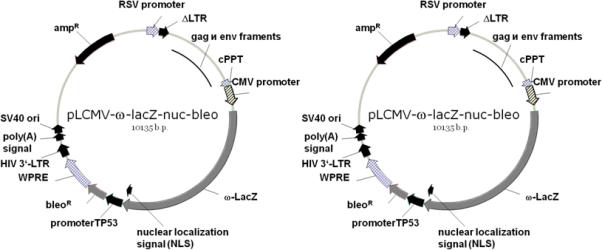
Schemes of lentiviral reporter constructs. (a) Lentiviral vector with the insert coding for the β-galactosidase ω fragment fused with the nuclear localization signal of the SV40 T antigen. The second expression cassette directs the bleo expression to confer phleomycin resistance. (b) Lentiviral vector coding for FOXO3A whose C end is fused with the β-galactosidase α fragment and the c-Myc antigenic epitope. The second expression cassette directs the neo expression to confer resistance to G418.
Constructs of the second type determine the production of the target protein fused with the short α fragment of β-galactosidase. The fusion protein additionally contains the c-Myc antigenic epitope (10 amino acid residues). The epitope makes it possible to quantify and localize the fusion protein by standard immunological methods, which is especially convenient when specific antibodies to the target protein are unavailable. The constructs express neo, conferring G418 resistance on infected cells. Figure 1b shows a scheme of the pLCMV-PMN-based construct that ensures the production of FOXO3A with the α-fragment and the c-Myc antigenic epitope at the C end. Since the activity of some proteins may be altered by changes in their C-terminal regions, we constructed the pLCMV-MPN vector, which is designed to produce proteins N fused with the antigenic epitope and the α-fragment.
As a control, we used several second-type constructs with the α-peptide of β-galactosidase fused with the heterologous enhanced green fluorescent protein (EGFP). EGFPs expressed from these constructs lacked an intracellular localization signal or were fused with signal peptides directing the fusion protein to various cell structures. EGFP is convenient, allowing visual analysis of its localization.
Generation of Stable Reporter Cell Lines to Study the Localization of Proteins
When a recombinant construct is introduced, transgene expression substantially varies among cells, depending on the transgene integration site, the regulatory elements occurring in its vicinity, and the epigenetic status of the total host region. To obtain an optimal reporter cell line, we selected individual cell clones producing the ω–peptide of β-galactosidase in certain structures. As an example, selection of a reporter cell line designed to study the nuclear localization of proteins is described below. Individual clones of RKO cells carrying pLCMV-ω-lacZ-nuc-bleo were infected with pLCMV-EGFP-PMN, pLCMV-EGFP-nuc-PMN, or pLCMV-EGFP-mem-PMN, which produced the EGFP-α peptide fusion without a localization signal or with a signal directing the fusion to the nucleus or plasma membrane. Cells were selected in the presence of G418, and the EGFP level was estimated by fluorescence microscopy (Fig. 2a). Clones expressing EGFP to approximately the same levels were selected and tested for α-galactosidase activity (Fig. 2b). An optimal clone was selected by the maximal difference in α-galactosidase activity between the cultures producing the nuclear and membrane ω fragments. Another important factor is the system sensitivity, which depends on the level of β-galactosidase activity. Hence, 24 primary clones were used to select two clones, NZ1 and NZ11, which displayed the highest enzymatic activity after the introduction of pLCMV-EGFP. Then, clone NZ1 was selected as displaying the highest difference in enzymatic activity after the introduction of the nuclear and membrane proteins. The high background activity in NZ11 cells could be explained by the presence of a fraction of the ωfragment outside the nucleus or the relatively high proliferative activity of cells, since nuclear and cytoplasmic proteins are mixed during meiosis.
Fig. 2.
