

1: ABSTRACT
MEN1, the gene responsible for the cancer predisposition syndrome multiple endocrine neoplasia type I, has been implicated in DNA repair, cell cycle control, and transcriptional regulation. It is unclear to what degree these processes are integrated into a single encompassing function in normal cellular physiology and how deficiency of the MEN1-encoded protein, “menin”, contributes to cancer pathogenesis. In this study, we found that loss of Men1 in mouse embryonic fibroblasts caused abrogation of the G1/S and intra-S checkpoints following ionizing radiation. The cyclin-dependent kinase inhibitor, p21, failed to be upregulated in the mutant although upstream checkpoint signaling remained intact. Menin localized to the p21 promoter in a DNA damage-dependent manner. The MLL histone methyltransferase, a positive transcriptional regulator, bound to the same region in the presence of menin but not in Men1−/− cells. Finally, p53 retained damage-responsive binding to the p21 promoter in the Men1 mutant. These data indicate that menin participates in the checkpoint response in a transcriptional capacity, upregulating the DNA damage-responsive target p21.
2: INTRODUCTION
The maintenance of genomic integrity is indispensable to cellular well-being. To ensure that genetic information is faithfully transmitted, cell division proceeds under the control of pathways dedicated to preserving such information. The functions of these pathways and their constituent proteins include direct repair of DNA lesions, cell cycle arrest, gene transcription, and even apoptosis should the damage prove irremediable (1). Many of these functions are in fact integrated; a central response to DNA damage involves the transcriptional activation of cell cycle inhibitors in order to halt the cell cycle.
A key regulator of cell cycle arrest is the cyclin-dependent kinase inhibitor (CDKI) p21WAF1/CIP1 (heretofore referred to as p21), which is required for the maintenance of arrest following genotoxic insult. p21 is the primary downstream target in p53-mediated damage response. Canonically, DNA damage activates the PIKK kinases ATM and ATR, which phosphorylate and activate the Chk effector kinases; these kinases go on to stabilize p53 via discrete phosphorylation events so that it may stimulate transcription of its target p21 (2). However, the stabilization of p53 alone cannot guarantee this upregulation. Transcriptional regulation of p21 requires additional factors, perhaps most importantly the alteration of chromatin environment in its promoter region (3), and the full complement of these ancillary complexes has not yet been elucidated.
The MEN1 gene, responsible for the autosomal dominant cancer predisposition syndrome multiple endocrine neoplasia type I, has been associated with both maintenance of genomic integrity and transcriptional regulation (4). Men1-deficient Drosophila and mouse embryonic fibroblasts (MEFs) are hypersensitive to crosslinking agents and ionizing radiation (5, 6); Drosophila are hypermutable in response to these agents. Conditional deletion of Men1 in the pancreatic islets in mice causes hyperplasia and dysregulation of cell cycle behavior at baseline (7). In addition, Men1-mutant Drosophila and MEFs continue to synthesize DNA after exposure to sufficient doses of ionizing radiation to cause S-phase arrest in wild-type cells (8). Taken together, these data strongly support a function for menin in DNA damage response. A role as a transcriptional comodulator is suggested by its association with transcription factors including JunD (9), NFκB (10), and Smad3 (11), as well as with two distinct histone modifying complexes, mSin3A histone deacetylase (HDAC) (12, 13) and the Mixed Lineage Leukemia (MLL) histone methyltransferase (HMT) (14, 15). The menin-containing MLL HMT complex has been shown to regulate the CDKIs p27 and p18 at baseline, thus linking the proliferation abnormalities with its transcriptional role (7, 16). However, no similar link between transcriptional regulation and its role in DNA damage response has yet been demonstrated.
In this work, we precisely characterize a cell cycle defect after DNA damage in Men1-mutant MEFs and show that menin functions through transcriptional regulation of p21 via the MLL HMT.
3: MATERIALS & METHODS
Cell Culture
Men1−/− (MEN1−/−) and Men1−/− MEFs complemented with wild-type Men1 cDNA (COMP) were a gift from the laboratory of Dr. Xianxin Hua (University of Pennsylvania, Philadelphia, PA) (6). Spontaneously immortalized WT MEFs (wti) were a gift from the laboratory of Dr. Tony Koleske (Yale University, New Haven, CT). E6/E7 immortalized WT MEFs (wt) were a gift from the laboratory of Dr. Bruce Magun (Oregon Health and Science University, Portland, OR). Men1−/−, Men1-complemented, and wild-type MEFs were isolated from mice from the 129/Sv background. Unless otherwise indicated, cells were grown in DMEM supplemented with 10% fetal bovine serum, 100 μg/mL streptomycin, 100 units/mL penicillin, 1% nonessential amino acids, and 1% L-glutamine (all from Gibco).
Cell Treatments with DNA Damaging Agents
For radiation treatment, exponentially or synchronously growing (as indicated) MEFs that had been passaged in tandem were irradiated in a 137Cs irradiator or by a germicidal UV lamp (NIS G30T8, 30 W, 80 cm from plates). For chemical mutagen treatment, exponentially growing MEFs of similar passage number were exposed to either 25uM cisplatin for 24 h (Sigma) or 25 ug/mL methyl methanesulfonate (Sigma) for 48 h before further treatment and collection.
Antibodies
For Western blots, anti-phospho-Chk1 (Ser354) (2341), anti-Chk1 (2345), and anti-phospho-p53 (Ser 20) (9287) were obtained from Cell Signaling Technology. Anti-p21 (M-19) (sc-471) and anti-actin (H-196) (sc-7210) were obtained from Santa Cruz Biotechnology. For chromatin immunoprecipitation, anti-menin (A300-105A) and anti-MLL1 (A300-086A) were obtained from Bethyl Laboratories, anti-p53 (DO-1 X) (sc-126X) was obtained from Santa Cruz Biotechnology, and normal mouse and rabbit IgGs were contained in the Upstate EZ-Chip kit (17–371). The following secondary antibodies were used: horseradish peroxidase (HRP)-conjugated anti-rabbit IgG (Amersham), and AlexaFluor 594-conjugated anti-rabbit IgG (Molecular Probes). For BrdU staining, FITC-conjugated anti-BrdU was obtained from Becton Dickinson.
