

Abstract
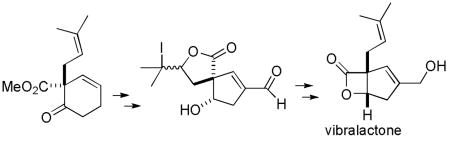
Reductive alkylation of methyl 2-methoxybenzoate with prenyl bromide and hydrolysis afforded methyl 6-oxo-1-prenyl-2-cyclohexenecarboxylate. Reduction of the ketone, hydrolysis, iodolactonization, ozonolysis and intramolecular aldol reaction provided a spiro lactone cyclopentenal. Retro-iodolactonization with activated Zn, formation of the β-lactone, and reduction of the aldehyde completed an efficient first synthesis of (±)-vibralactone. No protecting groups were used except for the novel use of an iodolactone to protect both the prenyl double bond and carboxylic acid.
Liu and coworkers recently reported the isolation of the unusual fused β-lactone vibralactone (1) from cultures of the Basidiomycete Boreostereum vibrans (see Scheme 1).1 The structure was assigned by detailed spectroscopic analysis and the absolute stereochemistry was assigned by computational methods. Vibralactone inhibits pancreatic lipase with an IC50 of 0.4 μg/mL. The pancreatic lipase inhibitor percyquinnin, which was originally assigned as a regioisomer of 1, 2 has the same planar structure as vibralactone (1).1 Pancreatic lipase inhibitors are clinically used for the treatment of obesity and improved drugs are needed.3 This prompted us to undertake the synthesis of vibralactone (1), a new lead structure that should be readily amenable to analogue synthesis.
Scheme 1.

Retrosynthesis of (±)-Vibralactone (1)
The instability of the β-lactone ring and the functional group density makes the synthesis of vibralactone a challenging problem despite its small size. We envisioned that the β-lactone ring of vibralactone (1) could be prepared from cis hydroxy acid 2 by activation of the acid group followed by β-lactonization with retention of stereochemistry.4 Alternatively, vibralactone should be accessible from trans hydroxy acid 3 by conversion of the alcohol to a good leaving group and β-lactonization by an SN2 reaction with inversion. This less common approach to β-lactone formation has recently been improved by Wu and Sun.5 The cyclopentenal moiety of 2 and 3 should be readily available by oxidative cleavage of the cyclohexene double bond of 5 and 6, respectively, followed by an intramolecular aldol reaction. Unfortunately, the cyclohexene double bond cannot be oxidatively cleaved in the presence of the more nucleophilic side chain double bond. This necessitated the protection of the side chain double bond of 5 or 6, which might be accomplished by formation of iodolactone 7.6 However, iodolactonization could occur at either double bond of 5 or 6. Even if iodolactonization occurs selectively as expected and required at the more nucleophilic side chain double bond, stereoisomeric mixtures of γ- and δ-lactones could be formed that would complicate product analysis. Furthermore, it was by no means certain that oxidative cleavage of the double bond and intramolecular aldol reaction to give cyclopentenal 4 could be accomplished in the presence of a reactive tertiary iodide. Retro-iodolactonization by zinc reduction of 4 would regenerate 2 or 3 with both the side chain double bond and the free acid needed for lactonization.
Alcohols 5 and 6 should be accessible by the diastereoselective reduction of ketone 9, which surprisingly proved to be the most challenging step in the synthesis. Ketone 9 can be easily prepared by reductive alkylation7 of methyl 2-methoxybenzoate (8) with prenyl bromide followed by acidic hydrolysis of the methyl enol ether.
Reduction of 8 with K in liquid NH3 at −78 °C, addition of LiI and prenyl bromide, and slow warming to 25 °C afforded the alkylated cyclohexadiene 10 in 77% yield (see Scheme 2).7 Hydrolysis of the enol ether with methanolic HCl gave ketone 9 in 84% yield. Reduction of 9 with NaBH4 in MeOH cleanly provided a 2:1 mixture of trans hydroxy ester 6 and cis hydroxy ester 5.8,9 Magnus reported similar ratios of products from the NaBH4 reduction of a related keto amide and also found that Zn(BH4)2 gave the trans hydroxy amide with >99:1 selectivity.10 Reduction of 9 with Zn(BH4)2 in ether11 afforded trans hydroxy ester 6 in 69% yield. Reduction with CaCl2 and NaBH4, which selectively provides trans hydroxy esters from 1-alkyl-2-oxocyclohexanone carboxylates,12 gave a 7.5:1 mixture of 6 and 5, from which 6 was isolated in 69% yield.
