

Abstract
Implicit solvent models are increasingly popular for estimating aqueous solvation (hydration) free energies in molecular simulations and other applications. In many cases, parameters for these models are derived to reproduce experimental values for small molecule hydration free energies. Often, these hydration free energies are computed for a single solute conformation, neglecting solute conformational changes upon solvation. Here, we incorporate these effects using alchemical free energy methods. We find significant errors when hydration free energies are estimated using only a single solute conformation, even for relatively small, simple, rigid solutes. For example, we find conformational entropy (TΔS) changes of up to 2.3 kcal/mol upon hydration. Interestingly, these changes in conformational entropy correlate poorly (R2 = 0.03) with the number of rotatable bonds. The present study illustrates that implicit solvent modeling can be improved by eliminating the approximation that solutes are rigid.
I. Introduction
Solvation and desolvation processes drive many important biological and chemical processes, including binding, adsorption, protein folding, protein–protein interactions, and membrane formation. Hence, modeling the solvation component is important in computational biology and chemistry. However, because such computer simulations often involve large numbers of atoms and long time scales, it is valuable to compute accurate aqueous solvation (hydration) free energies as quickly as possible.1–8 Because of this need, a common strategy is to use implicit solvent models, where water is treated as a continuum solvent, rather than in explicit molecular detail.
Perhaps the simplest and best-known such model is the Born model of ion solvation, which treats solvent as a high dielectric continuum and solves for the potential of a point charge in a spherical low dielectric cavity. It has proved relatively successful in estimating relative hydration free energies of ions with relatively few parameters9,10 (ionic radii and internal and external dielectric constants). Other approaches include numerical solution of the Poisson–Boltzmann (PB) equation,11–13 which describes the electrostatic interactions of charges embedded in a dielectric continuum, and generalized Born (GB) models, which approximate the PB equation and generalize the Born equation to situations other than a single ion in a uniform dielectric.14,15 A common feature of all of these approaches is that they require parametrization. At minimum, they require values for internal and external dielectrics and atomic radii. GB models often have additional parameters fit to maximize agreement with numerical solution of the PB equation on a test set, or with experimental solvation free energies.
Another class of solvation model is attributed to Cramer, Truhlar, and others and is based on semiempirical quantum mechanics on a solute in a dielectric continuum.16,17 Such models are empirically optimized to reproduce solvation free energies;16,17 these, too, require parametrization.
All of these solvation models have a number of adjustable parameters. Such parameters are obtained in different ways, including (1) optimizing the parameter set to minimize the discrepancy between hydration free energies computed with a single conformer and experimental hydration free energies (or transfer free energies between water and other solvents);10,16–22 (2) optimizing GB models to reproduce single-conformer PB solvation free energies23–27 (in some cases with additional adjustments); (3) optimizing implicit solvent models to reproduce single-conformer hydration or charging free energies calculated from free energy calculations in explicit solvent;28–30 and (4) optimizing implicit solvent models to reproduce forces and/or energies from explicit solvent simulations for a fixed molecular geometry.31
In the first three of these methods, each hydration free energy is computed with a single solute conformation. In reality, a solute adopts an ensemble of conformations in both vacuum and solvent, and that ensemble can differ in the two environments. Thus, using a single solute conformation is an approximation, and neglects the conformational entropy and enthalpy changes of the solute. This may be especially important for approaches 1 and 2, above, where single-conformation results are compared with experimental values. It is not obvious that single-conformation hydration “free energies” should agree with experimental hydration free energies, which include any conformational enthalpic and entropic changes of the solute.
There appears to have been little work on conformational enthalpic and entropic changes in implicit solvent models. One study, on a few compounds, found that computed single-conformation hydration free energies agreed poorly with experiment for two compounds (which were previously believed to change conformation upon solvation) until conformational changes were included.10 Another study found that averaging over multiple conformations did not significantly affect computed water-to-octanol transfer free energies,18 but did not test hydration free energies.
Here, our purpose is to learn the magnitude of errors incurred by fixing solutes into a single conformation when computing hydration free energies with implicit solvent models. We find that computed single-conformation hydration free energies, on average, vary over a range of 1.85 ± 0.08 kcal/mol depending on the solute conformation chosen, and differ substantially from true hydration free energies computed using the same parameters.
II. Theory
A. Hydration Free Energies in Implicit Solvent
We consider the transfer of a single solute molecule from gas (with a 1 M standard state) to water (1 M standard state), at standard pressure and a particular temperature T. Following the notation of Deng and Roux,32 we express the total potential energy U of the solute in water as
| (1) |
where U is the potential energy, and u and v denote solute and solvent, respectively. ru denotes the full set of coordinates of all atoms in the solute, including the system's volume, and rv denotes the similar quantity for the solvent. The classical hydration free energy, or excess chemical potential, is then given by
| (2) |
The integrals run over the full simulation volume. Here, β = 1/kBT, where kB is the Boltzmann constant, and T is temperature. The numerator inside the logarithm is the partition function for the solute in water, and the denominator is the product of the partition functions for the solute in vacuum and for pure water. A key point here is that the hydration free energy involves integrals over all of the relevant (low potential energy) conformations of the solute and the water bath.
The principal approximation made in implicit solvent models is the replacement of the integral over the water degrees of freedom with a solute–water interaction free energy, Gint, effectively integrating out the solvent degrees of freedom. Then the solute experiences an effective potential energy (which already includes solvent entropic effects):
| (3) |
where Ueff denotes an effective energy function that includes the solute potential energy and the interaction free energy (Gint). Gint itself is the sum of two components:
| (4) |
where Gpolar treats the electrostatic interactions of the solute with the solvent (i.e., using continuum electrostatics), and Gnonpolar includes the nonpolar component of solvation. Then eq 2 reduces to
| (5) |
Here, the integrals over the solvent degrees of freedom are already incorporated into the effective potential energy Ueff-(ru).
Equation 5 provides a rigorous way to compute hydration free energies within the framework of an implicit solvent model, including, in principle, solute conformational enthalpic and entropic changes. Essentially, it represents the free energy of turning on the implicit solvent term in an equilibrium ensemble, including any changes to the ensembles sampled as the solvent is turned on.
B. Single-Conformation Hydration Free Energies
It is common to estimate hydration free energies using single solute conformations10,16,18–22 (perhaps for speed, or because conformation is assumed to be unimportant). Because these approaches keep the solute fixed in a single conformation for the transfer process, these are approximate. We refer to this quantity here as a single-conformation hydration free energy, and reserve the term hydration free energy for free energies computed including proper sampling of solute degrees of freedom.
It follows from eq 5 and eq 3 that the single-conformation hydration free energy (eq 4) will be equal to the true hydration free energy in only two situations: (1) the solute prefers the same conformation in both environments, and this is the only relevant conformation, or (2) Ueff is independent of the solute conformation.
When computing the single-conformation hydration free energy, a complication is how to choose which single conformation of the solute should be used.60 We show below that different choices of solute conformations give different single-conformation hydration free energy estimates, but there is only one correct thermodynamic estimate (using eq 5).