Cell Cycle Analysis
For DNA content timecourse, cells were synchronized in DMEM+0.1%FBS for 48 hours, released into DMEM+10%FBS+200ng/mL nocodazole (Sigma), irradiated to 7.5 Gray (Gy) in a 137Cs irradiator or mock irradiated at 0 h, and collected by trypsinization at indicated timepoints. Following collection, cells were fixed in 70% ethanol, resuspended in 1X PBS, and treated with 10 ul of 1mg/mL RNAse A and 400 ul of 50ug/mL propidium iodide (Sigma). DNA content analysis was done on a BD FACSCalibur four-color analysis cytometer (Becton Dickinson) calibrated using the BD DNA QC kit (Becton Dickinson), and data were analyzed using FloJo software. For BrdU staining, exponentially growing MEFs were treated as described above. Twelve hours after IR and UV irradiation and 24 or 48 hours after exposure to chemical mutagens, medium was exchanged for DMEM plus 10 μL/mL Cell Proliferation Labeling Reagent (Amersham Biosciences) for 15 minutes. Cells were washed with ice-cold PBS, trypsinized, and fixed with 70% ethanol, and treated with 2 N HCl followed by PBS plus 0.3% Triton X-100, washed in PBS plus 0.5% Tween 20 plus 2% bovine serum albumin, and hybridized to FITC-conjugated anti-BrdU antibody (Becton Dickinson, San Jose, CA). Cells were analyzed for FITC fluorescence using a BD FACSCalibur four-color analysis cytometer (Becton Dickinson), and data were analyzed with CellQuest software.
Determination of protein and RNA levels
For western blotting, MEFs were irradiated to 15 Gy in a 137Cs irradiator, collected at indicated timepoints by trypsinization, washed in 1X PBS, and lysed in RIPA buffer + protease inhibitors (complete mini tablet, Roche) + 0.1 mM sodium orthovanadate. If specified, cells were synchronized by treatment with 200 ng/mL nocodazole (Sigma), released into DMEM+10%FBS at 0 hours, irradiated at 6 hours, and protein was collected as described above. Total protein concentration was quantified on a Pharmacia LKB Ultraspec III spectrophotometer, and 50 ug was loaded. Blots were probed using previously described antibodies according to manufacturer specifications. For immunofluorescence staining, cells were grown on glass slides, fixed in 4% formaldehyde, washed with PBS+0.3% Triton-X, hybridized with antibody at manufacturer-specified concentrations in a humidified chamber, and stained with 0.2% 4′,6-diamidino-2-phenylindole (DAPI) to visualize nuclei. For quantification, cell numbers from at least two separate viewfields from each of three independent experiments were counted (at least 1800 cells per sample). For RT-PCR analysis, RNA was extracted using TRIZOL reagent from Invitrogen, quantified via UV spectrophotometry, and used with the Titan One-Tube RT-PCR system (Roche). In MEFs, exponentially growing cells were irradiated to 15 Gy in a 137Cs irradiator or mock-irradiated, and collected at indicated timepoints by trypsinization. Primer sequences given as Supplementary Material.
Chromatin Immunoprecipitation
The EZ Chip Chromatin Immunoprecipitation Kit from Upstate (17–371) was used according to the manufacturer-specified protocol with the following exceptions. 2×106 Men1−/− and complemented MEFs were used per ChIP reaction, following no treatment or 2 hours following irradiation with 15 Gy in a 137Cs irradiator. Briefly, chromatin was extracted as such: cells were treated with 1% formaldehyde for 10 m, and glycine was added to a final concentration of 0.125 M for 5 minutes. Cells were then collected by trypsinization, washed in 1X PBS, treated with hypotonic buffer (20 mM Hepes pH 7.9, 10 mM KCl, 1mM EDTA pH 8, 10% glycerol, 1 mM DTT, 0.1 mM sodium orthovanadate) + protease inhibitors (complete mini tablet, Roche) on ice for 15 m, and Dounce homogenized 30 strokes on ice. Nuclei were collected by centrifugation and lysed in RIPA buffer + 1 mM DTT, 0.1 mM sodium orthovanadate) + protease inhibitors (complete mini tablet EDTA-free, Roche) on ice for 20 m. Nuclear lysate was sonicated with a Virsonic sonicator to yield fragments between 200–1500 bp. Sonicated samples were treated according to kit instructions. Semi-quantitative PCR was performed on purified DNA (33 cycles at Tm=59 for all primer sets in anti-menin and anti-MLL precipitations; 30 cycles at Tm=57 for all primer sets in anti-p53 precipitation) and band intensity was quantified using AlphaEase PC AccuFluor software. Primer sequences given as Supplementary Material.
Transfection & Expression of p21
Wild-type p21 on the Tet-repressible plasmid pUHD10 was a gift from the laboratory of Dr. Bernard Ducommun (University of Toulouse, Toulouse, France), pTet-Ttak transactivator plasmid was obtained from Life Technologies, and TK-Hyg plasmid was a gift from the laboratory of Dr. Peter Glazer (Yale University, New Haven, CT). Cells were co-transfected using Lipofectamine (Invitrogen) according to manufacturer protocol, clones were selected in the presence of Hygromycin B (30 ug/mL, Invitrogen) and tetracycline (2mM, Sigma), and screened by PCR and Western Blot as described above. To induce p21 from this construct 6 h following ionizing radiation, mimicking the wild-type response, tetracycline was removed from the media 18 h before IR treatment.
4: RESULTS
Men1-mutant MEFs display anomalous cell cycle arrest following DNA damage
MEFs deficient for menin fail to exhibit S-phase arrest in response to ionizing radiation (8). While wild-type cells exposed to 7.5 Gy of IR halt DNA synthesis, shown by an abrogation of BrdU incorporation, menin-mutant cells continue to synthesize DNA despite challenge, indicative of an S-phase arrest defect (8). To further characterize the nature of the cell cycle arrest defect in menin-deficient cells, we performed a timecourse analysis of DNA content in MEFs both at baseline and following ionizing radiation in cell populations with equivalent doubling times.
We synchronized menin-deficient cells and menin-deficient cells complemented with wild type menin by serum starvation. Immediately after release from starvation (0 hours) we irradiated the cells with 7.5 Gy and evaluated the DNA content at 4 hour intervals up to 24 hours (figure 1A). The menin-complemented MEFs exhibited a significant retention of cells within G1 and S populations (25 and 40 percent, respectively) through 24 hours following release, indicating a robust checkpoint response constraining cells within G1 and S. The menin-deficient MEFs, on the other hand, exhibited near-complete entry into G2 by 24 hours with only slight retention of G1 and S populations (7 and 13 percent, respectively). DNA content analysis in spontaneously immortalized wild-type cells with a doubling time of 24 hours revealed kinetics similar to those seen in the complemented cells, indicating that the behavior of menin-complemented cells is comparable to an independent wild-type line (Supplementary Figure 1). This time course indicates that loss of menin compromises both the intra-S checkpoint and the G1/S transition checkpoint.