Scheme 2.
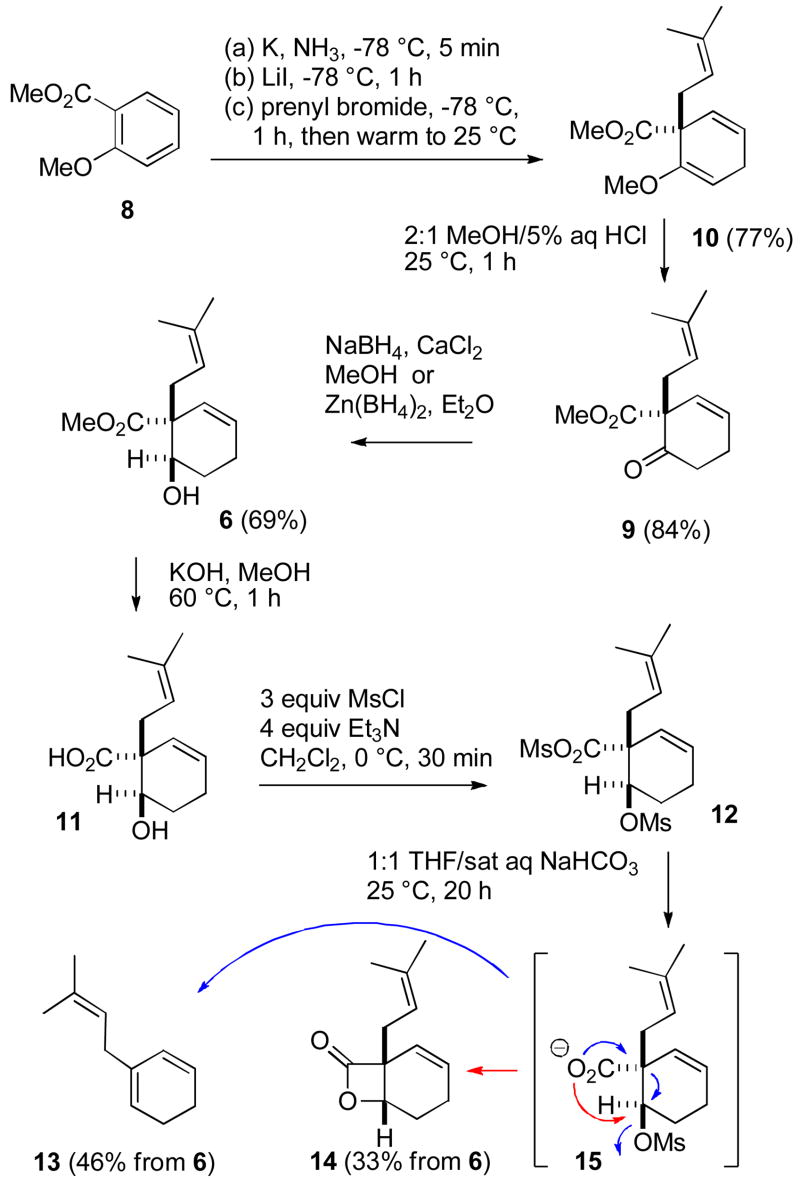
Synthesis of Model β-Lactone 14
We chose to carry out a model study exploring lactone formation with inversion by hydroxy group activation using the now readily available trans hydroxy ester 6. Hydrolysis of the methyl ester with KOH in MeOH at 60 °C provided the required trans hydroxy acid. In their studies with acyclic β-hydroxy acids, Wu and Sun needed to activate the alcohol without activating the carboyxlic acid because lactonization by acid activation will give the other stereoisomer.5 Trans hydroxy acid 11 would form a highly strained trans-fused β-lactone by reaction of the alcohol with an activated acyl group. Therefore there is no need to selectively activate the alcohol. Treatment of 11 with MsCl and Et3N in CH2Cl2 afforded the mesylate mixed anhydride 12. Hydrolysis of the more reactive mixed anhydride with NaHCO3 in aqueous THF gave mesylate carboxylate 15, which underwent an intramolecular SN2 reaction with inversion (red arrow) to provide the desired lactone 14 in 33% overall yield from hydroxy ester 6. Unfortunately, Grob fragmentation with loss of CO2 (blue arrows) to give triene 13 in 46% overall yield from 6 was the major reaction. An even greater percentage of 13 was obtained using DBU or solid K2CO3 as bases in THF. This fragmentation reaction was previously noted by Wu and Sun, but has not been widely observed in β-lactone synthesis.5 This fragmentation process is quite distinct from the well-known decarboxylation of β-lactones that occurs on heating;13 lactone 14 is stable under the reaction conditions. These results suggested that trans hydroxy esters 6 and 3 are not ideal precursors for vibralactone (1) synthesis.
