

Abstract
Vascular remodeling and atheromatous lesion formation are determined in part by the balance between apoptosis and survival of vascular smooth muscle cells (VSMCs). In the chronic stages, apoptosis of VSMCs in the atherosclerotic plaques contributes to the weakening and potential rupture of the plaque causing pathologies such as acute coronary syndrome. The higher incidence of apoptosis in the plaques of symptomatic than in asymptomatic patients has been demonstrated, but the expression of survival proteins, including the inhibitor of apoptosis proteins (IAPs), has not been thoroughly examined. The aim of this study was to investigate the immunohistochemical expression of cellular inhibitor of apoptosis protein-2 (cIAP2), x-linked inhibitor of apoptosis protein (XIAP), and survivin in normal carotid arteries, and carotid endarterectomy specimens of symptomatic and asymptomatic patients with carotid stenosis. The results demonstrated stronger immunopositivity to smooth muscle myosin heavy chain antigen (SM-MHC) (sm2), proliferating cell nuclear antigen (PCNA), and p50 subunit of NF-κβ in the asymptomatic plaques than in symptomatic plaques. Furthermore, there was higher expression of cIAP2, XIAP, and survivin in the symptomatic than in the asymptomatic plaques and this paralleled caspase-3 expression. The increased expression of IAPs in symptomatic plaques could be due to endogenous defense mechanism to protect against the pro-apoptotic effect of the inflammatory stimuli that are released in the plaques. This could be involved in the stabilization of symptomatic atheromatous plaques and may prove a potential therapeutic target.
Keywords: Apoptosis, Atherosclerosis, Carotid plaque, Carotid endarterectomy, Inhibitors of apoptosis, PCNA, Vascular smooth muscle cell
INTRODUCTION
Cardiovascular disease (CVD) is the leading cause of death in developed and underdeveloped nations, stroke being the third leading cause of death after ischemic heart disease and cancer. The morbidity of stroke can be devastating with 30% of patients dying within a year of having a stroke and 50% left permanently disabled. (Warlow et al., 2001). Time of diagnosis and increased risk of stroke are directly proportional with 5.5% within 48 hours, 8% within a week, 11–14% in 30 days, and 17–20% in 3 months (Warlow et al., 2001). In the cases of transient ischemic attacks, current diagnostic guidelines recommend carotid imaging for diagnostic confirmation and if indicated carotid endarterectomy is performed (Louridas and Junaid, 2005). Asymptomatic carotid artery stenosis is usually detected by auscultation of the carotid arteries and hearing bruits or accidentally during ultrasound of the neck. Among patients with carotid bruit, only 35% have significant lesions with 70% to 90% occlusion. Among patients with significant hemodynamic carotid stenosis, a bruit has been noted only in 50% cases during physical examination. Asymptomatic patients are characterized by significant stenosis of ≥60% with no symptoms and the defined criteria exclude patients who have suffered ipsilateral cerebrovascular events, vertebrobasilar distribution events, or contralateral symptoms within the previous 45 days (Biller et al., 1998). While carotid endarterectomy is the most common procedure to prevent stroke, recent studies suggest that carotid artery stenting is less invasive and better treatment for patients with high-risk stenosis (Yadav et al., 2004). Nevertheless, careful examination of carotid endarterectomy specimens provide invaluable snapshot into the cellular and molecular events leading to plaque rupture.
In the advanced stages of the atheroma, inflammation and apoptosis of the vascular smooth muscle cell (VSMCs) in the fibrous cap or shoulder region contributes to weakening and inevitable rupture of the plaque (Dhume and Agrawal, 2003; Dhume et al., 2003; Mitra and Agrawal, 2004). We have previously reported a higher number of CD68+ cells in the fibrous cap region in symptomatic than in asymptomatic plaques and that there was an increase in inflammatory cytokines, such as IL-12 and IFN-γ, released by these cells that can induce VSMC apoptosis (Dhume et al., 2004; Jia et al., 2006). Although the increased expression of Bcl-2, a member of the anti-apoptotic protein family, has been demonstrated in asymptomatic carotid plaques than in the symptomatic plaques (Artese et al., 2005; Dhume and Agrawal, 2006), the role of survival proteins such as the inhibitor of apoptosis protein (IAPs) in carotid plaques has not been evaluated.