Figure 1. Menin is required for G1/S and intra-S arrest in response to ionizing radiation.

A) Men1−/− MEFs and the same MEFs complemented with wild type MEN1 were synchronized, exposed to 7.5 Gy of ionizing radiation, and sampled for DNA content at 4 hour intervals. Complemented cells arrest normally while Men1−/− cells progress through the G1/S transition and through S phase. In the absence of radiation exposure, these two cell lines progress through the cell cycle with comparable kinetics. However, Men1−/− cells exhibit a slightly accelerated G1/S transition (13–20 h), compared to menin-complemented cells. B) Men1−/− cells exhibit compromised S-phase arrest in response to ionizing radiation and the crosslinking agent cisplatin (all p<0.02), but are competent for arrest in response to UV and the alkylating agent MMS. Menin-complemented cells are competent for arrest in response to all mutagens. All figures are representative of 3 independent replicates.
Cell cycle progression at baseline was largely comparable between mutant and menin-complemented cells. Both populations began to enter S phase at 13 hours post-release, indicated by spreading of the discrete G1 peak towards increased DNA content, and were largely in G2 by 20 hours. However, a slightly accelerated progression into S phase was observed in the menin-deficient cells (Figure 1A), consistent with what has been previously reported (7). Disappearance of the G1 peak consistently occurred by 17 hours in the mutant population, while the menin-complemented population exhibited a portion of cells remaining in G1 up to 19 or 20 hours following release. As above, an independent wild-type cell line showed kinetics comparable to the complemented line (Supplementary Figure 1).
The anomalous arrest phenotype in Men1 mutants is mutagen-specific
The hypersensitivity profile of menin deficiency is limited to ionizing radiation and crosslinking agents (5). We therefore questioned if the anomalous arrest phenotype was similarly mutagen-specific. Using BrdU incorporation as a readout for S-phase arrest, we compared the arrest responses of Men1−/− and menin-complemented MEFs in response to an array of genotoxic challenges: 7.5 Gy ionizing radiation, 1 minute UVC irradiation (see Methods), 25 uM cisplatin, or 25 ug/mL methyl methanesulfonate (MMS). In response to both ionizing radiation and cisplatin, Men1 mutant cells exhibited no significant decrease in BrdU incorporation, in contrast to a significant decrease in their menin-complemented counterparts, indicative of an arrest defect in response to these two mutagens. In response to UVC irradiation and the alkylating agent MMS, on the other hand, Men1−/− cells exhibited a comparable level of a decrease in BrdU incorporation to the menin-complemented cells, indicating that arrest in response to these other agents is intact in the mutant (Figure 1B). These data suggest that the mutagen specificity of the arrest defect in the menin mutant is the same as the hypersensitivity profile: exclusive sensitivity to ionizing radiation and crosslinking agents. Furthermore, E6/E7 immortalized wild-type MEFs are competent for arrest in response to all genotoxic challenges, illustrating that the arrest defect observed in menin-deficient cells is not due to an artifact of immortalization, and that complemented cells do not compensate for an E6/E7-dependent abrogation of arrest by overexpressing menin (Supplementary Figure 2).
The core ATR checkpoint pathway remains intact in menin mutants
The deficient arrest phenotype seemed to indicate a disruption of core arrest signaling pathways. Therefore, we first set out to determine if menin functioned upstream of these pathways, reconnoitering lesions or creating activating or recruiting marks on histones, by assaying the state of those pathways by Western blotting for known activation epitopes within the ATR-mediated axis of PIKK checkpoint response. We chose this specific pathway because while both ATM and ATR can respond to the type of damage caused by ionizing radiation and crosslinking agents, it is becoming increasingly clear that ATR participates in a specialized branch of crosslink-specific signaling and repair (17), making it an attractive candidate for a menin-involved pathway. In addition, cells with deficiencies in ATM signaling are characterized by deficiency in recombination and double-strand break repair (18), while models lacking menin are competent for both. Menin-mutant Drosophila perform meiotic recombination and are not sensitive to DSBs caused by P element excision (5), and Men1−/−MEFs form normal H2AX and Rad51 foci in response to IR and crosslinking agent treatment (Marek & Bale, unpublished data). We probed for phosphorylation of Chk1 at serine 345, an ATR-specific phosphorylation event (19) at 1 & 2 h following genotoxic insult; and phosphorylation of p53 at serine 20, a Chk1-specific phosphorylation event (20) at 2 & 4 h. Following 15 Gy ionizing radiation, a robust increase in phosphorylation was present in both mutant cells and cells complemented with wild-type menin. Indeed, mutants displayed comparable (p53) or even increased (Chk1) levels of phosphorylation on these residues compared to menin-complemented cells, and a low but consistent level of activation in the absence of damage which was not present in menin-complemented cells, both of which likely reflect accumulation of damage in the absence of an intact checkpoint (Figure 2A). p53 phosphorylation was not detected in complemented cells at lower doses of irradiation, but Chk1 phosphorylation was robust in mutant and complemented cells at a dose of 10 Gy as well (data not shown). Furthermore, while the radiation dose used for western blot analysis (15 Gy) was higher than that used for cell cycle analysis (7.5 Gy), menin-deficient MEFs exhibit radioresistant DNA synthesis even at the higher dose, indicating that failure to arrest is not a consequence of low-dose dependence (data not shown).
Figure 2. p21 is not upregulated post-damage in menin mutants.