On the other hand, lactone formation from cis hydroxy ester 5, the minor product from the NaBH4 reduction, proceeded cleanly and in high yield. Hydrolysis of the methyl ester with KOH in MeOH at 60 °C afforded the hydroxy acid, which was treated with TsCl in pyridine to form the mixed anhydride, which cyclized to give lactone 14 in 83% yield (see eq 1).
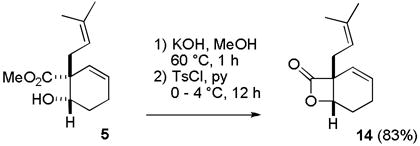 |
(1) |
We therefore turned our attention to methods to selectively prepare the cis hydroxy ester 5. As expected, Mitsunobu reaction with the hindered secondary alcohol 6 failed, even using procedures optimized for hindered alcohols.14 Fraga reported that reduction of ethyl 1-allyl-2-oxocyclohexanonecarboxylate with (n-Bu)4NBH4 in MeOH gave an 8.2:1 mixture favoring the cis hydroxy ester.12 Eventually, we found that reduction of 9 with Me4NBH4 in 1:1 THF/MeOH at 25 °C afforded a 3:2 mixture of 5 and 6 from which pure 5 was isolated in 42% yield along with a 1:2 mixture of 5 and 6 in 40% yield (see Scheme 3). This mixture can easily be recycled by oxidation with Dess-Martin periodinane to give ketone 9 in 94% yield.
Scheme 3.
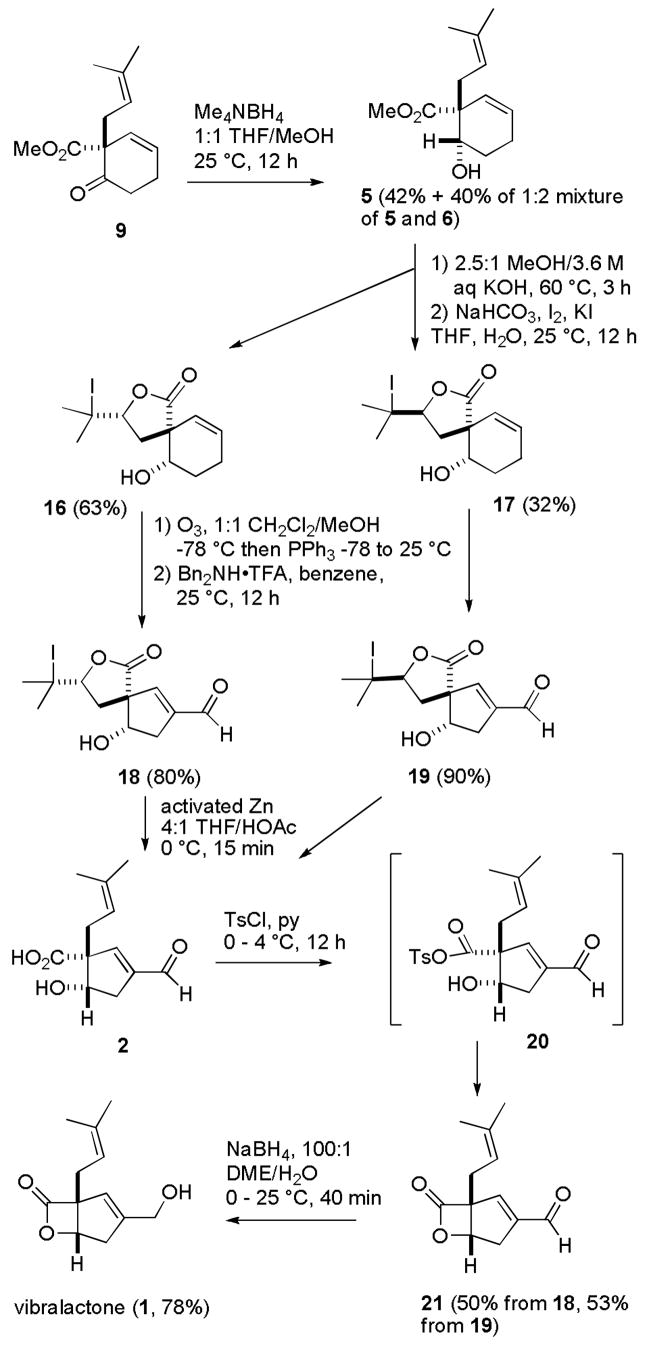
Synthesis of (±)-Vibralactone (1)
Hydrolysis of the methyl ester of 5 with KOH in MeOH at 60 °C provided the hydroxy acid, which was treated with NaHCO3, I2 and KI in aqueous THF to give the desired iodolactones 16 (63%) and 17 (32%). These iodolactones were separated to aid in characterization of intermediates, but can be carried through as a mixture because the sequence converges at cis hydroxy acid 2. Both isomers are γ-lactones with carbonyl absorptions at 1752 and 1749 cm−1, respectively. The methyl groups absorb between δ 1.92–2.0 as expected for CMe2I. X-ray crystal structure determination of the minor isomer 17 established the stereochemistry of the lactone ring and confirmed our stereochemical assignment of the hydroxy group.8 Ozonolysis at −78 °C followed by reduction of the ozonide with Ph3P afforded a dialdehyde, which was immediately subjected to Corey’s protocol (Bn2NH•TFA in benzene)15 to effect the intramolecular aldol reaction. This sequence converted cyclohexenes 16 and 17 to cyclopentenals 18 and 19 in 80% and 90% yields, respectively.
Completion of the synthesis required reduction of the aldehyde to the alcohol, retro-iodolactonization to regenerate the unsaturated acid, and β-lactone formation. Reduction of 18 or 19 with NaBH4 gave a complex mixture, probably as a result of the instability of the tertiary iodide. Fortunately, retro-iodolactonization of 18 and 19 under mild conditions with activated Zn16 in 4:1 THF/HOAc at 0 °C for 15 min regenerated