The IAPs are upregulated in response to cellular compromise and are involved in binding to and inhibiting activity of effector caspases 3, 7 and 9, thereby preventing or inhibiting apoptosis. Here, we hypothesized that with an increase in inflammation in the rupture-vulnerable symptomatic plaques; there would be increased expression of IAPs. The findings from this study provide insight into the expression of cIAP2, XIAP and survivin in normal carotid artery and in the atherosclerotic plaques of symptomatic and asymptomatic patients with carotid stenosis.
MATERIALS AND METHODS
Carotid Endarterectomy Specimens
Surgical specimens of human atherosclerotic plaques (8 plaques from asymptomatic patients, 71.0 ± 1.7 yr with 3 female, and 8 plaques from symptomatic patients, 77.0 ± 4.1 yr with 4 female) were obtained from patients undergoing carotid endarterectomy procedures. Two normal carotid arteries were collected from Nebraska Organ Retrieval System. The Institutional Review Board of Creighton University approved the research protocol and informed consent was obtained from the patients. The carotid endarterectomy specimens were categorized as either symptomatic or asymptomatic according to patients’ history and clinical examination. Symptoms included hemispheric transient ischemic attacks, amaurosis fugax or stroke. Carotid endarterectomy samples were collected and transported to the lab in the University of Wisconsin (UW) solution and maintained at 4°C. This solution has been proved to maintain the functional and morphological integrity of the vascular specimen for at least 24 hours (Abebe et al., 1993). Each specimen was transversely sectioned at 2 mm intervals and embedded separately into optimum cutting temperature (OCT), then frozen and stored at −80°C. Thin sections (6 μm) were cut using a cryostat and sections were mounted on electrostatically-coated slides and stored at −80°C before staining.
Plaque Evaluation
One section of each specimen was stained with hematoxylin-eosin and evaluated to define regions of the plaque. The dense staining of eosin differentiated the fibrous cap region and base from the necrotic core. The plaque regions were differentiated and categorized as follows: the fibrous cap (FC), necrotic core (NC), shoulder region of the plaque (S), and the base of the lesion (B) (Figure 1, g–i).
Figure 1.
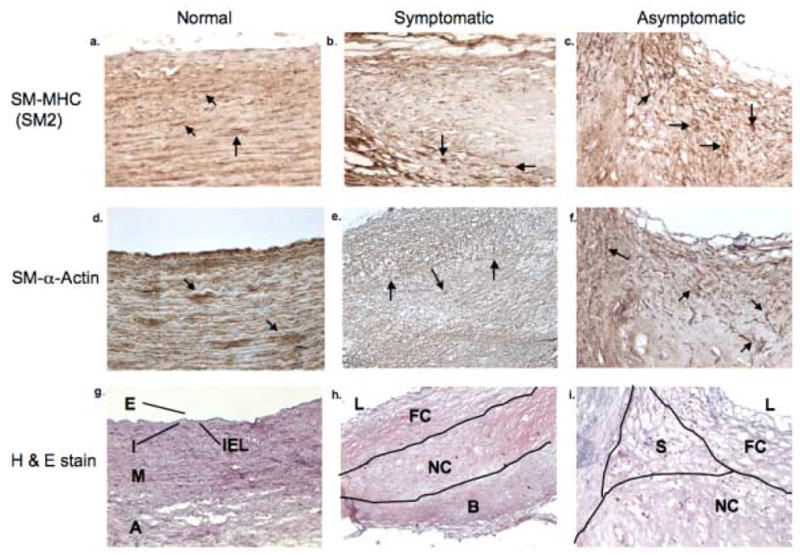
Immunohistochemistry to Smooth muscle myosin heavy chain (SM-MHC; SM2) (a–c), smooth muscle (SM)- α-Actin (d–f), and hematoxylin-eosin staining (g–i) in normal carotid artery, symptomatic carotid plaque, and the asymptomatic carotid plaque. Various regions in the cross section of a normal carotid artery are shown as the endothelium (E), the intima (I), the internal elastic lamina (IEL), the media (M), the adventitia (A). In the symptomatic and asymptomatic carotid plaques (h and i, respectively), regions are shown as the fibrous cap (FC), necrotic core (NC), shoulder region of the plaques (S), base of the lesion (B) and the lumen of the vessel (L). These results are representative of 2 normal carotid arteries, 8 symptomatic and 8 asymptomatic plaques (magnification 10x).