III. Previous Work
Alchemical free energy calculations are now commonly used with explicit solvent molecular mechanics models and force fields to compute hydration free energies (for recent examples, see refs 32–37). In the limit of sufficient sampling, these are equivalent to evaluating the ratio of partition functions in eq 2, for the given model and force field.
Alchemical free energy methods can, in principle, be used for implicit solvent hydration free energies. But we are not aware of a single case where this was done. Surprisingly, several studies comparing implicit solvent “hydration free energies” with those from explicit solvent also estimate free energies in this manner: the implicit solvent studies used single conformations, and the explicit solvent studies used alchemical methods.21,22,38
Implicit solvent hydration free energy calculations have typically used single solute conformations. In some cases, these used different single conformations in vacuum and in solvent. It is worth briefly summarizing some of the different approaches commonly used in order to facilitate comparison with our different single-conformation schemes. We have identified several basic approaches: (1) separately optimizing gas-phase and water geometries, and using these different conformations16 (but still neglecting changes in solute entropy); (2) performing quantum mechanical17,20,39 or molecular mechanics18,19 geometry optimizations in the gas phase, and using these single conformations; and (3) using “reasonable” or “low internal energy” solute configurations obtained from different sources, sometimes third parties.22–40 Sometimes single conformations are used with no explanation of their origin10 or some are taken from the Protein Data Bank.21 Finally, it is worth noting that several recent papers have suggested that any single “reasonable” or optimized gas phase or liquid geometry is sufficient.17,39
IV. The Solute Test Set and Methods
A. Overview
We constructed a test set of 504 neutral small molecules with experimentally measured hydration free energies. We ran molecular dynamics simulations for each solute from a variety of different starting conformations, both in a GB water model and in vacuum. We then estimated hydration free energies for these by analyzing the data using both alchemical free energy methods (which evaluate the ratio of partition functions as in eq 5), and by selecting single conformations and using eq 4, in the manner common in the literature.
B. Test Set Selection
We focus here only on neutral solute molecules, rather than on ions.61 For our test set, we took as a starting point the experimental data compiled by Rizzo et al.19 and removed the charged molecules. In an attempt to construct a set of all neutral compounds with known hydration free energies, we added (neutral) amino acid side chain analogues and other compounds from our previous studies in explicit solvent,22,35 and various small molecules from another set of 52 small molecules and associated references provided by J. Peter Guthrie,41 removing redundancies. The result is a set containing 504 neutral small molecules.
The full set of experimental solvation free energies (1 M gas to 1 M water) and associated references are available in the Supporting Information. Most of these solute molecules are relatively small and fairly rigid, with an average of only 1.6 rotatable bonds per molecule, and an average molecular weight of 112 Da. Histograms of the number of rotatable bonds and molecular weight are shown in the Supporting Information.
C. Molecule Preparation
Initial mol2 files for the small molecules in the Bordner et al. subset18 of the Rizzo et al. set were obtained from the Supporting Information from that paper. The remaining mol2 files were generated from their chemical names using the tools nam2mol, babel2, and Omega, from OpenEye Scientific Software. To ensure consistency, conformers for the mol2 files from the Bordner set were generated from the mol2 files using Omega, a conformer-generation program. All OpenEye tools used were those distributed with version 1.5.0 of the OpenEye toolkits.
As a check of the Bordner mol2 files, we converted those mol2 files into chemical names and SMILES strings and compared them with the desired chemical names and SMILES strings. We found that the mol2 files for iodoethane, iodomethane, and butanone actually contained slightly different molecules (iodopropane, iodoethane, and butan-2-one, respectively), so we generated new mol2 files containing the correct molecules.
In this work, protonation states were taken as the default generated by the OpenEye tools, and the protonation state was kept fixed throughout the simulations. It seems likely, however, that some of the molecules in this test set change protonation state upon solvation (acetic acid, for example, which has a pKa around 4.742), and others may have multiple relevant protonation states in solvent. Since protonation states are not the focus of this work, we simply use the default protonation states, and keep these fixed throughout our calculations. This issue should probably be revisited in future work.
D. Simulation Setup
For each molecule, partial charges were generated using the AM1-BCC method43,44 in ANTECHAMBER v1.27 using the first molecular conformation generated by Omega. Various charge models could have been used,35 and our purpose here is to compare results for a particular parameter set, not to optimize the parameters. Likewise, AM1-BCC charges depend somewhat on the solute conformation used to compute the charges, but we did not explore this here. Using other parameter sets or partial charges could change the quality of the overall agreement with experiment, but should not affect our main conclusions.
Following calculation of partial charges, we generated starting conformations using Omega. We subsequently performed five independent sets of simulations (each with a vacuum simulation and a water simulation) for each molecule, so we wanted independent starting conformations for each, when possible, to help assess convergence. When Omega generated fewer than five conformations, we reused some of the conformations. For example, if Omega generated three conformations, we used conformations one and two twice. Different starting velocities were assigned in every case, so even when conformations were reused, results were different.
Following the charge calculation and the generation of starting conformations, ANTECHAMBER was used to generate generalized Amber force field (GAFF)45 parameters for the small molecules and set them up for simulation in the Amber molecular dynamics package,46 version 9. For GB simulations, radii were set as recommended in the Amber manual (different implicit solvent models have different recommended settings). Radii and screening factors had to be added to the source code of tLeap for bromine and iodine; screening factors were set to the default. Radii for bromine and iodine for the “bondi”, “mbondi”, and “mbondi2” radii schemes were set to 1.85 and 1.98 Å, respectively, as in other work.19
E. Simulation Protocols
For each molecule, we ran five sets of simulations, with each set consisting of a separate GB and vacuum simulation. The GB model used was Amber's IGB=5, from Onufriev et al.,26 with no surface area term used during dynamics, although this was later added using reweighting. Each of these calculations was 10 ns long, using 2 fs time steps and a 16 Å nonbonded cutoff, and default RGBMAX. Temperature was regulated (to 300 K) using Langevin dynamics with a friction coefficient of 1/ps. Center of mass motion was removed every 100 time steps, and the RESPA47 algorithm was employed to evaluate long-range interactions only every two time steps. This means, according to the Amber source code, with RESPA, Born radii and interactions outside the inner cutoff of 8 Å are recomputed only every two steps. Trajectory snapshots were saved every 5000 steps (10 ps) for later reprocessing.
For each molecule, the total computational time was around 100 min on a 2.8 GHz Xeon processor (depending on the size of the molecule), including all five copies. For most applications, one copy would probably be sufficient; the extra copies simply provided a convergence test. Additionally, each of our calculations was 10 ns long; shorter simulations might suffice for many applications. Furthermore, as we note below, overlap was good enough between different implicit solvent models that we were able to compute hydration free energies in other implicit solvent models by just re-evaluating energies and computing the appropriate average (with a computational cost of less than a minute per molecule) rather than rerunning simulations. Thus, most parametrization efforts could probably be accomplished without having to rerun simulations.