prenyl acid 2 without pinacol coupling of the aldehyde.17 We chose to prepare the β-lactone prior to reduction of the aldehyde because this would not require the use of a protecting group for the primary alcohol. Treatment of 2 with TsCl in pyridine for 12 h at 0 to 4 °C afforded mixed anhydride 20, which cyclized to give β-lactone 21 in 50% overall yield from 18 and 53% overall yield from 19. Unfortunately, reduction of the aldehyde of 21 with NaBH4 in MeOH also hydrolyzed the β-lactone to the hydroxy methyl ester. This problem was easily solved using a procedure developed by Corey and Schreiber for reductions of ketones in the presence of the β-lactone of omuralide.18 Reduction of aldehyde 21 with NaBH4 in 100:1 DME/H2O for 40 min at 0 to 25 °C gave (±)-vibralactone (1) in 78% yield with spectral data identical to those reported.1
In conclusion, we have developed a ten-step synthesis of (±)-vibralactone (1) from methyl 2-methoxy benzoate and prenyl bromide that proceeds in 9% (higher if recycled 6 is included) overall yield. No protecting groups are used except for the novel use of an iodolactone to protect both the prenyl double bond and carboxylic acid. This synthesis is readily amenable to analogue preparation and we are currently adapting it to the preparation of optically pure vibralactone using the diastereoselective reductive alkylation of chiral benzamides.19
Supplementary Material
Complete experimental procedures, copies of 1H and 13C NMR spectral data, and X-ray crystallographic data (CIF file) for 17. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
We are grateful to the National Institutes of Health (GM-50151) for support of this work. We thank the National Science Foundation for the partial support of this work through grant CHE-0521047 for the purchase of an X-ray diffractometer. We thank Prof. Bruce M. Foxman, Brandeis University, for assistance with the X-ray structure determination.
References
- 1.Liu DZ, Wang F, Liao TG, Tang JG, Steglich W, Zhu HJ, Liu JK. Org Lett. 2006;8:5749–5752. doi: 10.1021/ol062307u. [DOI] [PubMed] [Google Scholar]
- 2.Hoppmann C, Kurz M, Müller G, Toti L. 1142886. Eur Pat Appl. 2001; Chem Abstr. 2001:287595. [Google Scholar]
- 3.Birari RH, Bhutani KK. Drug Discov Today. 2007;12:879–889. doi: 10.1016/j.drudis.2007.07.024. and references cited therein. [DOI] [PubMed] [Google Scholar]
- 4.For reviews on β-lactones, see: Pommier A, Pons JM. Synthesis. 1993:441–459.Pommier A, Pons JM. Synthesis. 1995:729–744.Lowe C, Vederas JC. Org Prep Proced Int. 1995;27:305–346.Yang HW, Romo D. Tetrahedron. 1999;55:6403–6434.Wang Y, Tennyson RL, Romo D. Heterocycles. 2004;64:605–658.
