

Abstract
The first enantioselective synthesis of biologically active 6-amino-5-cyanodihydropyrano[2,3-c]pyrazoles has been achieved through a cinchona alkaloid-catalyzed tandem Michael addition and Thorpe-Ziegler type reaction between 2-pyrazolin-5-ones and benzylidenemalononitriles. The reaction may also be carried out in a three-component or a four-component fashion via the in situ formation of these two components from simple and readily available starting materials. The desired products were obtained in excellent yields with mediocre to excellent enantioselectivities (up to >99% ee).
Keywords: Dihydropyrano[2, 3-c]pyrazole; Pyrazole; Dihydropyrane; Cinchona alkaloid; Organocatalysis; Multi-component reaction; Tandem reaction
Dihydropyrano[2,3-c]pyrazole derivatives have very important biological activities, such as anticancer,1a antimicrobial,1b anti-inflammatory,1c insecticidal,1d and molluscicidal activities.1e,f They are also potential inhibitors of human Chk1 kinase (Fig. 1).1g Due to their biological significance,1 there has been considerable interest in developing synthetic methods for 6-amino-5-cyanodihydro-pyrano[ 2,3-c]pyrazoles.2–6 These compounds may be readily obtained from the reaction of 4-arylmethylene-5-pyrazolone and malononitrile, 2,3 or 2-pyrazolin-5-ones and benzylidenemalononitriles.3 The overall reaction is a tandem Michael addition and a Thorpe-Ziegler type reaction (an enol addition to a cyano group) followed by tautomerization.3 It should be pointed out that these compounds may exist in the 1,4-dihydro or 2,4-dihydro tautomeric forms when the N1 position is unsubstituted. Although most studies assigned the 1,4-dihydro structure to these derivatives,2–4 recent X-ray crystallographic data prefer the 2,4-dihydro tautomer.5,6
Figure 1.
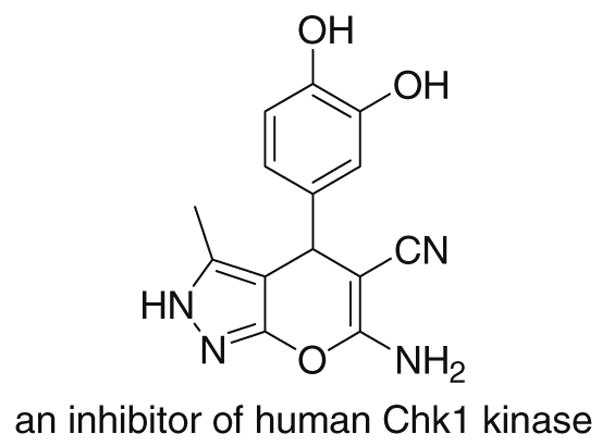
A biologically active 6-amino-5-cyanodihydropyrano[2,3-c]-pyrazole.
Since benzylidenemalononitriles may be synthesized in situ from aromatic aldehydes and malononitrile under the reaction conditions, these compounds may also be synthesized through a three-component reaction of 2-pyrazolin-5-ones, malononitrile, and aromatic aldehydes.4,5 Most recently, a four-component synthesis by using hydrazine hydrate, acetoacetate, malononitrile, and aromatic aldehydes has also been demonstrated.6 Nevertheless, to our knowledge, an enantioselective synthesis of these interesting compounds has not yet been realized.7
During our ongoing research in developing novel organocatalytic enantioselective methods for the synthesis of biologically active compounds,8 we became interested in the asymmetric synthesis of 6-amino-5-cyanodihydro-pyrano[2,3-c]pyrazoles. Herein we wish to report the first enantioselective synthesis of these derivatives through a tandem Michael addition-Thorpe-Ziegler reaction, using some readily available cinchona derivatives as the catalyst.9
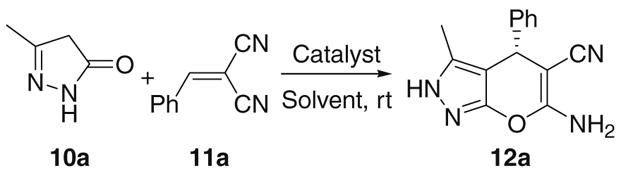
Initially we studied the synthesis with 3-methyl-2-pyrazolin-5-one (10a) and benzylidenemalononitrile (11a) as the model substrates. Several readily available cinchona alkaloid derivatives (Scheme 1) were screened as the catalysts. The results are summarized in Table 1.