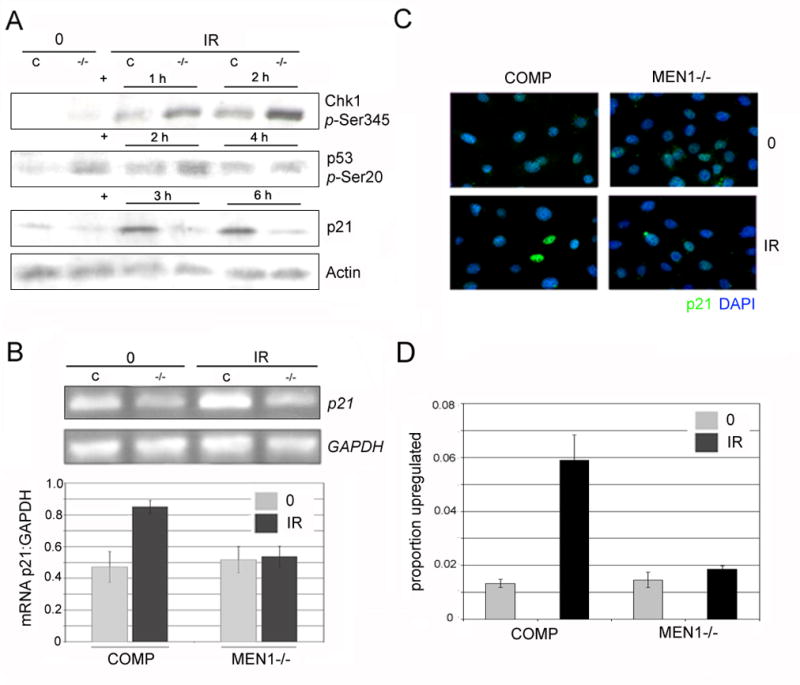
A. Men1−/− MEFs show normally activated ATR signaling but do not upregulate p21. Western blot for activation-specific phospho-epitopes following 15 Gy IR shows hyperphosphorylation of Chk1 (1 & 2 h post-IR) and normal phosphorylation of p53 (2 & 4 h post-IR) in Men1−/− (−/−) cells compared to those complemented with wild-type menin (C). At both 3 & 6 h post-IR, increased levels of p21 protein are observed in cells complemented with wild-type menin, but not in menin-deficient cells. B. Lack of p21 upregulation in Men1−/−MEFs occurs at the mRNA level. 2 h post-IR, cells complemented with wild-type menin exhibit increased levels of p21 mRNA; Men1−/− cells show no such upregulation. Quantitation of RT-PCR comparing p21 mRNA levels to Gapdh levels shows an increase in p21 mRNA in cells complemented with wild-type menin and no increase in Men1−/− cells. C. In complemented cells, p21 is robustly upregulated in single cells 4 h after IR; this increase is absent in Men1−/− cells. p21 protein levels were assayed by immunofluorescence, with p21 secondary antibody (AlexaFluor-594, green) overlaid on nuclear stain (DAPI, blue). D. Quantitation of p21 upregulation at the single cell level confirms that Men1−/− cells fail to upregulate p21 (p<.01); Men1-complemented (COMP) cells exhibit significant upregulation. All figures are representative of 3 independent replicates; at least 1800 cells were counted per sample.
Upregulation of p21 is decreased in menin mutants
The primary target of activated p53 in the DNA damage/checkpoint response is the cyclin-dependent kinase inhibitor p21 (2). As menin has been reported to affect the regulation of other CDK inhibitors - p18 and p27 - at baseline (16), we set out to determine if the damage-dependent upregulation of p21 was impaired in menin mutants. At the protein level, menin-complemented cells exhibit a marked increase in p21 levels 3 and 6 hours after treatment with 15 (Figure 2A) or 10 (data not shown) Gy ionizing radiation, while p21 levels in the menin mutant fail to increase at the same timepoints. At baseline, there is no significant reduction in p21 levels (Figure 2A). p21 protein is also upregulated 6 h following ionizing radiation in the E6/E7 immortalized wild-type cells to a level similar to that seen in complemented cells (Supplementary Figure 5). To distinguish between defects in transcription, translation, and protein stability, we performed semi-quantitative RT-PCR on total RNA extracted from mutant and menin-complemented cells 2 and 4 hours following ionizing radiation. At 2 h following 1.5 krad IR, p21 RNA levels increased as expected in control cells, while transcripts from menin mutant cells remained at levels close to those at baseline, indicating that the decrease in p21 stems from the transcriptional level (Figure 2B). The increased levels of p21 RNA in cells complemented with wild-type menin persisted at 4 h following IR, and were still absent in the mutant (data not shown).
Finally, to establish whether the difference in levels between menin-complemented and menin-deficient cells occurs due to a slight modulation in the population as a whole, or due to failure to drastically upregulate expression in single, damaged cells, we performed immunofluorescence staining on p21. In all cells, p21 exhibited the expected nuclear localization, with slight variation between cells at baseline likely indicative of cell cycle stage and a low level of p21 upregulation likely due to endogenously occurring damage. Following ionizing radiation, a discrete subset of cells complemented with wild-type menin exhibited a strong increase in staining. Nearly identical levels of upregulation to those found in menin-complemented cells were observed in E6/E7 immortalized wild-type cells (Supplementary Figure 3), ruling out the possibility that such upregulation is caused by individual complemented cells overexpressing menin, though complemented cells in fact express menin consistently between cells and at levels comparative to the wild-type line (Supplementary Figure 4A–B). The single cells with strong p21 signal were not found in the irradiated mutant sample, suggesting that in the absence of menin, damaged cells are unable to significantly upregulate p21 (Figure 2C, D).
Menin binds to the p21 gene in a DNA damage-dependent manner
To establish whether menin is a direct transcriptional regulator of p21, or if the decrease in p21 mRNA levels was due to a secondary effect of menin loss, we performed chromatin immunoprecipitation (ChIP) using an α-menin antibody on chromatin extracted from cells complemented with wild-type menin both at baseline and following damage. We created primers corresponding to the promoter region of p21, just upstream of the transcriptional start site, and to an early intragenic region (Figure 3A). At baseline, α-menin failed to precipitate DNA corresponding to these regions of p21.
Figure 3. Menin is a transcriptional regulator of p21 in response to DNA damage.

A. Primers to regions −208 to −12 upstream of the p21 transcriptional start site and at the end of the first intron were designed for PCR amplification. B. Menin binds to the probed regions of p21 in a damage-dependent manner. 2 h following 15 Gy IR or at baseline, chromatin was precipitated with either rabbit IgG or α-menin antibody. In cells complemented with wild-type menin, enrichment of p21 promoter sequences is observed after IR; in menin-null cells, no amplification is observed. C. Measurement of band intensity following semi-quantitative PCR of ChIP products and input chromatin shows significant enrichment of p21 promoter sequences in DNA precipitated using α-menin following IR in cells complemented with wild-type menin, and not in menin-null cells. All p<0.05, and all figures are representative of 3 independent replicates.
Experiments conducted on chromatin extracted 2 h following treatment with 15 Gy of ionizing radiation showed a marked increase in menin binding to these regions. No localization of menin to the Gapdh promoter was observed in either case, indicating that this binding is not due to either nonspecific interactions or a global association with transcriptionally active promoters. Similarly, no significant interaction of rabbit IgG with any assayed regions was observed (Figure 3B). Quantitation of amplification of p21 and Gapdh regions in immunoprecipitated samples compared to that of input showed a significant enrichment of p21 promoter sequences in menin-precipitated samples, specifically after IR, to a level of approximately 0.4% (Figure 3C), comparable to values found for menin binding to other CDKIs at baseline (15).