F. Reprocessing and Computation of Free Energy Differences
The Bennett acceptance ratio (BAR) method provides a minimum uncertainty estimate of free energy differences between two thermodynamic states.48,49 This method requires that potential energies of simulation snapshots from each state be evaluated in the other state. We obtained these potential energies by reprocessing stored snapshots using the SANDER module in reprocessing mode (as described in the manual, with imin=5, maxcyc=1). We reprocessed our original GB simulations in GB and in vacuum, and then reprocessed the vacuum simulations in GB and in vacuum to obtain the potential energy differences (including the GB solvation component, when using GB) needed for BAR.
Alchemical free energy calculations in explicit solvent typically require multiple intermediate alchemical states to transition between the solvated and vacuum states. Here, however, we found that overlap between the solvated and vacuum ensembles of the solute was sufficient that the distributions of forward and reverse work overlapped well, and it was unnecessary to add intermediate states to obtain reasonable statistical uncertainties.
Our initial simulations were run without a surface area penalty term, so molecular surface areas for each stored snapshot were calculated using OpenEye's ZAP, and a nonpolar term was added to the total energies in solvent prior to analysis, for the single-conformation schemes. For hydration free energies, the nonpolar contribution to the hydration free energy was computed using exponential averaging (the Zwanzig relation)50 of the nonpolar potential energies. The nonpolar term was evaluated as Gnonpolar = γSA + β, as in ref 19, with γ = 0.00542 kcal/(mol Å) 2 and β = 0.92 kcal/mol. A variety of recent work has suggested that this traditional form of the nonpolar term can be improved by including some treatment of dispersion interactions, 51–54 but we have not tested these alternate treatments here, as our focus is not on the accuracy of implicit solvent models, but on the method for computing hydration free energies given an implicit solvent model.
However, for comparison, we also reprocessed the GB simulations using two alternate GB models available in Amber: the model of Tsui and Case,25 and the GBn model of Mongan et al.27 This allowed us to rigorously compute hydration free energies for these models, as well, using exponential averaging,50 because phase space overlap was very good.
Also, we reprocessed our stored configurations in OpenEye's PB solver, ZAP, and used exponential averaging to compute solvation free energies using PB. ZAP settings were as default, with default radii, and an inner dielectric of 1 was used with a grid spacing of 0.5 Å. The same nonpolar component was used. We have made stored trajectories and parameters (in Amber format) available online (http://www.dillgroup.ucsf.edu/dmobley/paper_exports/trajectories_implicit_solvent.tar.gz).
In addition to computing the hydration free energy, we also computed the effective enthalpy and entropy of solvation, using TΔS = ΔH − ΔG, where ΔH = 〈UGB〈 − 〈Uvac〉, where S, H, and G are the entropy, enthalpy, and free energy, respectively, and UGB is the potential energy in GB solvent, while Uvac is the potential energy in vacuum. Since UGB is an effective potential energy that already implicitly includes solvent entropies (as in eq 3), the entropy of solvation computed here only includes changes in solute entropy. Solvent entropy changes, since they are built in to the energy function, get included in the effective enthalpy of solvation. Thus, the effective enthalpy of solvation includes the solvent terms of eq 4, plus an additional term that includes the solute enthalpy change on solvation, which will be especially important in the case of conformational change.
G. Single-Conformation Hydration Free Energies
For computing single-conformation hydration free energies, we used several different schemes for picking conformations: (1) the lowest potential energy conformation of the solute in solvent (the “BestSolv” scheme); (2) the lowest potential energy conformation of the solute in vacuum (the “BestVac” scheme); (3) the conformation of the solute in solvent that results in the most favorable hydration free energy estimate (the “BestG” scheme); and (4) the conformation of the solute in solvent that results in the least favorable hydration free energy estimate (the “WorstG” scheme). The BestSolv and BestG schemes are distinct, because internal energies are included in the potential energy when selecting the conformation for the BestSolv estimate, but are neglected in selecting the conformation for the BestG estimate (since internal energies always cancel out when computing single-conformation hydration free energies). Thus, BestG conformations can have larger internal strain energies.
In section III, we briefly discussed several different schemes for selecting conformations in previous implicit solvent studies. A number of studies used conformations that were geometry-optimized in vacuum, and thus are likely similar to those selected with the “BestVac” scheme here.
Reported single-conformation hydration free energies (below) were computed by selecting a single snapshot from each set of simulations for analysis. For each computed free energy, the overall value was taken as the average over the five separate sets of simulations.
H. Error Analysis
Statistical uncertainties were estimated for each simulation set using the asymptotic variance of the BAR method49 and Taylor expansion-based error propagation for exponential averaging. Standard error propagation was then used to combine the hydration free energy estimates from each simulation and compute the uncertainty in the final (mean) hydration free energy.
For comparison, we took the standard error in the mean of the estimated ΔGhyd over the five sets of simulations. Because these were started from different conformations, this was, in some cases, larger than the estimate from the approach discussed above from BAR. To be conservative, reported uncertainties below are the larger of these two estimated uncertainties.
For single-conformation hydration free energies, computed uncertainties were the standard error in the mean when averaging over the five different measurements. For example, we computed the BestVac estimate from each set of simulations, and then computed the uncertainty by taking the standard error in the mean from our five estimates of the BestVac single-conformation hydration free energy.
V. Results and Discussion
A. Comparison of True Hydration Free Energies with Single-Conformation Hydration Free Energies
Our main goal here is to compare different methods of estimating hydration free energies from implicit solvent models. As described in section IV, we used the GB model of Onufriev et al.26 As our gold standard, we computed hydration free energies using extensive molecular dynamics sampling of the vacuum and solvated phases, and the BAR. Thus, these results include any solute conformational enthalpic and entropic changes (as in eq 5). We compare these results to hydration free energies computed using fixed solute conformations (eq 4).
We tested several different choices of solute conformations for computing single-conformation hydration free energies, as discussed in section IV.G. The BestG and WorstG schemes span the range of single-conformation hydration free energies. We find that the average value for this range is 1.85 ± 0.08 kcal/mol across the whole test set of 504 small molecules. The distribution of ranges is shown in Figure 2. There are 314 small molecules with ranges larger than 1 kcal/mol; 110 with ranges larger than 2.5 kcal/mol; 31 with ranges larger than 5 kcal/mol, and 5 with ranges larger than 7.5 kcal/mol. This indicates that, for many molecules, single-conformation hydration free energies are quite sensitive to the conformation chosen. Furthermore, 187 small molecules have a range of values that is at least twice as large as the error relative to experiment, so the errors introduced by using single conformations are larger than those introduced by the force field in many cases. The full distribution of single-conformation hydration free energies is shown in Figure 3.
Figure 2.