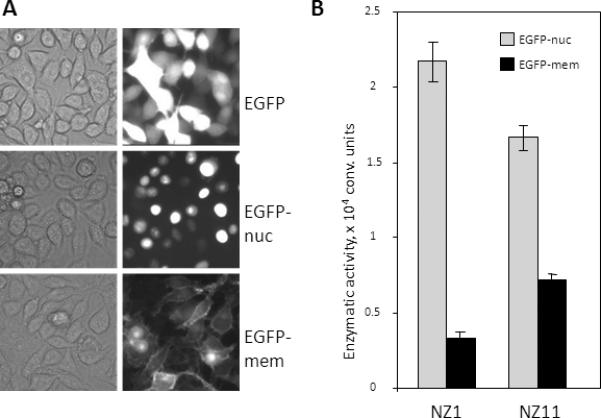
Selection of the optimal reporter RKO cell clone producing the ω fragment of β-galactosidase with the nuclear localization signal. (a) Visual analysis of the EGFP distribution in RKO cells producing EGFP without any localization signal (top), EGFP with the nuclear localization signal (center), or EGFP with the plasma membrane localization signal (bottom). (b) Comparative specificity of two RKO clones carrying pLCMV-ω-lacZ-bleo. Clones NZ1 and NZ11 were selected by maximal enzymatic activity after the introduction of the lentiviral construct expressing the α peptide fused with EGFP. In addition, the constructs expressing the α peptide fused with EGFP and the nuclear or membrane localization signal were introduced in the clones. After selection in the presence of G418, cell cultures were tested for β-galactosidase activity. Cell lysates were incubated with a fluorescent substrate, and fluorescence was measured with a plate reader.
A similar procedure was used to obtain the optimal reporter clones that produced the ω fragment fused with the mitochondrial, plasma membrane, or endoplasmic reticulum localization signal. The results indicate that the reporter system for proteins localized in closed cell structures, such as the nucleus and mitochondria, have a far greater dynamic range. The most plausible explanation is that the two components of the reporter complex are more likely to come close together in open structures even when the protein fused with the α peptide has another localization signal. However, even when the ω peptide was introduced in open structures with proper constructs, we observed a significant difference in activation of the reporter protein in both positive (the α peptide had the same localization) and negative (the α peptide was directed to another structure) controls (data not shown). This allows us to expect that such reporter systems are suitable for studying the intracellular translocations of proteins in open structures, provided that translocations involve a substantial portion of the target protein contained in the given structure.
The Reporter System Adequately and Quantitatively Detects FOXO3A Translocation into the Nucleus
The reporter cell lines were used to quantify the translocation of FOXO3A into the nucleus. In addition to pLCMV-FOXO3A-PMN, we obtained two control structures expressing FOXO3A fused with the α peptide and the nuclear or plasma membrane localization signal. Measuring the enzymatic activity after the introduction of the constructs into the RKO-NZ1 reporter cell line, we estimated the maximal and minimal possible content of FOXO3A in the nucleus. A major part of FOXO3A was outside the nucleus in RKO cells, because the α-galactosidase activity in cells carrying pLCMV-FOXO3A-PMN was only two-fold higher than in the negative control and eightfold lower than in cells producing the protein with the nuclear localization signal (Fig. 3a).
Fig. 3.
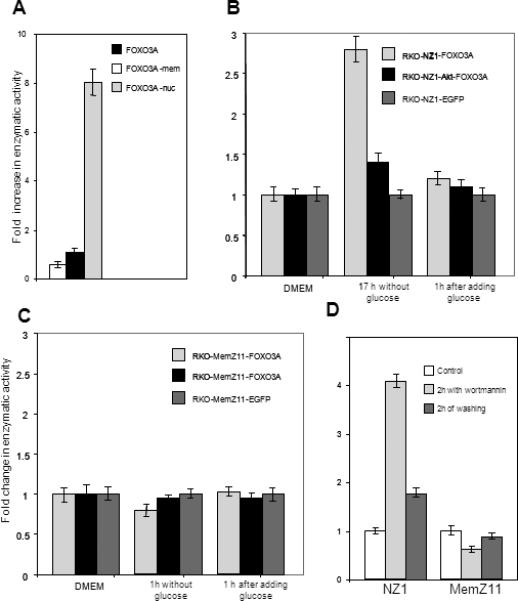
Enzymatic assay system reports the intracellular translocations of FOXO3A. (a) Relative enzymatic activity in RKO-NZ1 cells after the introduction of the constructs that produce FOXO3A fused with the α peptide and the nuclear or plasma membrane localization signal. The enzymatic activity observed in the presence of the protein lacking a localization signal was taken as unity. (b) Changes in enzymatic activity observed in reporter cell lines in response to the presence or absence of glucose in the culture medium. We used control RKO-NZ1-EGFP-PMN cells, reporter RKONZ1-FOXO3A-PMN cells, and RKO-NZ1-FOXO3A-PMN cells producing constitutively activated Akt kinase. Cells were incubated in glucose-free DMEM for 1 h; then, a portion of cells was further incubated in glucose-containing DMEM for 1 h. The β-galactosidase activity was measured with a plate reader. (c) A similar panel of cells was derived from the RKO-MemZ11 reporter line, containing the ω fragment with the plasma membrane localization signal. Cells were treated as in (b). (d) Effect of the PI3K inhibitor wortmannin on the FOXO3A localization in RKO-NZ1 and RKO-MemZ11 reporter cells. Before measuring the enzymatic activity, cells were treated with 50 nM wortmannin for 2 h and washed with the fresh medium for 2 h.