Immunohistochemistry
Frozen sections were allowed to dry, fixed and permeabilized in acetone before staining with peroxidase-conjugated reagents. The detection of immunohistochemical staining with Vectastain ABC reagents (Vector Laboratories, Burlingame, CA) was used. Following acetone treatment, sections were washed with phosphate-buffered saline, incubated with the relevant primary antibody overnight at 4°. Subsequently, the sections were labeled with biotinylated horse-anti-primary antibody (Vector Laboratories) for 1 hour followed by incubation with avidin:biotinylated-peroxidase complex (Vector Laboratories) for 30 minutes. Immunoreactivity was detected with diaminobenzidine/hydrogen peroxide for 4–8 minutes and a hematoxylin nuclear counterstain.
RESULTS
Morphology and Identification of VSMCs
VSMCs from normal carotid arteries, symptomatic and asymptomatic plaque samples were characterized by their positive immunoreactivity to smooth muscle myosin heavy chain (SM-MHC) (sm2) and smooth muscle (SM)-α-actin and were localized to the media and adventitia of the normal carotid. (Figure 1a, d). The distribution of VSMCs was detected in the necrotic core of both symptomatic and asymptomatic plaques; however, there was a greater preponderance of the smooth muscle cells in the asymptomatic plaques as compared to the symptomatic counterparts (Figure 1b–c). The positive immunoreactivity to sm2 and SM-α-actin was detected in the fibrous cap, necrotic core, the base and surrounding adventitia (Figures 1e–f). While, SM-α-actin was detected in the necrotic core of both symptomatic and asymptomatic plaques, sm2 antibody for SM-MHC showed greater immunopositivity in the asymptomatic than in the symptomatic plaque (Figures 1b–c and e–f).
Expression of Inflammatory Proteins in Symptomatic and Asymptomatic Plaques
Immunoreactivity to the NF-κβ regulatory p50 subunit (Figure 2c) was detected in the fibrous cap and necrotic core of asymptomatic plaques while a diffuse punctate immunopositivity was observed in the symptomatic plaques (Figure 2b). There was no immunopositivity to NF-κβ in the normal carotid artery (Figure 2a). Although there was no expression of caspase-3 in the normal carotid artery, greater expression of caspase-3 was observed in the fibrous cap and necrotic core of the symptomatic plaques as compared to the asymptomatic plaques (Figure 2e–f). The marker of proliferation, proliferating cell nuclear antigen (PCNA), was highly expressed in the fibrous cap, necrotic core and base of the asymptomatic plaque than the symptomatic plaques (Figure 2h–i).
Figure 2.
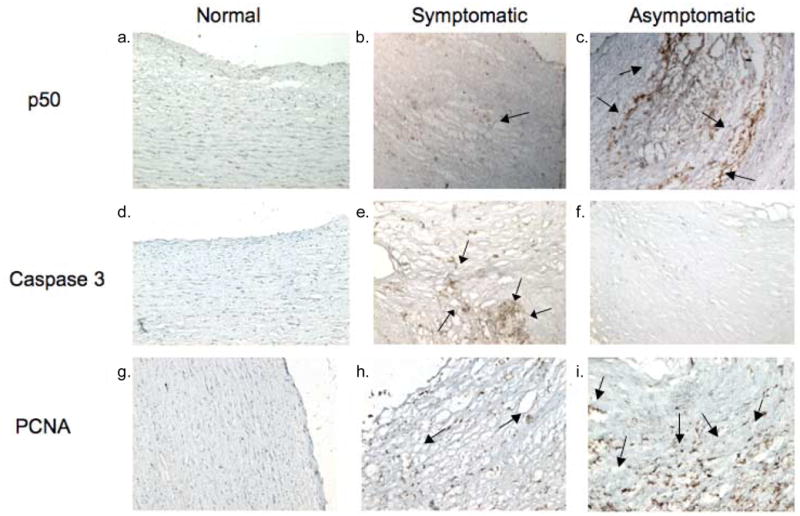
Representative cross sections (magnification 10x) of normal carotid artery and carotid endarterectomy plaques. Figure 2c shows stronger immunoreactivity to p50 NF-κβ, as shown by arrows, in asymptomatic plaques when compared to symptomatic plaques (Figure 2b). Immunostaining to caspase-3 (2d–f) showed strong reactivity in the symptomatic carotid plaque, as shown by arrows (Figure 2e), but not in the asymptomatic carotid plaque and no staining in the normal carotid. Immunostaining to PCNA (2g–i) shows stronger reactivity in the asymptomatic carotid plaque, as shown by arrows, than the symptomatic plaque with no staining in the normal carotid vessel. These results are representative of 2 normal carotid arteries, 8 symptomatic and 8 asymptomatic plaques.