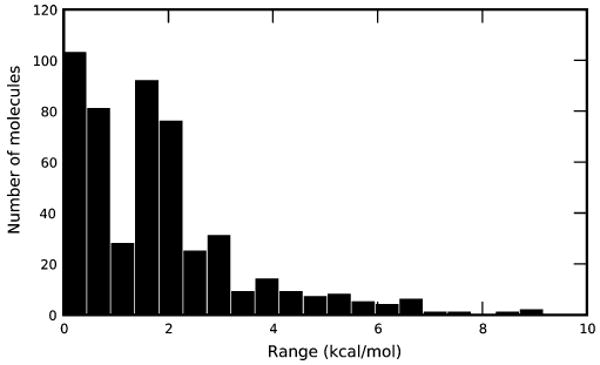
Distribution of range of single-conformation hydration free energies. Single-conformation hydration free energies are sensitive to the choice of conformation used. For each compound, the range is the difference between the minimum and maximum single-conformation hydration free energies obtained. Shown here is a histogram of the ranges for the 504 small molecules in the test set.
Figure 3.
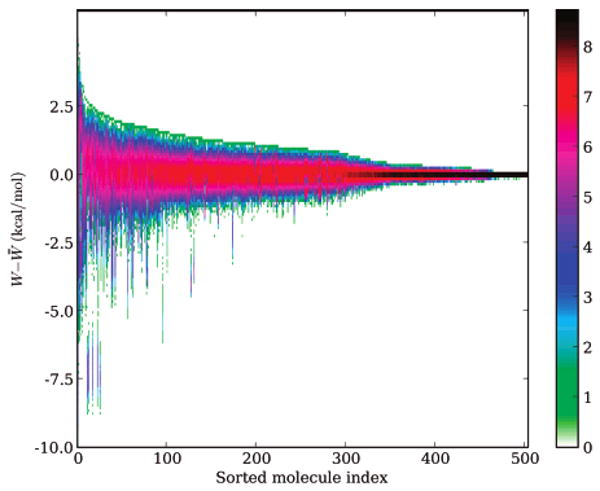
Distribution of single-conformation hydration free energies. Shown on the vertical axis is the distribution of single-conformation hydration free energies around the mean for each molecule; W denotes the single-conformation hydration free energy, and W̄) is the mean single-conformation hydration free energy. Colors denote the natural log of the binned probability of each energy. Molecules are sorted by the width of their single-conformation hydration free energy distributions.
We find that root-mean-square (RMS) differences between the computed hydration free energies and single-conformation hydration free energies can be up to 1.8 kcal/mol (Table 1, Figure 1), depending on how the fixed solute conformation is chosen for single-conformation hydration free energies. The full hydration free energies included both solute conformational enthalpic changes and entropic effects, so it was of interest to separate out the effects of entropic and enthalpic changes. We used the expression ΔG = ΔUeff − TΔS, where Ueff is the effective energy function discussed above and implicitly includes solvent entropy changes. Thus, ΔS here only includes solute entropic changes. We refer to ΔUeff as the effective enthalpy of hydration; it includes solute conformational enthalpic changes.
TABLE 1. RMS Errors of Single-Conformation Hydration Free Energies Relative to Computed Hydration Free Energiesa.
| scheme | RMS error | mean error |
|---|---|---|
| BestSolv | 0.390 ± 0.053 | −0.123 ± 0.016 |
| BestVac | 0.338 ± 0.014 | 0.154 ± 0.013 |
| BestG | 1.796 ± 0.043 | −1.126 ± 0.062 |
| WorstG | 0.882 ± 0.007 | 0.726 ± 0.022 |
Single-conformer hydration free energies differ from computed hydration free energies. Shown are RMS and mean errors, relative to the computed hydration free energies, depending on the scheme for choosing the conformer for the calculation of single-conformation hydration free energies.
Figure 1.
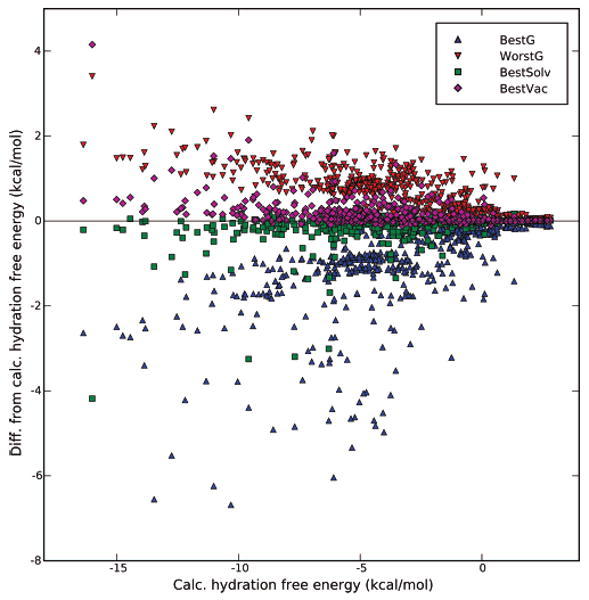
Calculated hydration free energies, versus error with single-conformation schemes. Calculated hydration free energies are plotted, versus the error from these values when using different choices of single-conformations for computing hydration free energies. The different schemes are as follows: BestG, the conformation resulting in the most favorable hydration free energy estimate; WorstG, the conformation resulting in the least favorable hydration free energy estimate; BestVac, the conformation with the lowest potential energy in vacuum; and BestSolv, the conformation with the lowest potential energy in solvent. The γ = 0 line indicates exact agreement between calculated single-conformation hydration free energies and calculated hydration free energies. Values below the line overestimate the affinity for water; those above the line underestimate the affinity for water.
We compared effective enthalpies of hydration with computed single-conformation hydration free energies (Table 2). We find that solute conformational enthalpic changes contribute more to the overall error than do solute entropic changes. The average TΔS of hydration over the entire test set is only 0.065 ± 0.001 kcal/mol.
TABLE 2. RMS Errors of Single-Conformation Hydration Free Energies Relative to Computed Effective Enthalpies of Hydrationa.
| scheme | RMS error | mean error |
|---|---|---|
| BestSolv | 0.519 ± 0.053 | −0.188 ± 0.022 |
| BestVac | 0.231 ± 0.022 | 0.089 ± 0.009 |
| BestG | 1.960 ± 0.049 | −1.190 ± 0.069 |
| WorstG | 0.789 ± 0.006 | 0.661 ± 0.019 |
Single-conformer hydration free energies differ from computed effective hydration enthalpies (which include solvent, but not solute, entropic effects). Shown are RMS and mean errors, relative to the computed effective hydration enthalpies, depending on the scheme for choosing the conformer for the calculation of single-conformation hydration free energies.
However, we cannot conclude that solute entropic changes are unimportant. We find that 17 of the small molecules have TΔS of hydration larger than 0.5 kcal/mol (Figure 4, and a table of the 17 in the Supporting Information). Interestingly, it appears to be hard to predict which molecules will have large entropic changes upon solvation. We find that there is essentially no correlation of TΔS with the number of rotatable bonds (R2 = 0.03 ± 0.01) (in agreement with recent work from the Gilson laboratory 55); correlations with the number of polar hydrogens and hydroxyl groups are also quite poor (R2 = 0.07 ± 0.03 and 0.18 ± 0.07, respectively).