Scheme 1.

Structure of the screened catalysts.
Table 1.
Catalyst screening and reaction condition optimization for the two-component reactiona
 | |||||
|---|---|---|---|---|---|
| Entry | Catalyst | Solvent | Time (h) | Yieldb (%) | eec (%) |
| 1 | 1 | CH2Cl2 | 27 | 80 | 23 |
| 2 | 2 | CH2Cl2 | 24 | 92 | 96 |
| 3 | 3 | CH2Cl2 | 24 | 91 | 65 |
| 4 | 4 | CH2Cl2 | 27 | 80 | 0 |
| 5 | 5 | CH2Cl2 | 27 | 88 | 14 |
| 6 | 6 | CH2Cl2 | 24 | 79 | 11 |
| 7 | 7 | CH2Cl2 | 27 | 96 | 10 |
| 8 | 8 | CH2Cl2 | 27 | 92 | 22d |
| 9 | 9 | CH2Cl2 | 24 | 83 | 6d |
| 10 | 2 | CHCl3 | 20 | 87 | 92 |
| 11 | 2 | THF | 20 | 74 | 60 |
| 12 | 2 | Et2O | 20 | 94 | 62 |
| 13 | 2 | Benzene | 20 | 79 | 82 |
| 14 | 2 | CH3CN | 20 | 91 | 24 |
All reactions were carried out with 10a (0.10 mmol), 11a (0.10 mmol), and the catalyst (5 mol %) in the indicated solvent (2.0 mL) at rt.
Yield of isolated product after chromatography.
Determined by HPLC analysis on a ChiralPak AS column.
The S enantiomer was obtained as the major product.
As shown in Table 1, when quinine (1) was used as the catalyst in CH2Cl2 at rt, a yield of 80% of the product 12a was obtained with a low ee value of 23% (entry 1). In contrast, when cupreine (2) was used as the catalyst, product 12a was obtained in high yield of 92% with an excellent ee value of 96% (entry 2). Nevertheless, 9-epi-cupreine (3) leads to a lower ee value of 65% (entry 3). When 9-epi-amino-9-deoxyquinine (4) was applied as the catalyst, a racemic product was obtained (entry 4). Similarly, poor enantioselectivities were achieved with quinine-derived thiourea catalysts 5–7 (entries 5–7). A low ee value of 22% for the opposite enantiomer was also obtained when quinidine (8) was used as the catalyst (entry 8). It is most surprising that cupreidine (9), the pseudo-enantiomer of cupreine (2), also leads to poor enantioselectivity of the other enantiomer (6%, entry 9). Thus, this screening identified cupreine (2) as the best catalyst for the reaction. The results also suggest that the reaction is very sensitive to the subtle changes in the catalyst structure. Further screening of the reaction conditions revealed that chloroform is also a good solvent for this reaction (entry 10), while THF, ether, benzene, and acetonitrile are worse ones (entries 11–14). Lowering the reaction temperature to 0 °C shows no improvement in the enantioselectivity (data not shown). Furthermore, control experiments also indicate that the product does not racemize under the reaction conditions (data not shown).
The absolute configuration of the major enantiomer obtained in Table 1, entry 2 was determined to be R according to the X-ray crystallographic analysis of the product 12a (Fig. 2).10 Our data also indicate that the product exists in the 2,4-dihydro tautomer form5,6 in the solid state.
Figure 2.