Expression of IAPs in symptomatic and asymptomatic plaques
Immunohistochemical analysis of cIAP revealed a basal expression in normal carotid artery (Figure 3a). There was increased cIAP2 expression in the fibrous cap, shoulder region and base of the symptomatic plaques when compared to the asymptomatic plaques (Figures 3b–c, e–f). XIAP (Figure 3d) and survivin (Figure 3g) did not show any immunoreactivity in the normal carotid arteries. However, there was an enhanced expression of both proteins in the fibrous cap region of the symptomatic carotid plaque when compared to the asymptomatic carotid plaque (Figure 3e–f, h–i). XIAP expressed punctate areas of immunoreactivity in the asymptomatic plaque. Survivin showed minor immunoreactivity in the necrotic core of the asymptomatic plaque (Figure 3i).
Figure 3.
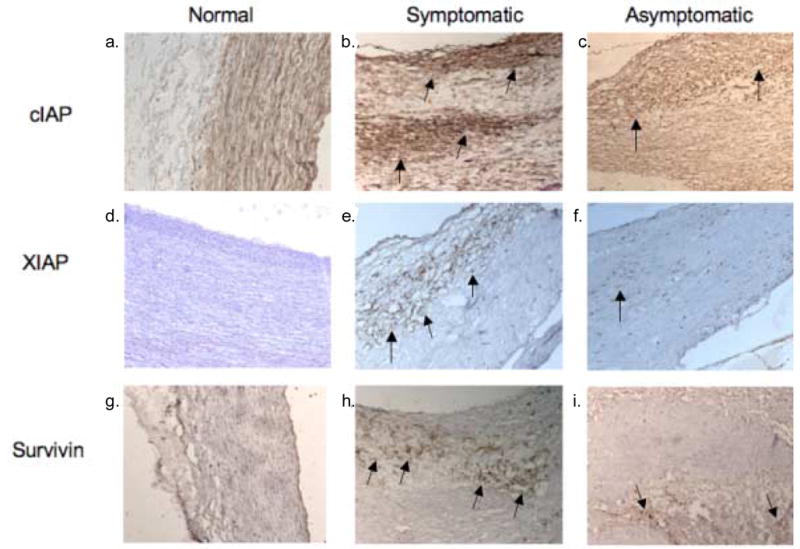
Representative cross sections (magnification 10x) of normal carotid artery and carotid endarterectomy plaques. In Figure 3a–c, immunoreactivity to cIAP-2 shows ubiquitous staining in the normal artery with an increased immunoreactivity in the symptomatic carotid plaque, as shown by arrows, when compared to the asymptomatic carotid plaque. In Figure 3d–f, immunoreactivity to XIAP shows stronger reactivity in the symptomatic carotid plaque, as shown by arrows, than the asymptomatic plaque with no staining in the normal carotid artery. In Figure 3g–i, immunoreactivity to survivin shows stronger reactivity in symptomatic plaques, as shown by arrows, when compared to asymptomatic plaques with no expression in the normal carotid artery. These results are representative of 2 normal carotid arteries, 8 symptomatic and 8 asymptomatic plaques.