Figure 4.
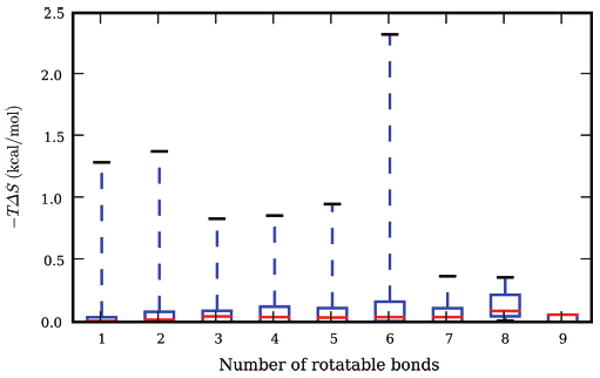
Number of rotatable bonds versus –TΔS of solvation. Shown is a box plot of the number of rotatable bonds versus –TΔS of solvation. The red line shows the median; the box shows the bounds of the upper and lower quartiles, and the dashed lines show the full range of –TΔS. The correlation between –TΔS and the number of rotatable bonds is only R2 = 0.03, and there are 17 small molecules with –TΔS larger than 0.5 kcal/mol.
Solute entropic changes are interesting, because they also end up being a strong predictor of the range of possible single-conformation hydration free energies. In particular, the range of single-conformer hydration free energies correlates only very weakly with the number of rotatable bonds (R2 = 0.05 ± 0.01) and the number of polar hydrogens (R2 = 0.19 ± 0.01), but more strongly with the TΔS of hydration (R2 = 0.53 ± 0.20). We suspect this is because molecules that undergo conformational changes on hydration are often those that form strong internal electrostatic interactions in vacuum (for example, internal hydrogen bonding), which are often less favorable in solvent. In addition to these conformational changes, these molecules often have very low entropy in their vacuum conformations due to the strength of these internal electrostatic interactions, and so entropy gains on hydration are particularly large.
It is interesting to note that, of the single-conformation approaches, the approach that uses conformations that are low potential energy in vacuum gave the lowest errors relative to full hydration free energies (Table 1). Presumably, this is because the choice of conformation is less important in solvent than in vacuum because of the roughness of the energy landscape.
Figure 5 shows examples of some of the changes in preferred conformation we observe on hydration, along with the resulting single-conformation hydration free energies. At least with these parameters, acetic acid undergoes a conformational transition upon solvation that seems fairly straightforward: in vacuum, the molecule prefers to put positive charge near negative charge and reduce the total potential energy, but in solvent, atoms can interact with their image charges in solvent, making the alternate conformation more favorable; electrostatic interactions are also less strong in solvent.62 Methyl formate is very similar; for both of these molecules, the BestG and BestSolv conformations are similar to one another, as are the BestVac and WorstG conformations. 2-Methoxyphenol also shows a conformational change upon solvation, but in this case, only the BestG conformation has the hydroxyl hydrogen pointing out (Figure 5c, right). This latter conformation is extremely unfavorable in vacuum because of the proximity of the two oxygens, both with negative partial charges. Since the single-conformation hydration free energy is computed as the difference between the potential energy of the conformation in vacuum and the effective energy in solvent, molecules can achieve very favorable single-conformation hydration free energies by adopting conformations that are very unfavorable in vacuum (and thus minimizing their apparent affinity for vacuum). This, of course, is purely an artifact of using single conformations to estimate hydration free energies, and is unphysical, resulting in a range of computed single-conformation hydration free energies of 6.4 kcal/mol for 2-methoxyphenol with these conformations.
Figure 5.
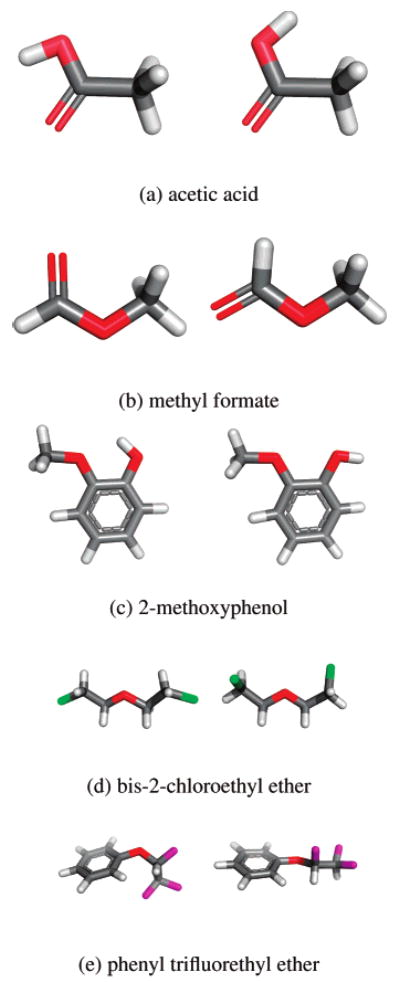
Sample conformations for molecules with significant ranges in computed hydration free energies. For each molecule, the left shows the lowest potential energy conformation in vacuum (BestVac) scheme from one simulation. (a) The BestVac conformation (left) yields a single-conformation hydration free energy of −8.95 kcal/mol; the BestSolv conformation (right) yields −17.75 kcal/mol. Worst and BestG conformations are similar to these two, respectively. (b) The BestVac conformation (left) yields −9.72 kcal/mol; the BestSolv conformation (right) yields −14.83 kcal/mol. WorstG and BestG conformations are similar to these two, respectively. (c) The BestVac conformation (left) yields −8.33 kcal/mol; the BestG conformation (right) yields −14.73 kcal/mol. BestSolv and WorstG conformations are similar to BestVac. (d) The BestVac conformation (left) yields −8.84 kcal/mol; the BestSolv conformation (right) yields −11.38 kcal/mol. WorstG and BestG conformations are similar to these two, respectively. (e) The BestVac conformation (left) yields −8.67 kcal/mol and the BestSolv conformation (right) yields −11.38 kcal/mol. The WorstG and BestG conformations are similar to these two, respectively. Images were made with PyMOL.59
Bis-2-chloroethyl ether and phenyl trifluoroethyl ether are also shown in Figure 5. In vacuum, bis-2-chloroethyl ether minimizes electrostatic clashes between the chlorine atoms, while in solvent, it can adopt other conformations. Phenyl trifluoroethyl ether appears similar, although it is less straightforward to interpret.
The key point here is that conformational changes can often occur upon solvation, and when they occur, single-conformation “hydration free energies” are substantially incorrect, because they always incorrectly represent either the vacuum or solvent conformation. When using conformations appropriate for vacuum, these single-conformation hydration free energies will tend to underestimate the affinity for solvent (Figure 1, Table 1), while the opposite happens when using conformations appropriate for solvent.