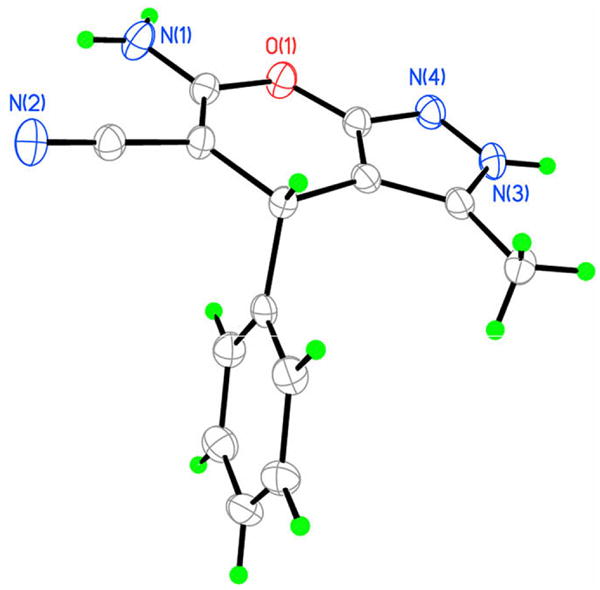
ORTEP drawing of the product 12a.
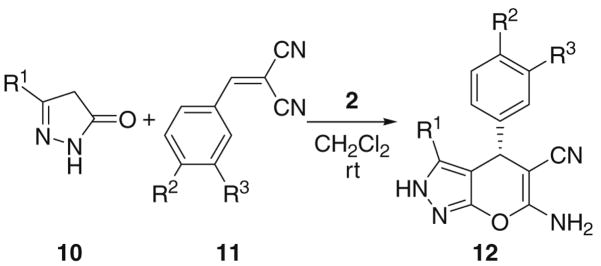
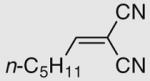
The scope and limitation of this enantioselective synthesis were next examined under the optimized conditions (with 5 mol % catalyst 2 in CH2Cl2 at rt).10 The results are listed in Table 2. As shown by the results in Table 2, besides 11a (entry 1), other benzylidenemalononitriles also participate in this reaction. However, the enantioselectivity of the reaction drops considerably if there is a substituent on the phenyl ring of the benzylidenemalononitrile. For example, the reaction of para-halogen-substituted benzylidenemalononitriles produces the expected products in high yields, but the ee values of the obtained products are only mediocre (48–62% ee, entries 2–5). Other electron-withdrawing groups at the para-position, such as, cyano and nitro groups, also lead to low ee values of the products (entries 6 and 7). Electron-donating groups (Me and MeO) at para-position also diminish the enantioselectivity of this reaction (entries 8 and 9). By comparing the results in entry 4 and entry 10, it is evident that moving the substituent to the meta-position leads to even worse enantioselectivity of the product. These results hint that the enantioselectivity of this reaction is most likely governed by steric factors instead of electronic factors. Moreover, replacing the methyl group in 3-methyl-2-pyrazolin-5-one (10a) with a larger ethyl group (10b) also leads to much poorer ee value of the product 12k (38% ee vs 96% ee, entries 1 and 11). Similar results were obtained with the product 12l of 3-phenyl-2-pyrazolin-5-one (10c, entry 12). The use of hexylidenemalononitrile (entry 13) instead of benzylidenemalononitriles also led to a poor ee value (28%) of the product 12m.
Table 2.
Enantioselective two-component reaction for the synthesis of pyranopyrazoles with catalyst 2a
 | |||||||
|---|---|---|---|---|---|---|---|
| Entry | Compound | R1 | R2 | R3 | Time (h) | Yieldb (%) | eec (%) |
| 1 | 12a | Me | H | H | 27 | 92 | 96 |
| 2 | 12b | Me | F | H | 7.5 | 96 | 58 |
| 3d | 12c | Me | Cl | H | 11 | 95 | 62 |
| 4 | 12d | Me | Br | H | 8 | 89 | 50 |
| 5d | 12e | Me | I | H | 11 | 94 | 48 |
| 6 | 12f | Me | CN | H | 15 | 92 | 40 |
| 7 | 12g | Me | NO2 | H | 14 | 88 | 36 |
| 8 | 12h | Me | Me | H | 15 | 96 | 20 |
| 9 | 12i | Me | OMe | H | 20 | 96 | 36 |
| 10 | 12j | Me | H | Br | 14 | 92 | 26 |
| 11 | 12k | Et | H | H | 12 | 84 | 38 |
| 12 | 12l | Ph | H | H | 17 | 89 | 48 |
| 13e | 12m | Me | 19 | 79 | 28 | ||
| |||||||
Unless otherwise indicated, all reactions were carried out with 10a (0.10 mmol), 11a (0.10 mmol), and the catalyst (5 mol %) in CH2Cl2 (2.0 mL) at rt.