DISCUSSION
It has been suggested that vascular remodeling and lesion formation are determined in part by the balance between apoptosis and proliferation or survival of VSMCs (Cho et al., 1997). Disruption of this balance in the fibrous cap or shoulder region of the lesion could lead to an increase in apoptosis and subsequent plaque rupture (Cho et al., 1997). Apoptosis is a pivotal regulator of cell number in the vessel wall. In the early pathogenesis, migration and proliferation of the VSMCs into the intima lead to the thickening of the fibrous cap, which stabilizes the atheroma. However, the thinning of the fibrous cap, inflammatory infiltration into the fibrous cap and shoulder regions, ulceration and rupture are characteristics of symptomatic plaques and are attributed to apoptosis of the VSMCs (Dhume and Agrawal, 2003; Morioka et al., 2004).
Three major parameters in atheromatous plaques were evaluated in this study: inflammation, proliferation, and apoptosis. Inflammatory processes mark all stages of atheroma development and progression. NF-κβ is a major transcription factor that regulates various aspects of inflammatory responses; however, it is also involved in the regulation of many inflammatory genes, and proliferation, migration and apoptosis of the cells. NF-κβ signaling has been reported to be involved in all stages of the pathogenesis of atheromas (de Winther et al., 2005). In our study we used NF-κβ (p50 subunit) as an indicator of inflammatory events in atheromatous carotid plaques obtained from patients undergoing carotid endarterectomy. Interestingly, expression of p50 NF-κβ was found to be stronger in asymptomatic than in symptomatic plaques. The more dense areas of immunoreactivity were localized to the fibrous cap and the necrotic core. This suggests that NF-κβ could be upregulated in response to VSMCs proliferation due to mitogen and cytokine activation.
There is a direct correlation between the thickness and stability of the plaque where the thickness of the fibrous cap is significantly greater in the asymptomatic plaques than in symptomatic plaques (Dhume et al., 2003; Mitra and Agrawal, 2004). In the development of the atheroma, activated VSMCs will rapidly migrate to and proliferate in the intima of the vessel. Increased immunoreactivity to PCNA has been reported in the intima of the carotid plaque when compared to the media (Gordon et al., 1990). We, therefore, assessed the expression of proliferating cell nuclear antigen (PCNA) and found a greater expression in the fibrous cap and necrotic core of the asymptomatic plaque when compared to the symptomatic plaque. The pronounced expression of NF-κβ correlated with the enhanced expression of PCNA.
The enhanced proliferation can be attributed by an increased mitogenic expression present in the atheroma. Insulin-like growth factor-1 (IGF-1) is a 70-amino acid peptide that mediates most of its biological effects through the IGF-1 receptor (IGF-1R), which is involved in differentiation, cell-cycle regulation, cell proliferation and apoptosis inhibition (Rose et al., 2004). We have previously reported that IGF-1 plays a major role in the survival of VSMCs (Balaram et al., 1997; Allen et al., 2005a, b). An increased sensitivity to IGF-1Rα could induce migration and proliferation of VSMCs in the fibrous cap of the asymptomatic plaque and contribute to the stability of atherosclerotic lesions. An increase in inflammation with the infiltration of macrophages in the plaques of symptomatic as compared to asymptomatic subjects would have an increased presence of inflammatory cytokines that may decrease the density and/or affinity of IGF-1Rα. We have previously shown a decrease in immunoreactivity to IGF-1Rα in the necrotic core, fibrous cap and base of the lesion in the symptomatic plaque (Jia et al., 2006). A decreased response to IGF-1 and an increase in inflammatory mediators released by macrophages would shift the balance of the VSMC survival versus apoptosis resulting into plaque instability.