B. Implications for Parametrization
As noted, single-conformation hydration free energies computed here using single conformations depend strongly on the solute conformation chosen. This dependence on conformation has significant implications for parametrization of implicit solvent models using single solute conformations. In particular, it raises an alarming possible scenario that is best illustrated by example. Suppose we selected the lowest potential energy conformation for each molecule in some existing solvent model (BestSolv scheme, here), computed experimental hydration free energies, and then were able to derive a new set of parameters (radii, etc.) that reproduced the experimental hydration free energies exactly. Then, we generated a new set of conformations for the same molecules using the scheme resulting in the most favorable hydration free energy (BestG scheme, here) and recomputed single-conformation hydration free energies; computed values would no longer agree completely with experiment. In fact, here, the RMS difference between the BestSolv and BestG schemes is 1.56 kcal/mol, with the same parameters! Given this, it seems reasonable to be somewhat skeptical of implicit solvent models that are parametrized using single conformations.63 Given that the full hydration free energies reported here are relatively inexpensive to compute, these should probably be used in future parametrization efforts.
One could conclude, on the basis of the data presented here, that as long as BestVac or BestSolv conformations are used, single-conformation hydration free energies are good enough: although the RMS difference between the full hydration free energies and these single-conformation estimates is 0.3–0.4 kcal/mol, the overall RMS error relative to experiment (roughly 2 kcal/mol) is hardly different from that with the full free energy approach, for the solvent models considered here. Essentially, the overall performance of the solvent models here is poor enough that using single solute conformations cannot make the results much worse. However, a number of solvent models have been developed that have reported RMS or mean errors as low as 0.3–0.5 kcal/mol.10,18,39,40 With RMS errors this low, the single-conformation approximation could cause much more significant problems.
C. Comparison with Experiments
So far, we have compared single-conformation hydration free energies with hydration free energies computed using extensive sampling of solute conformations. This comparison tests only the importance of sampling, our main interest here, and does not test the underlying force field by comparing with experiment. Here, we provide a brief comparison with experimental results.
For Amber's implicit solvent model IGB=5 (Onufriev et al.), we find that all of the approaches give relatively poor overall agreement with experiments (RMS errors from 2 to 2.4 kcal/mol), but still correlate well with experimental values (as observed previously on an overlapping test set19), so overall performance is mixed. Statistics are shown in Table 3. It is interesting, however, to note that the full free energy approach agrees somewhat better with experiment (in terms of RMS error, mean error, and correlation coefficient) than either approach that selects good conformations from the solvent simulations. That is, it is substantially better than the BestG scheme in every respect, and has small, but statistically significant, improvements in RMS error and mean error over the BestSolv scheme. The full free energy approach also results in roughly comparable agreement to the best of the other approaches (R2 relative to experiment of 0.698 ± 0.001 with the full free energy approach, versus R2 = 0.691 ± 0.002 with the best of the other approaches). Computed free energies are plotted versus experiment in Figure 6.
TABLE 3. Agreement with Experiment Using Different Schemesa.
| approach | RMS error (kcal/mol) |
mean error (kcal/mol) |
correlation coefficient (R2) |
|---|---|---|---|
| ΔGhyd | 2.145 ± 0.003 | −0.654 ± 0.091 | 0.698 ± 0.001 |
| BestSolv | 2.287 ± 0.017 | −0.777 ± 0.096 | 0.689 ± 0.005 |
| BestVac | 2.055 ± 0.006 | −0.500 ± 0.089 | 0.691 ± 0.002 |
| BestG | 3.300 ± 0.031 | −1.780 ± 0.124 | 0.671 ± 0.004 |
| WorstG | 1.947 ± 0.004 | 0.072 ± 0.087 | 0.671 ± 0.002 |
Shown are the RMS and mean differences from experiment, and the correlation with experiment, for the implicit solvent hydration free energy calculations (ΔGhyd) and the various single-conformation approaches.
Figure 6.
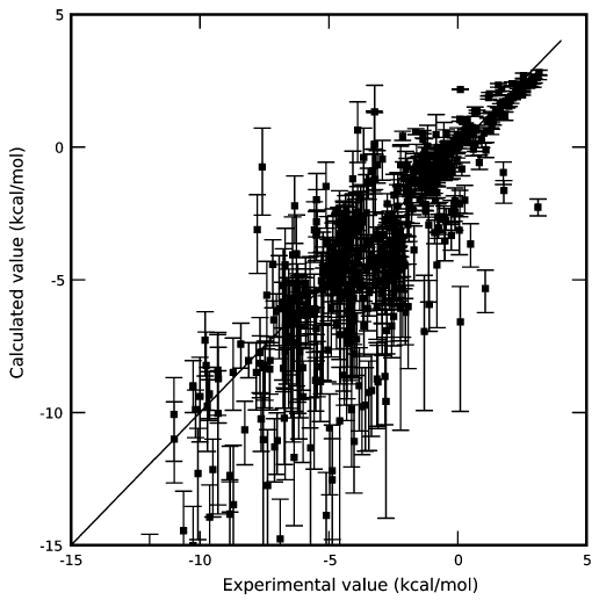
Computed hydration free energies, and range of single-conformation interaction energies, versus experiment. Computed hydration free energies are shown as black squares; vertical bars denote the range of single-conformation hydration free energies possible depending on the choice of solute conformation. The diagonal line is the x = y line.
We did not perform the same detailed comparison of different single-conformation schemes for the other implicit solvent models tested. However, we did compute hydration free energies using the full approach; the statistics are shown in Table 4 and results by compound, for the entire set, are available in the Supporting Information. Overall, the PB solver (OpenEye's ZAP) performs marginally better than the GB methods, which is not too surprising, since these particular GB approaches (unlike many implicit solvent models) were parametrized (with some adjustments) to match PB results.
TABLE 4. Agreement with Experiment Using Different Implicit Solvent Modelsa.
| solvent model | RMS error (kcal/mol) |
mean error (kcal/mol) |
correlation coefficient (R2) |
|---|---|---|---|
| IGB=526 | 2.145 ± 0.003 | −0.654 ± 0.091 | 0.698 ± 0.001 |
| PB (ZAP) | 2.014 ± 0.008 | −0.796 ± 0.082 | 0.774 ± 0.001 |
| IGB=125 | 2.137 ± 0.006 | −0.645 ± 0.091 | 0.737 ± 0.001 |
| IGB=727 | 2.433 ± 0.002 | −1.056 ± 0.098 | 0.685 ± 0.001 |
Shown are the RMS and mean differences from experiment, and the correlation with experiment, for the implicit solvent hydration free energy calculations using various different implicit solvent models (the IGB=x models are different GB models in Amber), as described in the text.
It is interesting to note that mean hydration free energies with all schemes and all solvent models are negative (too favorable for water; Tables 3 and 4), suggesting there may be systematic errors in the implicit solvent model, or that a different treatment of the nonpolar component of solvation may be more appropriate. For example, the coefficients in the nonpolar term could be adjusted to make the mean error essentially zero, but this would increase the overall RMS error.