Yield of isolated product after chromatography.
Determined by HPLC analysis on a ChiralPak AS column.
Carried out at 0 °C.
Determined by HPLC analysis on a ChiralPak AD-H column.
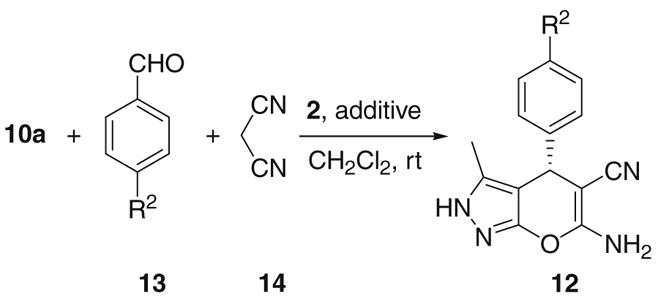
Multi-component reactions involving domino processes allow molecular complexity and diversity to be created by the formation of several new covalent bonds in a one-pot transformation. This methodology has emerged as a powerful synthetic strategy.12 Most recently, this approach also found many applications in organocatalysis. 13 Since benzylidenemalononitriles (11) may be formed in situ from aromatic aldehydes and malononitrile under the reaction conditions,4,5 we also studied the three-component reaction of 10a, an aromatic aldehyde (13), and malononitrile (14). The results are listed in Table 3.11
Table 3.
Enantioselective three-component reaction for the synthesis of pyranopyrazoles with catalyst 2a
 | |||||
|---|---|---|---|---|---|
| Entry | 12 | Additive | Time (h) | Yieldb (%) | eec (%) |
| 1 | 12a | — | 18 | 80 | 96 |
| 2 | 12a | Na2SO4d | 25 | 69 | 99 |
| 3 | 12a | MS(4 Å)e | 21 | 72 | 94 |
| 4 | 12c | — | 6 | 54 | 42 |
| 5 | 12c | Na2SO4d | 6 | 50 | 34 |
| 6 | 12c | MS(4 Å)e | 6 | 50 | 70 |
| 7 | 12d | — | 6.5 | 74 | 66 |
| 8 | 12d | Na2SO4d | 23 | 88 | 25 |
| 9 | 12d | MS(4 Å)e | 22 | 89 | 58 |
All reactions were carried out with 10a (0.10 mmol), 13 (0.10 mmol), 14 (0.10 mmol), and the catalyst (5 mol %) in CH2Cl2 (2.0 mL) at rt.
Yield of isolated product after chromatography.
Determined by HPLC analysis on a ChiralPak AS column.
Na2SO4 (0.10 mmol) was added.
Molecular sieves (40 mg) were added.
As shown by the results in Table 3, indeed, when cupreine (2) was used as the catalyst, the desired product 12a may be obtained in 80% yield and 96% ee by using 10a, 14, and benzaldehyde (13a) as the substrates (entry 1). Since 1 equiv of water was formed under the three-component reaction conditions, some drying agents were intentionally added to the reaction mixture to evaluate their effects on the enantioselectivity of this reaction. When 1 equiv of Na2SO4 was used, the ee value of the product was improved to 99% ee (entry 2). However, adding 4 Å molecular sieves as the drying agent led to slightly inferior ee value of 94% (entry 3). The yields were also slightly lower in both cases as compared to the reaction without drying agents. Nonetheless, the effects of these additives are more complicated. For example, with p-chlorobenzaldehyde (13c), molecular sieves prove to give the highest ee value (70%, entry 6) of the product 12c, which is much higher than those obtained without the additive or with Na2SO4 (entries 4 and 5). However, with p-bromobenzaldehyde (13d), both additives give worse enantioselectivities of the product 12d (entries 8 and 9) than the reaction without these additives (entry 7). Under these individually optimized conditions, higher ee values of the products may be obtained by using the three-component reaction than by using the two-component reaction (Table 3, entry 2 vs Table 2, entry 1; Table 3, entry 6 vs Table 2, entry 3; Table 3, entry 7 vs Table 2, entry 4).