MLL binds to the p21 gene in a DNA damage-dependent and menin-dependent manner
Menin has been shown to act as a transcriptional comodulator, acting in concert with other transcription factors or complexes to regulate gene transcription, and is not thought to influence transcription alone. It has been previously shown that menin can recruit the MLL HMT, and that the CDKIs p27 and p18 are regulated at baseline in this manner. We therefore set out to determine if the MLL complex was mediating menin’s action at the p21 locus by performing ChIP in cells complemented with menin using an α-MLL antibody. We observed MLL binding to the p21 locus following ionizing radiation, but not at baseline, identical to the gross binding kinetics observed for menin (Figure 4). To determine if this association was menin dependent, we repeated the ChIP experiments in menin-null cells, and observed near-abrogation of MLL binding to the promoter region (Fragment 1) and the early intragenic region (Fragment 2), suggesting that menin is required for robust MLL binding to these regions (Figure 4A). As above, no amplification of the Gapdh promoter was observed, and precipitation with rabbit IgG yielded no significant amplification with any primer pairs. Quantitation of amplification of p21 and Gapdh regions in immunoprecipitated samples compared to that of input showed a significant increase in p21 promoter sequences in MLL-precipitated samples from IR-treated, menin-complemented cells: close to 0.4% of input (Figure 4B), consistent with reported values for MLL-binding to CDKI loci at baseline in menin-mutant cells complemented with wild-type menin. MLL expression has been shown to be decreased in the menin mutant compared to wild-type; however, in stable menin-complemented lines, this difference is greatly attenuated (16), and so lack of MLL binding in menin-null cells is likely a true abrogation due to menin-dependent targeting and not merely an artifactual consequence of lower MLL levels.
Figure 4. MLL binds the p21 promoter post-damage in a menin-dependent manner.

A. Following IR, MLL binds to p21. In Men1−/− cells, the association is lost at the promoter and early intragenic regions of p21. 2 h following 15 Gy IR or at baseline, chromatin was precipitated with either rabbit IgG or α-MLL antibody. In cells complemented with wild-type menin, enrichment of p21 promoter sequences is observed after IR; in menin-null cells, no significant amplification is observed. B. Measurement of band intensity following semi-quantitative PCR of ChIP products and input chromatin shows significant enrichment of p21-specific sequences in DNA precipitated using α-MLL following IR in cells complemented with wild-type menin, and not in menin-null cells. All p<0.05, and all figures are representative of 3 independent replicates.
p53 binding to p21 is unchanged in the mutant
It was, however, unclear whether disrupted MLL localization to the p21 promoter was affecting upstream signaling in the form of p53 binding, or downstream transcriptional events. To distinguish between these possibilities, we used ChIP to assay for p53 binding. In the mouse, there is a distal consensus binding sequence for p53 at −2.85 kb from the transcriptional start site (TSS) of p21 (21). In menin-complemented cells, we observed an enrichment in p53 binding at this site following ionizing radiation, as would be expected. In menin-mutant cells, we observed a similar pattern, indicating that p53 binding of p21 is retained in the mutant. Significant amplification was observed for neither Gapdh in any IP’ed sample, nor IP with normal IgG for any primer pair (Figure 5A). Quantitation of amplification of immunoprecipitated samples compared to that of input showed equivalent binding levels between cells mutant for and complemented with wild-type menin (Figure 5B). These data suggest that loss of MLL binding to p21 in the absence of menin, and the resulting abnormality in promoter architecture, does not affect the upstream signaling events through p53 stabilization and binding. These data are consistent with both menin and MLL binding near the TSS, while p53 activation involves binding further upstream of this region.
Figure 5. p53 retains the ability to bind the p21 regulatory sequences in menin-deficient cells.

A. Following IR, p53 binds −2.8 kb upstream of the p21 transcriptional start site, an established consensus binding site, in both cells complemented with wild-type menin and those deficient for menin. No binding was seen at baseline for either cell type. Chromatin immunoprecipitation was performed on these cells at baseline and 3 hours following 15 Gy IR using mouse IgG and α-p53 antibodies. B. Measurement of band intensity following semi-quantitative PCR of ChIP products and input chromatin shows comparable enrichment of p21 promoter sequences in samples precipitated using α-p53 following IR in both wild-type and Men1−/− cells. All p<0.05, and all figures are representative of 3 independent replicates.
Restoring p21 expression restores arrest in the mutant
To ensure that loss of menin does not affect p21 levels anywhere downstream of transcription, we set out to determine if Men1-mutant cells were capable of arrest when supplemented with exogenous p21 expression. We created a system to artificially upregulate p21 via a tetracycline (Tet)-repressible promoter contemporaneously with IR treatment, mimicking the wild-type p21 response. 24 hours following subtraction of tetracycline from the culture medium, Men1−/− cells containing Tet-repressible wild-type p21 exhibited p21 protein levels comparable to those of both E6/E7 immortalized wild-type and Men1-complemented cells 6 hours after exposure to ionizing radiation (Supplementary Figure 5). We then questioned if this “upregulation” could restore the arrest response in the mutant. Indeed, if p21 was induced in the mutant by subtracting tetracycline from the media 18 h prior to IR treatment, RDS was eliminated and mutant cells showed a decrease in BrdU incorporation to levels similar to that found in menin-complemented cells (Figure 6). These data are not only consistent with low levels of p21 having a causative effect on the arrest defect, but also suggest that menin has no posttranscriptional role.
Figure 6. Restoration of p21 levels following IR rescues arrest in the menin mutant.