VI. Conclusions
Our focus here is on how to parametrize implicit solvent models. In many cases, parameters are derived by selecting a single (presumably dominant) solute conformation and adjusting parameters to reproduce experimental vacuum-to-water transfer (hydration) free energies. Here, we performed molecular dynamics simulations of solutes in vacuum and water and computed hydration free energies using free energy methods (thus including entropic and conformational enthalpic changes). We then compared with results computed using single solute conformations, as a test of the single-conformation approximation.
We find that the single-conformation approaches lead to average errors between 0.23 ± 0.02 and 1.96 ± 0.05 kcal/mol (compared to the full free energy approach), depending on how the single solute conformation is chosen. These errors result from two factors. First, the solute samples an ensemble of conformations in each phase. Since the ensemble can change upon hydration, there can be solute entropic changes that are not included in the single-conformation approaches. Second, the dominant conformation(s) of the solute can differ in the two phases. When this happens, no single conformation is appropriate for both phases. We find that the errors introduced by using single conformations are larger than those introduced by the force field in many cases. Moreover, errors introduced by using single conformations do not correlate strongly with the number of rotatable bonds, although errors do correlate more strongly with changes in entropy upon solvation.
In view of the results presented here, parametrizing single-conformation hydration free energy estimates to match experimental hydration free energies seems unwise. However, in addition to the alternative discussed here (parametrizing full hydration free energy estimates to match experiment), there is another sensible option that has been tried by several groups: parametrizing single-conformation hydration free energy estimates in implicit solvent to match single-conformation estimates from explicit solvent simulations.28–30 In this case, both computed free energies neglect solute conformational change, so at least this is an internally consistent way to derive parameters, and bypasses the conformation dependence problems described here.
Overall, we demonstrated an approach for computing hydration free energies in implicit solvent using free energy methods. This approach includes changes in solute conformational entropy and enthalpy that have often been neglected in implicit solvent studies of the solvation of small molecules. While it does not greatly improve the agreement between hydration free energy estimates and experiment, it does not degrade results, and at least provides a consistent way to treat solute conformational changes on solvation. We believe this approach should be used in future efforts to parametrize implicit solvent models.
Supplementary Material
Acknowledgments
We thank Vijay S. Pande and Edgar Luttmann (Stanford) for helpful discussions and comments on the manuscript. We also thank Nathan Baker (Washington University) and Jason Wagoner (Stanford) for comments on the manuscript. We appreciate the support of NIH grant R01 GM 64592. J.D.C. acknowledges support from Vijay Pande (Stanford) through an NSF grant for cyberinfrastructure (NSF CHE-0535616).
Footnotes
Supporting Information Available: Coordinate files (mol2) with AM1-BCC partial charges for the small molecules in the test set used here; list of computed values for each compound with each of the implicit solvent models, and for the model of Onufriev, Bashford, and Case, using different conformations and different analysis methods; experimental solvation free energies (1M vacuum to 1M water) and references for the molecules in the test set; histograms of molecular weight and number of rotatable bonds for the test set; an alternative version of Figure 1; and a table of the 17 small molecules with TΔS larger than 0.5 kcal/mol. This information is available free of charge via the Internet at http://pubs.acs.org.
References and Notes
- 1.Shoichet BK, Leach AR, Kuntz ID. Proteins Struct Funct Genet. 1999;34:4–16. doi: 10.1002/(sici)1097-0134(19990101)34:1<4::aid-prot2>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 2.Kalyanaraman C, Bernacki K, Jacobson MP. Biochemistry. 2005;44:2059–2071. doi: 10.1021/bi0481186. [DOI] [PubMed] [Google Scholar]
- 3.Camacho CJ, Ma H, Champ PC. Proteins Struct Funct Bioinf. 2006;63:868–877. doi: 10.1002/prot.20932. [DOI] [PubMed] [Google Scholar]
- 4.Ferrara P, Gohlke H, Price DJ, Klebe G, Brooks CL., III J Med Chem. 2004;47:3032–3047. doi: 10.1021/jm030489h. [DOI] [PubMed] [Google Scholar]
- 5.Huang N, Kalyanaraman C, Jacobson MPJ. Chem Inf Model. 2006;46:243–253. doi: 10.1021/ci0502855. [DOI] [PubMed] [Google Scholar]
- 6.Ozkan SB, Wu GA, Chodera JD, Dill KA. Proc Natl Acad Sci USA. 2007;104:11987–11992. doi: 10.1073/pnas.0703700104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zagrovic B, Sorin EJ, Pande VS. J Mol Biol. 2001;313:151–169. doi: 10.1006/jmbi.2001.5033. [DOI] [PubMed] [Google Scholar]
- 8.Pande VS, Baker I, Chapman J, Elmer SP, Khaliq S, Larson SM, Rhee YM, Shirts MR, Snow CD, Sorin EJ, Zagrovic B. Biopolymers. 2003;68:91–109. doi: 10.1002/bip.10219. [DOI] [PubMed] [Google Scholar]
- 9.Rashin AA, Honig B. J Phys Chem. 1985;89:5588–5593. [Google Scholar]
- 10.Sitkoff D, Sharp KA, Honig B. J Phys Chem. 1994;98:1978–1988. [Google Scholar]
- 11.Warwicker J, Watson HC. J Mol Biol. 1982;157:671–679. doi: 10.1016/0022-2836(82)90505-8. [DOI] [PubMed] [Google Scholar]
- 12.Zauhar RJ, Morgan RS. J Mol Biol. 1985;186:815–820. doi: 10.1016/0022-2836(85)90399-7. [DOI] [PubMed] [Google Scholar]
- 13.Gilson MK, Honig BH. Proteins Struct Funct Genet. 1988;3:32–52. doi: 10.1002/prot.340030104. [DOI] [PubMed] [Google Scholar]
- 14.Bashford D, Case DA. Annu Rev Phys Chem. 2000;51:129–152. doi: 10.1146/annurev.physchem.51.1.129. [DOI] [PubMed] [Google Scholar]