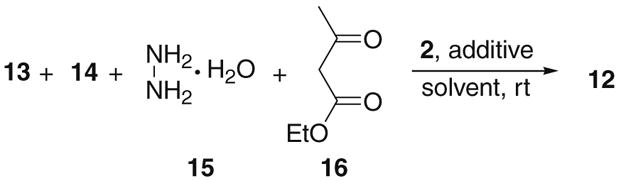
Next the four-component reaction was studied with cupreine (2) by using hydrazine hydrate (15) and acetoacetate (16) as the precursors for the in situ formation of compound 10a. The results are listed in Table 4. Benzaldehyde (13a) leads to formation of expected 12a in 28% yield and 16% ee (entry 1). Again various drying agents were evaluated for their effects on the stereoselectivity. Much improved ee values were obtained after adding 1 equiv of MgSO4 or Na2SO4, or molecular sieves (entries 2–4) to the reaction mixture, with Na2SO4 giving the best results (entry 3). By adding 2 equiv of Na2SO4 and carrying out the reaction at 0 °C, a single enantiomer of 12a may be obtained (entry 5). Similar results may also be achieved in other solvents, such as chloroform (entry 6), acetonitrile (entry 7), and THF (entry 8), except for benzene (entry 9). Whereas this four-component reaction leads to the highest ee value of product 12a, the yield is considerably lower than the two-component or the three-component reaction. Higher yields may be achieved for other aldehyde substrates, such as p-chloro (13c), p-bromo (13d), p-nitro (13g), and p-methoxybenzaldehyde (13i), but the enantioselectivities obtained were only low to mediocre (entries 10–13).
Table 4.
Enantioselective four-component reaction for the synthesis of pyranopyrazoles with catalyst 2a
 | ||||||
|---|---|---|---|---|---|---|
| Entry | 12 | Additive (equiv) | Solvent | Time (h) | Yieldb (%) | eec (%) |
| 1 | 12a | — | CH2Cl2 | 23 | 28 | 16 |
| 2 | 12a | MgSO4 (1) | CH2Cl2 | 27 | 21 | 89 |
| 3 | 12a | Na2SO4 (1) | CH2Cl2 | 27 | 25 | 96 |
| 4 | 12a | MS(4 Å)d | CH2Cl2 | 27 | 20 | 90 |
| 5 | 12a | Na2SO4 (2)e | CH2Cl2 | 28 | 43 | >99 |
| 6 | 12a | Na2SO4 (2) | CHCl3 | 24 | 28 | >99 |
| 7 | 12a | Na2SO4 (2) | MeCN | 24 | 24 | >99 |
| 8 | 12a | Na2SO4 (2) | THF | 24 | 30 | 98 |
| 9 | 12a | Na2SO4 (2) | Benzene | 24 | <5 | nd |
| 10 | 12c | Na2SO4 (2) | CH2Cl2 | 17 | 75 | 23 |
| 11 | 12d | — | CH2Cl2 | 21 | 73 | 58 |
| 12 | 12g | Na2SO4 (2) | CH2Cl2 | 17 | 80 | 2 |
| 13 | 12i | Na2SO4 (2) | CH2Cl2 | 18 | 82 | 34 |
Unless otherwise indicated, all reactions were carried out with 13 (0.10 mmol), 14 (0.10 mmol), 15 (0.10 mmol), 16 (0.10 mmol), and the catalyst (5 mol %) in the indicated solvent (2.0 mL) at rt.
Yield of isolated product after chromatography.
Determined by HPLC analysis on a ChiralPak AS column.
Molecular sieves (40 mg) were added.