Activation of caspase-3, which is involved in the execution phase of apoptosis, can occur through two pathways. The extrinsic pathway is triggered by the binding of ligands of TNF superfamily and receptor binding, such as the Fas/CD95 receptor (Chaudhari et al., 2006). The intrinsic pathway is caused by insults that induce the release of cytochrome c from mitochondria with further activation of caspase-9 through the interaction with Apaf-1 (Chaudhari et al., 2006). Since both pathways converge on caspase-3, it is considered the main effector caspase during apoptotic events. During apoptosis there is increased release of IAPs which could be problematic for the patient due to the weakening of the fibrous cap leading to plaque rupture. In this study procaspase/caspase-3 positive staining showed increased density in the fibrous cap and necrotic core in the symptomatic plaque as compared to the cells of the fibrous cap and necrotic core in the asymptomatic plaque, which displayed punctate areas of reactivity. Their upregulation is most likely due to the increased number of inflammatory cells present in the symptomatic plaque. This could be supported by our previous report where we found a significant increase in the expression of CD68+ positive macrophages in the symptomatic plaque, with the greatest expression in the fibrous cap, necrotic core and base of the lesion (Dhume et al., 2003). Hutter and colleagues (2004) noted the co-localization of active caspase-3 and macrophages in carotid and coronary atherosclerotic plaques. Also, administration of caspase-3 inhibitor in vivo in hyperlipidemic mice decreased the expression of both caspase-3 activity and number of macrophages (Hutter et al., 2004). This suggests that macrophages are attracted to the site in response to increased caspase activity and apoptosis of cells. This makes sense since macrophages are not only capable of releasing several cytokines and inflammatory mediators such as IL-18 and IFN-γ that contribute to the overall pathogenesis of the plaques (Mitra and Agrawal, 2004; Jia et al., 2006), but they are also primary phagocytic cells to engulf apoptotic bodies.
Other factors that contribute to the balance between apoptosis and proliferation or survival are the survival proteins. The expression of anti-apoptotic Bcl-2 family of proteins, including Bcl-2 and Bcl-xL, is increased in the asymptomatic carotid plaque than in the symptomatic plaques (Dhume and Agrawal, 2006). IAPs, such as cIAP2, XIAP and survivin, can bind to and inhibit the enzymatically active caspase-3, 7 and 9 (Roy et al., 1997, Deveraux et al., 1997, Ambrosini et al., 1997). The cIAP1/2, XIAP, and survivin are expressed in VSMCs of the atheroma (Simosa et al., 2005, von Wnuck et al., 2006, Li et al., 2005). It is, however, uncertain as to how IAP expression may respond to inflammatory cytokines and mitogens in atheromatous VSMCs. In this study the increased expression of the IAPs in the symptomatic carotid plaque paralleled caspase-3 expression. Our results indicate an increased expression of the IAPs in the symptomatic carotid plaque when compared to the asymptomatic. This could be contributed to an increase in inflammatory cytokines released by macrophages in the symptomatic plaque. In response to the apoptotic stimuli there is an activation of the caspases and this could result in subsequent upregulation of the IAPs. An increase in inflammation is necessary for upregulation of IAP expression as indicated by the lack of expression in the normal carotid arteries and the increased expression of caspase 3. An increase in apoptosis and apoptotic signaling may have an effect on the activity of IAP expression, leading to sustained survival of the VSMCs.
In summary, we for the first time report higher expression of cIAP2, XIAP, and survivin in symptomatic than in asymptomatic plaques of patients with carotid stenosis. The increased expression of IAPs paralleled with caspase-3. Since apoptosis of VSMCs has been reported in atheromatous plaques of symptomatic patients with carotid stenosis contributing to the rupture of the plaque, the increased expression of IAPs in symptomatic plaques could be a defense mechanism to stabilize plaque and prevent acute coronary events such as stroke. Additional studies are warranted to further define the role of IAPs in plaque stability.