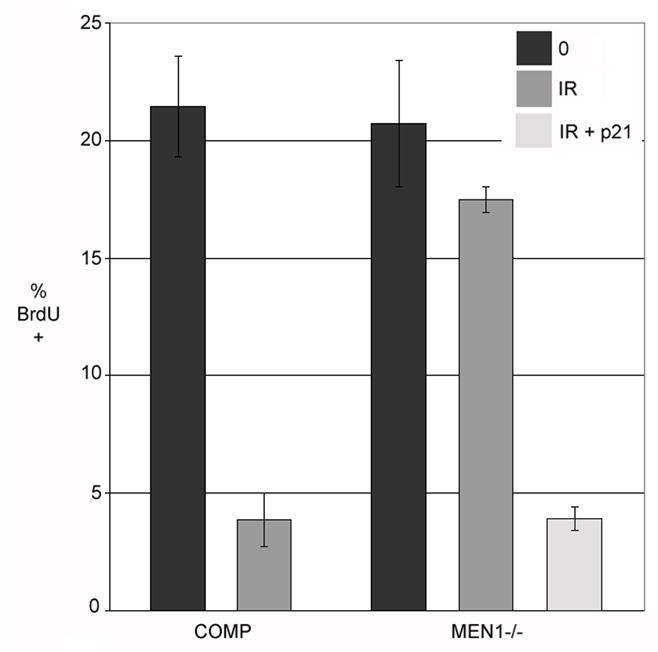
A decrease in BrdU incorporation, indicative of S-phase arrest, is observed in the p21-expressing Men1−/− cells, comparable to cells complemented with wild-type menin. Quantitation of the number of BrdU positive cells in cells complemented with wild-type menin, cells null for menin, and cells null for menin but expressing exogenous p21 following IR, indicates effective rescue by p21 (all p<0.02).
5: DISCUSSION
Studies of menin have suggested that it can play several, possibly overlapping roles in the cell including transcriptional control, chromatin modification, and DNA damage response. The connection between these processes has yet to be fully elucidated. In various cell culture and model organism systems, menin has been shown to bind and modulate the activity of numerous transcription factors and both histone deacetylase and histone methyltransferase complexes. Its role in the DNA damage response is supported by marked dysregulation of replication and repair in its absence. Both Drosophila and MEFs null for menin exhibit mild hypersensitivity to ionizing radiation, extreme hypersensitivity to crosslinking agents, hypermutability in response to both agents, and radioresistant DNA synthesis.
In this work, we show that menin deficiency causes a pervasive arrest defect in response to ionizing radiation, with challenged cells progressing through both the G1/S transition and DNA replication and halting only in G2. Further, the mutagen specificity of the arrest defect – in response only to ionizing radiation and crosslinking agents – is identical to the hypersensitivity profile. Abrogated arrest suggests a defect in checkpoint signaling; however, all of the upstream phosphorylation events probed were intact in the menin mutant. Indeed, hyperphosphorylation of both Chk1 and p53 was observed. This likely reflects an accumulation of damage in the mutant, consistent with the hypermutability phenotype seen in Drosophila (5, 22). It is unclear whether such damage accumulation results from the abrogation of G1 and S checkpoints demonstrated in this work, or from a separate repair function; however, the robust pathway activation suggests that in either case there is no role for menin upstream of core ATR signaling. Although p53 in menin-deficient cells is stabilized by posttranslational modification as in menin-complemented cells, its target p21 is not upregulated at the transcriptional level following damage. Accordingly, p53 retains its ability to bind its consensus site upstream of p21 even in the absence of menin, establishing a model in which p53, commonly mutated in cancer, remains active but cannot promote translation of its key target in the checkpoint transcriptional program.
Chromatin immunoprecipitation experiments placed menin physically at the promoter region of p21 specifically following ionizing radiation, indicating that the decrease in p21 levels is due to a direct transcriptional function rather than a secondary effect. MLL also associated with the same regulatory regions of p21 following IR, and in a menin-dependent manner, consistent with menin being an activating or targeting cofactor of this complex, though it remains unclear if menin targeting is sequence specific, and if so, how that specificity is altered by DNA damage. A compromised damage-dependent upregulation of p21 has been demonstrated in MLL leukemic fusions (23), and while these fusions retain a high level of transcriptional activity, the dampening effect on p21 expression has been attributed to a direct inhibition of p53 acetylation by p300. Destabilization of p53 in this model would likely abrogate p21 expression even in the presence of an active menin-MLL complex.
Our previous work suggested a role for CHES1 and its associated histone modifying complex, HDAC, in checkpoint response, since overexpression of CHES1 in Drosophila rescued the radioresistant DNA synthesis caused by loss of menin. CHES1 has been implicated as a HDAC inhibitor (24). p21 expression is sensitive to HDAC inhibition, and nonspecifically inhibiting HDACs will derepress the p21 promoter even in the absence of other activating stimuli like H3K4 methylation (25). Addition of trichostatin A, a potent HDAC inhibitor, increased p21 transcription and induced arrest even in the menin-deficient model (Kottemann and Bale, unpublished data). It is possible that the observed rescue in the CHES1 overexpression model is due to such nonspecific effects, and that the physical interaction reported is due either to promiscuous binding by menin or to a physical association due to some other function not biologically relevant to the checkpoint phenotype. However, it is also possible that menin, which also interacts with HDAC complexes, coordinates histone modification in damage response through multiple complexes, integrating all of these complexes into a coherent transcriptional response.
It is likely that p21, while certainly a key player in checkpoint maintenance, is not the only relevant target of menin in transcriptional response to DNA damage. That additional targets are likely important is underscored by the coincidence of phenotypes between mammalian and fly models – radioresistant DNA synthesis and hypersensitivity – when we consider that the fly p21 homolog and sole identified CDKI, dacapo, is not known to respond to DNA damage ((26), Kottemann and Bale, unpublished data). Similarly, the Drosophila homolog of p53, Dmp53, is primarily a regulator of apoptosis in the damage response and seems to be dispensable for inducing cell cycle arrest (27). The major transcriptional regulator of cell cycle in Drosophila is the Rb/E2F pathway (28). It seems likely that damage-dependent histone modification can influence transcription in this context as it does with p53-based transcription in the mammalian system, and in this sense perhaps the function of menin is conserved throughout metazoans if not the precise targets. Further exploration of menin’s involvement in chromatin modification in Drosophila will be necessary to address this conundrum.
Although a comprehensive view of menin’s role within a range of disparate and connected processes remains to be seen, this work establishes a link between its functions in transcriptional regulation and DNA damage response. It is possible that the mechanism of p21 transcription may in fact influence the hypermutability seen in menin-mutant models as well. p21 can regulate the activity of error-prone translesion synthesis polymerases via PCNA binding, and a decrease in p21 levels causes an increase in mutation load (29). Menin deficiency causes a mutation spectrum reminiscent of that incurred due to dysregulated translesion synthesis (21); perhaps menin’s DNA damage-dependent upregulation of p21 underlies both the cell cycle abnormality and the hypermutability observed in its absence.