Carried out at 0 °C.
In summary, we have developed the first enantioselective method for the synthesis of 6-amino-5-cyanodihydropyrano[2,3-c]pyrazoles via a two-component, a three-component, or a fourcomponent reaction using cupreine as the catalyst. The enantioselectivity of this reaction was found to be highly dependent on the reaction conditions and on the structure of the catalysts and the substrates.
Supplementary Material
Acknowledgments
This research is financially supported by the Welch Foundation (Grant No. AX-1593) and partly by the National Institute of General Medical Sciences (Grant No. 1SC1GM082718-01A1), for which the authors are most grateful. The authors also thank Dr. Hadi Arman for the help with the X-ray analysis.
Footnotes
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.tetlet.2009.02.210.
References and notes
- 1.(a) Wang JL, Liu D, Zhang ZJ, Shan S, Han X, Srinivasula SM, Croce CM, Alnemri ES, Huang Z. Proc Natl Acad Sci USA. 2000;97:7124–7129. doi: 10.1073/pnas.97.13.7124. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) El-Tamany ES, El-Shahed FA, Mohamed BH. J Serb Chem Soc. 1999;64:9–18. [Google Scholar]; (c) Zaki MEA, Soliman HA, Hiekal OA, Rashad AE. Z Naturforsch C. 2006;61:1–5. doi: 10.1515/znc-2006-1-201. [DOI] [PubMed] [Google Scholar]; (d) Ismail ZH, Aly GM, El-Degwi MS, Heiba HI, Ghorab MM. Egypt J Biotech. 2003;13:73–82. [Google Scholar]; (e) Abdelrazek FM, Metz P, Metwally NH, El-Mahrouky SF. Arch Pharm. 2006;339:456–460. doi: 10.1002/ardp.200600057. [DOI] [PubMed] [Google Scholar]; (f) Abdelrazek FM, Metz P, Kataeva O, Jaeger A, El-Mahrouky SF. Arch Pharm. 2007;340:543–548. doi: 10.1002/ardp.200700157. [DOI] [PubMed] [Google Scholar]; (g) Foloppe N, Fisher LM, Howes R, Potter A, Robertson AGS, Surgenor AE. Bioorg Med Chem. 2006;14:4792–4802. doi: 10.1016/j.bmc.2006.03.021. [DOI] [PubMed] [Google Scholar]
- 2.(a) Otto HH. Arch Pharm. 1974;307:444–447. doi: 10.1002/ardp.19743070609. [DOI] [PubMed] [Google Scholar]; (b) Otto HH, Schmelz H. Arch Pharm. 1979;312:478–486. [Google Scholar]
- 3.Sharanin YA, Sharanina LG, Puzanova VV. Zh Org Khim. 1983;19:2609–2615. [Google Scholar]
- 4.For examples, see: Lehmann F, Holm M, Laufer S. J Comb Chem. 2008;10:364–367. doi: 10.1021/cc800028m. and references cited therein.
- 5.Shestopalov AM, Yakubov AP, Tsyganov DV, Emel’yanova Yu M, Nesterov VN. Chem Heterocycl Compd. 2002;38:1180–1189. [Google Scholar]
- 6.Vasuki G, Kumaravel K. Tetrahedron Lett. 2008;49:5636–5638. [Google Scholar]
- 7.For related examples, see: Wang XS, Yang GS, Zhao G. Tetrahedron: Asymmetry. 2008;19:709–714.Alemán J, Milelli A, Cabrera S, Reyes E, Jørgensen KA. Chem-Eur J. 2008;14:10958–10966. doi: 10.1002/chem.200802030.