Acknowledgments
This study was supported by National Institute of Health grants RO1HL070885 and RO1HL07349.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abebe W, Cavallari N, Agrawal DK. Functional and morphological assessment of rat aorta stored in University of Wisconsin and Eurocollins solutions. Transplantation. 1993;56:808–16. doi: 10.1097/00007890-199310000-00006. [DOI] [PubMed] [Google Scholar]
- Allen RT, Krueger KD, Dhume A, Agrawal DK. Sustained Akt/PKB activation and transient attenuation of c-Jun N-terminal kinase in the inhibition of apoptosis by IGF-1 in vascular smooth muscle cells. Apoptosis. 2005a;10:525–535. doi: 10.1007/s10495-005-1882-3. [DOI] [PubMed] [Google Scholar]
- Allen RT, Krueger KD, Hunter WJ, Agrawal DK. Evidence that IGF-1 requires PKC-ε, PI3-kinase and MAP kinase pathways to protect human and rat vascular smooth muscle cells from apoptosis. Immunology and Cell Biology. 2005b;83:651–667. doi: 10.1111/j.1440-1711.2005.01387.x. [DOI] [PubMed] [Google Scholar]
- Ambrosini G, Adida C, Altieri DC. A novel anti-apoptosis gene, survivin, expressed in cancer and lymphoma. Nat Med. 1997;8:917–21. doi: 10.1038/nm0897-917. [DOI] [PubMed] [Google Scholar]
- Artese L, Ucchino S, Piattelli A, Piccirilli M, Perrotti V, Mezzetti A, Cipollone F. Factors associated with apoptosis in symptomatic and asymptomatic carotid atherosclerotic plaques. Int J Immunopathol Pharmacol. 2005;4:645–53. doi: 10.1177/039463200501800405. [DOI] [PubMed] [Google Scholar]
- Balaram SK, Agrawal DK, Allen RT, Kuszynski CA, Edwards JD. Cell adhesion molecules and insulin-like growth factor-1 in vascular disease. J Vasc Surg. 1997;5:866–76. doi: 10.1016/s0741-5214(97)70216-7. [DOI] [PubMed] [Google Scholar]
- Biller J, Feinberg WM, Castaldo JE, Whittemore AD, Harbaugh RE, Dempsey RJ, Caplan LR, Kresowik TF, Matchar DB, Toole JF, Easton JD, Adams HP, Jr, Brass LM, Hobson RW, 2nd, Brott TG, Sternau L. Guidelines for carotid endarterectomy: a statement for healthcare professionals from a Special Writing Group of the Stroke Council, American Heart Association. Circulation. 1998;97:501–9. doi: 10.1161/01.cir.97.5.501. Review. [DOI] [PubMed] [Google Scholar]
- Chaudhari BR, Murphy RF, Agrawal DK. Following the TRAIL to apoptosis. Immunologic Res. 2006;35(3):1–14. doi: 10.1385/IR:35:3:249. [DOI] [PubMed] [Google Scholar]
- Cho A, Mitchell L, Koopmans D, Langille BL. Effects of changes in blood flow rate on cell death and cell proliferation in carotid arteries of immature rabbits. Circ Res. 1997;81:328 –337. doi: 10.1161/01.res.81.3.328. [DOI] [PubMed] [Google Scholar]
- de Winther MP, Kanters E, Kraal G, Hofker MH. Nuclear factor kappaB signaling in atherogenesis. Arterioscler Thromb Vasc Biol. 2005;5:904–14. doi: 10.1161/01.ATV.0000160340.72641.87. [DOI] [PubMed] [Google Scholar]
- Deveraux QL, Takahashi R, Salvesen GS, Reed JC. X-linked IAP is a direct inhibitor of cell-death proteases. Nature. 1997;388(6639):300–4. doi: 10.1038/40901. [DOI] [PubMed] [Google Scholar]
- Dhume AS, Agrawal DK. Inability of vascular smooth muscle cells to proceed beyond S phase of cell cycle, and increased apoptosis in symptomatic carotid artery disease. J Vasc Surg. 2003;38:155–61. doi: 10.1016/s0741-5214(02)75463-3. [DOI] [PubMed] [Google Scholar]
- Dhume AS, Agrawal DK. Apoptosis of vascular smooth muscle cells via intrinsic and extrinsic pathways in symptomatic carotid artery plaques. J Clin Pathology. 2006 In press. [Google Scholar]
- Dhume AS, Soundararajan K, Hunter WJ, III, Agrawal DK. Comparison of vascular smooth muscle cell apoptosis and fibrous cap morphology in symptomatic and asymptomatic carotid artery disease. Ann Vasc Surg. 2003;17:1–8. doi: 10.1007/s10016-001-0331-1. [DOI] [PubMed] [Google Scholar]