We studied the effect of the culture medium on the FOXO3A redistribution between the nucleus and cytoplasm. Glucose starvation is known to inhibit PKB and, consequently, to suppress phosphorylation and nuclear export of FOXO3A. This adaptive trans-location of FOXO3A from the nucleus must be suppressed upon constitutive PKB activation, which can be simulated by introducing the activated Akt oncogene. We introduced constitutively activated Akt in RKO-NZ1 cells via lentivirus infection and used the resulting line as a control. Incubation in glucose-free DMEM for 1 h increased the enzymatic activity almost twofold in control RKO-NZ1 cells and by no more than 20% in cells producing activated Akt (Fig. 3b). When the normal glucose concentration in the medium was restored, the enzymatic activity reached the initial level in 1 h, suggesting partial translocation of FOXO3A from the nucleus. Changes in glucose concentration did not affect the enzymatic activity in a control cell line producing EGFP in place of FOXO3A (Fig. 3b). Enzymatic activity tended to decrease in response to glucose starvation and was restored in the presence of glucose in reporter RKO-MemZ11 cells, which expressed the ω fragment in the plasma membrane (Fig. 3c). Another way to change the intracellular distribution of FOXO3A is to treat cells with the PI3K inhibitor wortmannin, which suppresses PKB and thereby prevents FOXO3A translocation into the cytoplasm [25, 26]. After incubation with 40 nM wortmannin for 2 h, the enzymatic activity increased fourfold in RKO-NZ1-FOXO3A-PMN cells and decreased by 40% in RKO-MemZ11-FOXO3A-PMN cells. When wortmannin was washed out with the fresh medium for 2 h, the enzymatic activity decreased, suggesting FOXO3A translocation from the nucleus (Fig. 3d).
To verify that the observed changes in enzymatic activity were not due to an increase or decrease in the total FOXO3A level in the cell, the FOXO3A level was estimated by Western blotting with antibodies against the C-terminal c-Myc epitope, fused with FOXO3A. The FOXO3A level was much the same in control cells, cells treated with wortmannin, or washed from wortmannin (Fig. 4). It is clear that the changes in β-galactosidase activity were associated with a redistribution of the recombinant protein within the cell and consequent changes in its accessibility for the functional interaction with the ωfragment incorporated in certain cell structures.
Fig. 4.

Production of recombinant FOXO3A in (a) control nontreated RKO-NZ1-FOXO3A-PMN cells, (b) cells incubated with 50 nM wortmannin for 2 h, and (c) cells washed with the fresh medium for 2 h after treatment with wortmannin. Western blotting was carried out with antibodies against the C-terminal epitope of c-Myc.
Our enzymatic assay system, designed to study the localization of recombinant proteins, reliably reports a redistribution of a minor portion (5−10%) of the protein of interest. When several reporter cell lines are used to simultaneously study the protein localization in different cell structures, it is possible to quantify the redistribution of the protein in the cell and to monitor its translocations in response to changes in culture conditions with a sufficient accuracy. The most appealing application of our reporter system is searching for new potential drugs affecting the signaling cascades that involve functionally important translocations of proteins within the cell. The use of our method in place of laborious non-quantitative visual analysis of the cellular localization will increase the throughput and accuracy of screenings of chemical compounds, peptides, and genetic constructs by several orders of magnitude.