- 8.(a) Samanta S, Zhao CG. J Am Chem Soc. 2006;128:7442–7443. doi: 10.1021/ja062091r. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Dodda R, Zhao CG. Org Lett. 2006;8:4911–4914. doi: 10.1021/ol062005s. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Mandal T, Samanta S, Zhao CG. Org Lett. 2007;9:943–945. doi: 10.1021/ol070209i. [DOI] [PubMed] [Google Scholar]; (d) Dodda R, Goldman JJ, Mandal T, Zhao CG, Broker GA, Tiekink ERT. Adv Synth Catal. 2008;350:537–541. doi: 10.1002/adsc.200700331. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Dodda R, Mandal T, Zhao CG. Tetrahedron Lett. 2008;49:1899–1902. doi: 10.1016/j.tetlet.2008.01.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.For some leading examples of chincona alkaloid-catalyzed tandem reactions, see: Tan B, Chua PJ, Li Y, Zhong G. Org Lett. 2008;10:2437–2440. doi: 10.1021/ol8007183.Biddle MM, Lin M, Scheidt KA. J Am Chem Soc. 2007;129:3830–3831. doi: 10.1021/ja070394v.Zu LS, Wang J, Li H, Xie HX, Jiang W, Wang W. J Am Chem Soc. 2007;129:1036–1037. doi: 10.1021/ja067781+.Wang B, Wu F, Wang Y, Liu X, Deng L. J Am Chem Soc. 2007;129:768–769. doi: 10.1021/ja0670409.Wang Y, Liu XF, Deng L. J Am Chem Soc. 2006;128:3928–3930. doi: 10.1021/ja060312n.Dudding T, Hafez AM, Taggi AE, Wagerle TR, Lectka T. Org Lett. 2002;4:387–390. doi: 10.1021/ol017087t.Tan B, Chua PJ, Zeng X, Lu M, Zhong G. Org Lett. 2008;10:3489–3492. doi: 10.1021/ol801273x.Tan B, Shi Z, Chua PJ, Zhong G. Org Lett. 2008;10:3425–3428. doi: 10.1021/ol801246m.For an excellent review on organocatalyzed tandem reactions, see: Enders D, Grondal C, Hüttl MRM. Angew Chem, Int Ed. 2007;46:1570–1581. doi: 10.1002/anie.200603129.
- 10.CCDC 719284 contains the supplementary crystallographic data for 12a. These data can be obtained free of charge from the Cambridge Crystallographic Data centre via www.ccdc.cam.ac.uk/data_request/cif
- 11.For experimental details, please see the Supplementary data.
- 12.For excellent reviews, see: Tietze LF. Chem Rev. 1996;96:115–136. doi: 10.1021/cr950027e.Yus M, Ramón DJ. Angew Chem, Int Ed. 2005;44:1602–1634. doi: 10.1002/anie.200460548.
- 13.For some leading examples, see: Ramachary DB, Barbas CF., III Chem Eur J. 2004;10:5323–5331. doi: 10.1002/chem.200400597.Enders D, Huttl C, Grondal MRM, Raabe G. Nature. 2006;441:861–863. doi: 10.1038/nature04820.Chen XH, Zhang WQ, Gong LZ. J Am Chem Soc. 2008;130:5652–5653. doi: 10.1021/ja801034e.Jiang J, Yu J, Sun XX, Rao QQ, Gong LZ. Angew Chem, Int Ed. 2008;47:2458–2462. doi: 10.1002/anie.200705300.Chen XH, Xu XY, Liu H, Cun LF, Gong LZ. J Am Chem Soc. 2006;128:14802–14803. doi: 10.1021/ja065267y.Rueping M, Antonchick AP. Angew Chem, Int Ed. 2008;47:5836–5838. doi: 10.1002/anie.200801435.Zhang GW, Wang L, Nie J, Ma JA. Adv Syn Catal. 2008;350:1457–1463.Marigo M, Schulte T, Franzen J, Jørgensen KA. J Am Chem Soc. 2005;127:15710–15711. doi: 10.1021/ja055291w.Ramachary DB, Chowdari NS, Barbas CF., III Angew Chem, Int Ed. 2003;42:4233–4237. doi: 10.1002/anie.200351916.Casas J, Engqvist M, Ibrahem I, Kaynak B, Córdova A. Angew Chem, Int Ed. 2005;44:1343–1345. doi: 10.1002/anie.200461400.For a review, see: Guillena G, Ramón DJ, Yus M. Tetrahedron: Asymmetry. 2007;18:693–700.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.