Supplementary Material
An independent, spontaneously immortalized line of wild-type MEFs (wti) progress through the cell cycle at baseline and following ionizing radiation with comparable kinetics to the menin-complemented cells. These wild-type cells arrest following treatment with 7.5 Gy ionizing radiation at 0 hours, with retention of cells with G1 (30%) and S (35%) DNA content at 24 hours. During baseline S phase (13–20 h), spontaneously immortalized wild-type cells exhibit a G1/S transition similar to that seen in complemented cells, with Men1−/− cells progressing faster. Cells were synchronized by serum starvation, and collected and analyzed at indicated intervals after release.
A wild-type cell line immortalized by E6/E7, as are the menin and menin-complemented cells, is competent for S phase arrest in response to IR, cisplatin, UVC irradiation, and MMS treatments, comparable to the behaviour of menin-complemented cells.
Immunofluorescence analysis of p21 upregulation post-IR in menin-complemented and E6/E7 immortalized wild-type cells shows a comparable increase in p21-positive cells following damage in the two lines, in contrast to the absence of upregulation seen in the menin mutant cells.
A. Protein immunoprecipitated from Men1-mutant, E6/E7 immortalized wild-type, and Men1-complemented cell extracts, then blotted with anti-menin, shows both specificity of the antibody and comparable levels of menin in wild-type and Men1-complemented cells. B. Immunofluorescence visualization of menin levels in Men1-mutant, wild-type, and Men1-complemented cells shows that complemented cells express menin at similar levels on a cell-to-cell basis, comparable to wild-type.
Inducing exogenous p21 expression in the Men1-mutant from a tetracycline-repressible promoter contemporaneously with IR treatment yields p21 upregulation comparable to that induced endogenously in E6/E7 immortalized wild-type cells and complemented cells following IR treatment.
Acknowledgments
Grant Support: NIH grants R01-GM66079 (A.E. Bale), and T32-HD07149-29 (M.C. Kottemann)
We thank Drs. Xianxin Hua, Tony Koleske, and Bruce Magun for kind gifts of MEF cell lines, Dr. Bernard Ducommun for gift of p21 constructs, and Allison Clark and Drs. Mark Solomon and David Stern for helpful discussion.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
6: REFERENCES
- 1.Zhou BB, Elledge SJ. The DNA damage response: putting checkpoints in perspective. Nature. 2000;408:433–439. doi: 10.1038/35044005. [DOI] [PubMed] [Google Scholar]
- 2.Bartek J, Lukas J. Mammalian G1- and S-phase checkpoints in response to DNA damage. Curr Opin Cell Biol. 2001;13:738–747. doi: 10.1016/s0955-0674(00)00280-5. [DOI] [PubMed] [Google Scholar]
- 3.Gartel AL, Tyner AL. Transcriptional regulation of the p21((WAF1/CIP1)) gene. Exp Cell Res. 1999;246:280–289. doi: 10.1006/excr.1998.4319. [DOI] [PubMed] [Google Scholar]
- 4.Yang Y, Hua X. In search of tumor suppressing functions of menin. Mol Cell Endocrinol. 2007;265–266:34–41. doi: 10.1016/j.mce.2006.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Busygina V, Suphapeetiporn K, Marek LR, Stowers RS, Xu T, Bale AE. Hypermutability in a Drosophila model for multiple endocrine neoplasia type 1. Hum Mol Genet. 2004;13:2399–2408. doi: 10.1093/hmg/ddh271. [DOI] [PubMed] [Google Scholar]
- 6.Jin S, Mao H, Schnepp RW, Sykes SM, Silva AC, D’Andrea AD, Hua X. Menin associates with FANCD2, a protein involved in repair of DNA damage. Cancer Res. 2003;63:4204–4210. [PubMed] [Google Scholar]
- 7.Schnepp RW, Chen YX, Wang H, Cash T, Silva A, Diehl JA, Brown E, Hua X. Mutation of tumor suppressor gene Men1 acutely enhances proliferation of pancreatic islet cells. Cancer Res. 2006;66:5707–5715. doi: 10.1158/0008-5472.CAN-05-4518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Busygina V, Kottemann MC, Scott KL, Plon SE, Bale AE. Multiple endocrine neoplasia type 1 interacts with forkhead transcription factor CHES1 in DNA damage response. Cancer Res. 2006;66:8397–8403. doi: 10.1158/0008-5472.CAN-06-0061. [DOI] [PubMed] [Google Scholar]
- 9.Agarwal SK, Guru SC, Heppner C, Erdos MR, Collins RM, Park SY, Saggar S, Chandrasekharappa SC, Collins FS, Spiegel AM, Marx SJ, Burns AL. Menin interacts with the AP1 transcription factor JunD and represses JunD-activated transcription. Cell. 1999;96:143–152. doi: 10.1016/s0092-8674(00)80967-8. [DOI] [PubMed] [Google Scholar]
- 10.Heppner C, Bilimoria KY, Agarwal SK, Kester M, Whitty LJ, Guru SC, Chandrasekharappa SC, Collins FS, Spiegel AM, Marx SJ, Burns AL. The tumor suppressor protein menin interacts with NF-kappaB proteins and inhibits NF-kappaB-mediated transactivation. Oncogene. 2001;20:4917–4925. doi: 10.1038/sj.onc.1204529. [DOI] [PubMed] [Google Scholar]
- 11.Kaji H, Canaff L, Lebrun JJ, Goltzman D, Hendy GN. Inactivation of menin, a Smad3-interacting protein, blocks transforming growth factor type beta signaling. Proc Natl Acad Sci U S A. 2001;98:3837–3842. doi: 10.1073/pnas.061358098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gobl AE, Berg M, Lopez-Egido JR, Oberg K, Skogseid B, Westin G. Menin represses JunD-activated transcription by a histone deacetylase-dependent mechanism. Biochim Biophys Acta. 1999;1447:51–56. doi: 10.1016/s0167-4781(99)00132-3. [DOI] [PubMed] [Google Scholar]
- 13.Kim H, Lee JE, Cho EJ, Liu JO, Youn HD. Menin a tumor suppressor represses JunD-mediated transcriptional activity by association with an mSin3A-histone deacetylase complex. Cancer Res. 2003;63:6135–6139. [PubMed] [Google Scholar]