ACKNOWLEDGMENTS
The work was supported by grants from NIH (CA104903 and AG025278 to P.M.C.), the Russian Foundation for Basic Research (to P.M.C., J.E.K., E.I.F., and V.S.P.), the program Molecular and Cell Biology (to J.E.K. and P.M.C.), the Howard Hughes Medical Institute (to P.M.C.).
REFERENCES
- 1.Lever JE. The use of membrane vesicles in transport studies. CRC Crit. Rev. Biochem. 1980;7:187–246. doi: 10.3109/10409238009105462. [DOI] [PubMed] [Google Scholar]
- 2.van Roessel P, Brand AH. Imaging into the future: Visualizing gene expression and protein interactions with fluorescent proteins. Nature Cell Biol. 2002;4:E15–E20. doi: 10.1038/ncb0102-e15. [DOI] [PubMed] [Google Scholar]
- 3.Lakowicz JR. Principles of frequency-domain fluorescence spectroscopy and applications to cell membranes. Subcell. Biochem. 1988;13:89–126. doi: 10.1007/978-1-4613-9359-7_3. [DOI] [PubMed] [Google Scholar]
- 4.van Rheenen J, Langeslag M, Jalink K. Correcting confocal acquisition to optimize imaging of fluorescence resonance energy transfer by sensitized emission. Biophys. J. 2004;86:2517–2529. doi: 10.1016/S0006-3495(04)74307-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Balla T, Varnai P. Visualizing cellular phosphoinositide pools with GFP-fused protein modules. Sci. STKE. 20022002:PL3. doi: 10.1126/stke.2002.125.pl3. [DOI] [PubMed] [Google Scholar]
- 6.Ullmann A, Jacob F, Monod J. Characterization by in vitro complementation of a peptide corresponding to an operator-proximal segment of the β-galactosidase structural gene of Escherichia coli. J. Mol. Biol. 1967;24:339–343. doi: 10.1016/0022-2836(67)90341-5. [DOI] [PubMed] [Google Scholar]
- 7.Moosmann P, Rusconi S. αcomplementation of LacZ in mammalian cells. Nucl. Acids Res. 1996;24:11711172. doi: 10.1093/nar/24.6.1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Blakely BT, Rossi FM, Tillotson B, et al. Epidermal growth factor receptor dimerization monitored in live cells. Nature Biotechnol. 2000;18:218–222. doi: 10.1038/72686. [DOI] [PubMed] [Google Scholar]
- 9.Rossi FM, Blakely BT, Charlton CA, Blau HM. Monitoring protein-protein interactions in live mammalian cells by β-galactosidase complementation. Meth. Enzymol. 2000;328:231–251. doi: 10.1016/s0076-6879(00)28400-0. [DOI] [PubMed] [Google Scholar]
- 10.Wehrman TS, Casipit CL, Gewertz NM, Blau HM. Enzymatic detection of protein translocation. Nature Methods. 2005;2:521–527. doi: 10.1038/nmeth771. [DOI] [PubMed] [Google Scholar]
- 11.Caron MC, Caruso M. A nuclear localization signal in the matrix of spleen necrosis virus (SNV) does not allow efficient gene transfer into quiescent cells with SNV-derived vectors. Virology. 2005;338:292–296. doi: 10.1016/j.virol.2005.05.024. [DOI] [PubMed] [Google Scholar]
- 12.Lewis PF, Emerman M. Passage through mitosis is required for oncoretroviruses but not for the human immunodeficiency virus. J. Virol. 1994;68:510–516. doi: 10.1128/jvi.68.1.510-516.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Roe T, Reynolds TC, Yu G, Brown PO. Integration of murine leukemia virus DNA depends on mitosis. EMBO J. 1993;12:2099–2108. doi: 10.1002/j.1460-2075.1993.tb05858.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lewis P, Hensel M, Emerman M. Human immunodeficiency virus infection of cells arrested in the cell cycle. EMBO J. 1992;11:3053–3058. doi: 10.1002/j.1460-2075.1992.tb05376.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yamashita M, Emerman M. Retroviral infection of non-dividing cells: Old and new perspectives. Virology. 2006;344:88–93. doi: 10.1016/j.virol.2005.09.012. [DOI] [PubMed] [Google Scholar]