- Gordon D, Reidy MA, Benditt EP, Schwartz SM. Cell proliferation in human coronary arteries. Proc Natl Acad Sci U S A. 1990;87:4600–4. doi: 10.1073/pnas.87.12.4600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutter R, Valdiviezo C, Sauter BV, Savontaus M, Chereshnev I, Carrick FE, Bauriedel G, Luderitz B, Fallon JT, Fuster V, Badimon JJ. Caspase-3 and Tissue Factor Expression in Lipid-Rich Plaque Macrophages: Evidence for apoptosis as link between inflammation and atherothrombosis. Circulation. 2004;109:2001–8. doi: 10.1161/01.CIR.0000125526.91945.AE. [DOI] [PubMed] [Google Scholar]
- Jia G, Cheng G, Agrawal DK. Differential effects of insulin-like growth factor-1 and atheroma-associated cytokines on cell proliferation and apoptosis in plaque smooth muscle cells of symptomatic and asymptomatic patients with carotid stenosis. Immunology and cell Biology. 2006;84:1–8. doi: 10.1111/j.1440-1711.2006.01449.x. [DOI] [PubMed] [Google Scholar]
- Li H, Telemaque S, Miller RE, Marsh JD. High glucose inhibits apoptosis induced by serum deprivation in vascular smooth muscle cells via upregulation of Bcl-2 and Bcl-xl. Diabetes. 2005;54:540–5. doi: 10.2337/diabetes.54.2.540. [DOI] [PubMed] [Google Scholar]
- Louridas G, Junaid A. Management of carotid artery stenosis. Update for family physicians. Can Fam Physician. 2005;5:1984–89. Review. [PMC free article] [PubMed] [Google Scholar]
- Mitra AK, Dhume AS, Agrawal DK. Vulnerable plaques: Ticking of the time bomb. Can J Physiol and Pharmacol. 2004;82:860–871. doi: 10.1139/y04-095. [DOI] [PubMed] [Google Scholar]
- Morioka M, Hamada J, Hashiguchi A, Hasegawa Y, Todaka T, Yano S, Kai Y, Miura M, Fujioka S, Ushio Y. Contribution of angiotensin-converting enzyme and angiotensin II to ischemic stroke: their role in the formation of stable and unstable carotid atherosclerotic plaques. Surg Neurol. 2004;62:292–301. doi: 10.1016/j.surneu.2004.02.027. discussion 301–3. [DOI] [PubMed] [Google Scholar]
- Rose A, Froment P, Perrot V, Quon MJ, LeRoith D, Dupont J. The luteinizing hormone-releasing hormone inhibits the anti-apoptotic activity of insulin-like growth factor-1 in pituitary alphaT3 cells by protein kinase Calpha-mediated negative regulation of Akt. J Biol Chem. 2004;279:52500–16. doi: 10.1074/jbc.M404571200. [DOI] [PubMed] [Google Scholar]
- Roy N, Deveraux QL, Takahashi R, Salvesen GS, Reed JC. The c-IAP-1 and c-IAP-2 proteins are direct inhibitors of specific caspases. EMBO J. 1997;116:6914–25. doi: 10.1093/emboj/16.23.6914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simosa HF, Wang G, Sui X, Peterson T, Narra V, Altieri DC, Conte MS. Survivin expression is up-regulated in vascular injury and identifies a distinct cellular phenotype. J Vasc Surg. 2005;41:682–90. doi: 10.1016/j.jvs.2005.01.006. [DOI] [PubMed] [Google Scholar]
- von Wnuck Lipinski K, Keul P, Lucke S, Heusch G, Wohlschlaeger J, Baba HA, Levkau B. Degraded collagen induces calpain-mediated apoptosis and destruction of the X- chromosome-linked inhibitor of apoptosis (xIAP) in human vascular smooth muscle cells. Cardiovasc Res. 2006;69:697–705. doi: 10.1016/j.cardiores.2005.08.005. [DOI] [PubMed] [Google Scholar]
- Yadav JS, Wholey MH, Kuntz RE, Fayad P, Katzen BT, Mishkel GJ, Bajwa TK, Whitlow P, Strickman NE, Jaff MR, Popma JJ, Snead DB, Cutlip DE, Firth BG, Ouriel K. Stenting and angioplasty with protection in patients at high risk for endarterectomy investigators. Protected carotid-artery stenting versus endarterectomy in high-risk patients. N Engl J Med. 2004;351:1493–501. doi: 10.1056/NEJMoa040127. [DOI] [PubMed] [Google Scholar]
- Warlow CP, Dennis MS, van Gijn J, Sandrock PA, Bamford JM, Wardlaw J, editors. Stroke: a practical guide to management. Blackwell Science; Oxford, Engl: 2001. pp. 223–300. [Google Scholar]