- 14.Yokoyama A, Wang Z, Wysocka J, Sanyal M, Aufiero DJ, Kitabayashi I, Herr W, Cleary ML. Leukemia proto-oncoprotein MLL forms a SET1-like histone methyltransferase complex with menin to regulate Hox gene expression. Mol Cell Biol. 2004;24:5639–5649. doi: 10.1128/MCB.24.13.5639-5649.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hughes CM, Rozenblatt-Rosen O, Milne TA, Copeland TD, Levine SS, Lee JC, Hayes DN, Shanmugam KS, Bhattacharjee A, Biondi CA, Kay GF, Hayward NK, Hess JL, Meyerson M. Menin associates with a trithorax family histone methyltransferase complex and with the hoxc8 locus. Mol Cell. 2004;13:587–597. doi: 10.1016/s1097-2765(04)00081-4. [DOI] [PubMed] [Google Scholar]
- 16.Milne TA, Hughes CM, Lloyd R, Yang Z, Rozenblatt-Rosen O, Dou Y, Schnepp RW, Krankel C, Livolsi VA, Gibbs D, Hua X, Roeder RG, Meyerson M, Hess JL. Menin and MLL cooperatively regulate expression of cyclin-dependent kinase inhibitors. Proc Natl Acad Sci U S A. 2005;102:749–754. doi: 10.1073/pnas.0408836102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mladenov E, Tsaneva I, Anachkova B. Activation of the S phase DNA damage checkpoint by mitomycin C. J Cell Physiol. 2007;211:468–476. doi: 10.1002/jcp.20957. [DOI] [PubMed] [Google Scholar]
- 18.Lavin MF, Birrell G, Chen P, Kozlov S, Scott S, Gueven N. ATM signaling and genomic stability in response to DNA damage. Mutat Res. 2005;569:123–132. doi: 10.1016/j.mrfmmm.2004.04.020. [DOI] [PubMed] [Google Scholar]
- 19.Zhao H, Piwnica-Worms H. ATR-mediated checkpoint pathways regulate phosphorylation and activation of human Chk1. Mol Cell Biol. 2001;21:4129–4139. doi: 10.1128/MCB.21.13.4129-4139.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shieh SY, Ahn J, Tamai K, Taya Y, Prives C. The human homologs of checkpoint kinases Chk1 and Cds1 (Chk2) phosphorylate p53 at multiple DNA damage-inducible sites. Genes Dev. 2000;14:289–300. [PMC free article] [PubMed] [Google Scholar]
- 21.Macleod KF, Sherry N, Hannon G, Beach D, Tokino T, Kinzler K, Vogelstein B, Jacks T. p53-dependent and independent expression of p21 during cell growth, differentiation, and DNA damage. Genes Dev. 1995;9:935–944. doi: 10.1101/gad.9.8.935. [DOI] [PubMed] [Google Scholar]
- 22.Marek LR, Kottemann MC, Glazer PM, Bale AE. MEN1 and FANCD2 mediate distinct mechanisms of DNA crosslink repair. DNA Repair (Amst) 2008 doi: 10.1016/j.dnarep.2007.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wiederschain D, Kawai H, Shilatifard A, Yuan ZM. Multiple mixed lineage leukemia (MLL) fusion proteins suppress p53-mediated response to DNA damage. J Biol Chem. 2005;280:24315–24321. doi: 10.1074/jbc.M412237200. [DOI] [PubMed] [Google Scholar]
- 24.Scott KL, Plon SE. Loss of Sin3/Rpd3 histone deacetylase restores the DNA damage response in checkpoint-deficient strains of Saccharomyces cerevisiae. Mol Cell Biol. 2003;23:4522–4531. doi: 10.1128/MCB.23.13.4522-4531.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gui CY, Ngo L, Xu WS, Richon VM, Marks PA. Histone deacetylase (HDAC) inhibitor activation of p21WAF1 involves changes in promoter-associated proteins, including HDAC1. Proc Natl Acad Sci U S A. 2004;101:1241–1246. doi: 10.1073/pnas.0307708100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brodsky MH, Weinert BT, Tsang G, Rong YS, McGinnis NM, Golic KG, Rio DC, Rubin GM. Drosophila melanogaster MNK/Chk2 and p53 regulate multiple DNA repair and apoptotic pathways following DNA damage. Mol Cell Biol. 2004;24:1219–1231. doi: 10.1128/MCB.24.3.1219-1231.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jaklevic BR, Su TT. Relative contribution of DNA repair, cell cycle checkpoints, and cell death to survival after DNA damage in Drosophila larvae. Curr Biol. 2004;14:23–32. doi: 10.1016/j.cub.2003.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Neufeld TP, de la Cruz AF, Johnston LA, Edgar BA. Coordination of growth and cell division in the Drosophila wing. Cell. 1998;93:1183–1193. doi: 10.1016/s0092-8674(00)81462-2. [DOI] [PubMed] [Google Scholar]
- 29.Livneh Z. Keeping mammalian mutation load in check: regulation of the activity of error-prone DNA polymerases by p53 and p21. Cell Cycle. 2006;5:1918–1922. doi: 10.4161/cc.5.17.3193. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
An independent, spontaneously immortalized line of wild-type MEFs (wti) progress through the cell cycle at baseline and following ionizing radiation with comparable kinetics to the menin-complemented cells. These wild-type cells arrest following treatment with 7.5 Gy ionizing radiation at 0 hours, with retention of cells with G1 (30%) and S (35%) DNA content at 24 hours. During baseline S phase (13–20 h), spontaneously immortalized wild-type cells exhibit a G1/S transition similar to that seen in complemented cells, with Men1−/− cells progressing faster. Cells were synchronized by serum starvation, and collected and analyzed at indicated intervals after release.
A wild-type cell line immortalized by E6/E7, as are the menin and menin-complemented cells, is competent for S phase arrest in response to IR, cisplatin, UVC irradiation, and MMS treatments, comparable to the behaviour of menin-complemented cells.
Immunofluorescence analysis of p21 upregulation post-IR in menin-complemented and E6/E7 immortalized wild-type cells shows a comparable increase in p21-positive cells following damage in the two lines, in contrast to the absence of upregulation seen in the menin mutant cells.
A. Protein immunoprecipitated from Men1-mutant, E6/E7 immortalized wild-type, and Men1-complemented cell extracts, then blotted with anti-menin, shows both specificity of the antibody and comparable levels of menin in wild-type and Men1-complemented cells. B. Immunofluorescence visualization of menin levels in Men1-mutant, wild-type, and Men1-complemented cells shows that complemented cells express menin at similar levels on a cell-to-cell basis, comparable to wild-type.
Inducing exogenous p21 expression in the Men1-mutant from a tetracycline-repressible promoter contemporaneously with IR treatment yields p21 upregulation comparable to that induced endogenously in E6/E7 immortalized wild-type cells and complemented cells following IR treatment.