- 5.(a) Wu Y, Sun YP. Chem Commun. 2005:1906–1908. doi: 10.1039/b416383d. [DOI] [PubMed] [Google Scholar]; (b) Sun YP, Wu Y. Synlett. 2005:1477–1479. [Google Scholar]; (c) Wu Y, Sun YP. J Org Chem. 2006;71:5748–5751. doi: 10.1021/jo060844m. [DOI] [PubMed] [Google Scholar]
- 6.For a review, see: Dowle MD, Davies DI. Chem Soc Rev. 1979;8:171–197.
- 7.(a) Hook JM, Mander LN, Woolias M. Tetrahedron Lett. 1982;23:1095–1098. [Google Scholar]; (b) Schultz AG, Dittami JP, Lavieri FP, Salowey C, Sundararaman P, Szymula MB. J Org Chem. 1984;49:4429–4440. [Google Scholar]
- 8.The stereochemistry of alcohols 5 and 6 was established by an IR study in dilute (0.1 M) CCl4 solution and a 1D NOESY experiment. Trans hydroxy ester 6 showed two equally intense peaks at 1734 cm−1 (free carbonyl) and 1711 cm−1 (hydrogen bonded carbonyl) while cis hydroxy ester 5 showed one medium peak at 1734 cm−1 (free carbonyl) and a very strong peak at 1716 cm−1 (hydrogen bonded carbonyl).9 In a 1D NOESY experiment, irradiation of the CHOH hydrogen at δ 4.14 in 6 showed NOEs to the hydroxy hydrogen at δ 2.77 and the ring hydrogens at δ 1.88–1.76. Irradiation of the CHOH hydrogen at δ 3.79 in 5 showed NOEs to the hydroxy hydrogen at δ 3.28 and the allylic side chain methylene group at δ 2.52–2.40 establishing that the hydrogen is cis to the prenyl side chain.
- 9.Castells J, Palau J. J Chem Soc. 1964:4938–4941. [Google Scholar]
- 10.Magnus P, Tavares F, Westwood N. Tetrahedron Lett. 1997;38:1341–1344. [Google Scholar]
- 11.Nakata T, Tani Y, Hatozaki M, Oishi T. Chem Pharm Bull. 1984;32:1411–1415. [Google Scholar]
- 12.Fraga CAM, Teixeira LHP, Menezes CM, de S, Sant’Anna CMR, Ramos M, da CKV, de Aquino Neto FR, Barreiro EJ. Tetrahedron. 2004;60:2745–2755. [Google Scholar]
- 13.Mulzer J, Pointner A, Chucholowski A, Brüntrup G. J Chem Soc, Chem Commun. 1979:52–54. [Google Scholar]
- 14.(a) Martin SF, Dodge JA. Tetrahedron Lett. 1991;32:3017–3020. [Google Scholar]; (b) Saïah M, Bessodes M, Antonakis K. Tetrahedron Lett. 1992;33:4317–4320. [Google Scholar]
- 15.Corey EJ, Danheiser RL, Chandrasekaran S, Siret P, Keck GE, Gras JL. J Am Chem Soc. 1978;100:8031–8034. [Google Scholar]
- 16.Hannick SM, Kishi Y. J Org Chem. 1983;48:3833–3835. [Google Scholar]
- 17.Hara A, Sekiya M. Chem Pharm Bull. 1972;20:309–312. [Google Scholar]
- 18.(a) Fenteany G, Standaert RF, Lane WS, Choi S, Corey EJ, Schreiber SL. Science. 1995;268:726–731. doi: 10.1126/science.7732382. [DOI] [PubMed] [Google Scholar]; (b) Macherla VR, Mitchell SS, Manam RR, Reed KA, Chao TH, Nicholson B, Deyanat-Yazdi G, Mai B, Jensen PR, Fenical WF, Neuteboom STC, Lam KS, Palladino MA, Potts BCM. J Med Chem. 2005;48:3684–3687. doi: 10.1021/jm048995+. [DOI] [PubMed] [Google Scholar]
- 19.Schultz AG. Acc Chem Res. 1990;23:207–213. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Complete experimental procedures, copies of 1H and 13C NMR spectral data, and X-ray crystallographic data (CIF file) for 17. This material is available free of charge via the Internet at http://pubs.acs.org.