- 16.Blomer U, Naldini L, Kafri T, et al. Highly efficient and sustained gene transfer in adult neurons with a lentivirus vector. J. Virol. 1997;71:6641–6649. doi: 10.1128/jvi.71.9.6641-6649.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Miyoshi H, Takahashi M, Gage FH, Verma IM. Stable and efficient gene transfer into the retina using an HIV-based lentiviral vector. Proc. Natl. Acad. Sci. USA. 1997;94:10319–10323. doi: 10.1073/pnas.94.19.10319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Burgering BM, Kops GJ. Cell cycle and death control: Long live Forkheads. Trends Biochem. Sci. 2002;27:352–360. doi: 10.1016/s0968-0004(02)02113-8. [DOI] [PubMed] [Google Scholar]
- 19.Kaestner KH, Knochel W, Martinez DE. Unified nomenclature for the winged helix/forkhead transcription factors. Genes Dev. 2000;14:142–146. [PubMed] [Google Scholar]
- 20.Biggs WH, 3rd, Meisenhelder J, Hunter T, et al. Protein kinase B/Akt-mediated phosphorylation promotes nuclear exclusion of the winged helix transcription factor FKHR1. Proc. Natl. Acad. Sci. USA. 1999;96:7421–7426. doi: 10.1073/pnas.96.13.7421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brownawell AM, Kops GJ, Macara IG, Burgering BM. Inhibition of nuclear import by protein kinase B (Akt) regulates the subcellular distribution and activity of the forkhead transcription factor AFX. Mol. Cell. Biol. 2001;21:3534–3546. doi: 10.1128/MCB.21.10.3534-3546.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brunet A, Bonni A, Zigmond MJ, et al. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. 1999;96:857–868. doi: 10.1016/s0092-8674(00)80595-4. [DOI] [PubMed] [Google Scholar]
- 23.Kops GJ, de Ruiter ND, de Vries-Smits AM, et al. Direct control of the Forkhead transcription factor AFX by protein kinase B. Nature. 1999;398:630–634. doi: 10.1038/19328. [DOI] [PubMed] [Google Scholar]
- 24.Rena G, Guo S, Cichy SC, et al. Phosphorylation of the transcription factor forkhead family member FKHR by protein kinase B. J. Biol. Chem. 1999;274:1717917183. doi: 10.1074/jbc.274.24.17179. [DOI] [PubMed] [Google Scholar]
- 25.Brunet A, Kanai F, Stehn J, et al. 14−3−3 transits to the nucleus and participates in dynamic nucleo-cytoplasmic transport. J. Cell. Biol. 2002;156:817–828. doi: 10.1083/jcb.200112059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Muslin AJ, Xing H. 14−3−3 proteins: Regulation of subcellular localization by molecular interference. Cell Signal. 2000;12:703–709. doi: 10.1016/s0898-6568(00)00131-5. [DOI] [PubMed] [Google Scholar]
- 27.Rizzuto R, Brini M, Pizzo P, et al. Chimeric green fluorescent protein as a tool for visualizing subcellular organelles in living cells. Curr. Biol. 1995;5:635–642. doi: 10.1016/s0960-9822(95)00128-x. [DOI] [PubMed] [Google Scholar]
- 28.Fliegel L, Burns K, MacLennan DH, et al. Molecular cloning of the high affinity calcium-binding protein (calreticulin) of skeletal muscle sarcoplasmic reticulum. J. Biol. Chem. 1989;264:21522–21. 528. [PubMed] [Google Scholar]
- 29.Skene JH, Virag I. Posttranslational membrane attachment and dynamic fatty acylation of a neuronal growth cone protein, GAP-43. J. Cell Biol. 1989;108:613–624. doi: 10.1083/jcb.108.2.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lanford RE, Kanda P, Kennedy RC. Induction of nuclear transport with a synthetic peptide homologous to the SV40 T antigen transport signal. Cell. 1986;46:575–582. doi: 10.1016/0092-8674(86)90883-4. [DOI] [PubMed] [Google Scholar]
- 31.Guryanova OA, Makhanov M, Chenchik AA, et al. Optimization of a genome-wide disordered lentivector-based short hairpin RNA library. Mol. Biol. 2006;40:448–459. doi: 10.1134/S002689330603006X. [DOI] [PMC free article] [PubMed] [Google Scholar]