- 15.Still WC, Tempczyk A, Hawley RC, Hendrickson T. J Am Chem Soc. 2000;90:6127–6129. [Google Scholar]
- 16.Cramer CJ, Truhlar DG. J Am Chem Soc. 1991;113:8305–8311. [Google Scholar]
- 17.Thompson JD, Cramer CJ, Truhlar DG. J Phys Chem A. 2004;108:6532–6542. [Google Scholar]
- 18.Bordner AJ, Cavasotto CN, Abagyan RA. J Phys Chem B. 2002;106:11009–11015. [Google Scholar]
- 19.Rizzo RC, Aynechi T, Case DA, Kuntz ID. J Chem Theory Comput. 2006;2:128–139. doi: 10.1021/ct050097l. [DOI] [PubMed] [Google Scholar]
- 20.Lim C, Bashford D, Karplus M. J Phys Chem. 1991;95:5610–5620. [Google Scholar]
- 21.Mohan V, Davis ME, McCammon JA, Pettitt BM. J Phys Chem. 1992;96:6428–6431. [Google Scholar]
- 22.Nicholls A, Mobley DL, Guthrie JP, Chodera JD, Pande VS. J Med Chem. doi: 10.1021/jm070549+. in press. [DOI] [PubMed] [Google Scholar]
- 23.Jayaram B, Sprous D, Beveridge DL. J Phys Chem B. 1998;102:9571–9576. [Google Scholar]
- 24.Dominy BN, Brooks CL., III J Phys Chem B. 1999;103:3765–3773. [Google Scholar]
- 25.Tsui V, Case DA. Biopolymers (Nucleic Acid Sci) 2001;56:275–291. doi: 10.1002/1097-0282(2000)56:4<275::AID-BIP10024>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 26.Onufriev A, Bashford D, Case DA. Proteins Struct Funct Bioinf. 2004;55:383–394. doi: 10.1002/prot.20033. [DOI] [PubMed] [Google Scholar]
- 27.Mongan J, Simmerling C, McCammon JA, Case DA, Onufriev A. J Chem Theory Comput. 2007;3:156–169. doi: 10.1021/ct600085e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang LY, Gallicchio E, Friesner RE, Levy RM. J Comput Chem. 2001;22:591–607. [Google Scholar]
- 29.Nina M, Beglov D, Roux B. J Phys Chem B. 1997;101:5239–5248. [Google Scholar]
- 30.Swanson JMJ, Adcock SA, McCammon JA. J Chem Theory Comput. 2005;1:484–493. doi: 10.1021/ct049834o. [DOI] [PubMed] [Google Scholar]
- 31.Swanson JMJ, Wagoner JA, Baker NA, McCammon JA. J Chem Theory Comput. 2007;3:170–183. doi: 10.1021/ct600216k. [DOI] [PubMed] [Google Scholar]
- 32.Deng Y, Roux B. J Phys Chem B. 2004;108:16567–16576. [Google Scholar]
- 33.Shirts MR, Pitera JW, Swope WC, Pande VS. J Chem Phys. 2003;119:5740–5761. [Google Scholar]
- 34.Shirts MR, Pande VS. J Chem Phys. 2005;122:134508. doi: 10.1063/1.1877132. [DOI] [PubMed] [Google Scholar]
- 35.Mobley DL, Dumont È, Chodera JD, Dill KA. J Phys Chem B. 2007;111:2242–2254. doi: 10.1021/jp0667442. [DOI] [PubMed] [Google Scholar]
- 36.Hess B, van der Vegt NFA. J Phys Chem B. 2006;110:17616–17626. doi: 10.1021/jp0641029. [DOI] [PubMed] [Google Scholar]
- 37.Xu Z, Luo HH, Tieleman DP. J Comput Chem. 2007;28:689–697. doi: 10.1002/jcc.20560. [DOI] [PubMed] [Google Scholar]
- 38.Jean-Charles A, Nicholls A, Sharp K, Honig B, Tempczyk A, Hendrickson TF, Still WC. J Am Chem Soc. 1991;113:1454–1455. [Google Scholar]
- 39.Kelly CP, Cramer CJ, Truhlar DG. J Chem Theory Comput. 2005;1:1133–1152. doi: 10.1021/ct050164b. [DOI] [PubMed] [Google Scholar]
- 40.Gallicchio E, Zhang LY, Levy RM. J Comput Chem. 2002;23:517–529. doi: 10.1002/jcc.10045. [DOI] [PubMed] [Google Scholar]
- 41.Guthrie JP. University of Western Ontario, London; Ontario, Canada: To be submitted for publication. [Google Scholar]
- 42.Fasman GD. Handbook of Biochemistry and Molecular Biology. Crc Press; Boca Raton, FL: 1977. [Google Scholar]
- 43.Jakalian A, Bush BL, Jack DB, Bayly CI. J Comput Chem. 2000;21:132–146. [Google Scholar]
- 44.Jakalian A, Jack DB, Bayly CI. J Comput Chem. 2002;23:1623–1641. doi: 10.1002/jcc.10128. [DOI] [PubMed] [Google Scholar]
- 45.Wang J, Wolf RM, Caldwell JW, Kollman PA, Case DA. J Comput Chem. 2004;25:1157–1174. doi: 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- 46.Case DA, Cheatman TE, III, Darden T, Gohlke H, Luo R, Merz KM, J, Onufriev A, Simmerling C, Wang B, Woods R. J Comput Chem. 2005;26:1668–1688. doi: 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tuckerman M, Berne BJ, Martyna GJ. J Chem Phys. 1992;97:1990–2001. [Google Scholar]
- 48.Bennett CH. J Comput Phys. 1976;22:245–268. [Google Scholar]
- 49.Shirts MR, Bair E, Hooker G, Pande VS. Phys Rev Lett. 2003;91:140601. doi: 10.1103/PhysRevLett.91.140601. [DOI] [PubMed] [Google Scholar]
- 50.Zwanzig RW. J Chem Phys. 1954;22:1420–1426. [Google Scholar]
- 51.Gallicchio E, Levy RM. J Comput Chem. 2004;25:479–499. doi: 10.1002/jcc.10400. [DOI] [PubMed] [Google Scholar]
- 52.Pitera JW, van Gunsteren WF. J Am Chem Soc. 2001;123:3163–3164. doi: 10.1021/ja0057474. [DOI] [PubMed] [Google Scholar]
- 53.Choudhury N, Pettit BM. Mol Simul. 2005;31:457–463. [Google Scholar]
- 54.Wagoner JA, Baker NA. Proc Natl Acad Sci USA. 2006;103:8331–8336. doi: 10.1073/pnas.0600118103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chang CEA, Chen W, Gilson MK. Proc Nat Acad Sci USA. 2007;104:1534. doi: 10.1073/pnas.0610494104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schurhammer R, Engler E, Wipff G. J Phys Chem B. 2001;105:10700–10708. [Google Scholar]
- 57.Srinivasan J, Cheatman TE, III, Cieplak P, Kollman PA, Case DA. J Am Chem Soc. 1998;120:9401–9409. [Google Scholar]
- 58.Tsui V, Case DA. J Am Chem Soc. 2000;122:2489–2498. [Google Scholar]
- 59.The PyMOL Molecular Graphics System. [June 2007]; http://www.pymol.org.
- 60.The assumption may be that the solute conformation is irrelevant.
- 61.Experimental hydration free energies for ions are really measured for ion pairs, and then separated into hydration free energies for individual ions using extrathermodynamic assumptions like the so-called TATB hypothesis, which assumes that the hydration free energies of two isosteric large hydrophobic ions of opposite sign are equal. Recent work has called this hypothesis, and related assumptions, into question.56 At the very least, the issue of comparing with experimental ion hydration free energies is a complicated one, and one we choose to avoid here.
- 62.As noted in the Methods section, acetic acid is one of the molecules in this test set that is likely to change protonation state upon hydration. In this study, protonation states were kept fixed, in keeping with common practices in parameterizing implicit solvent models.
- 63.In some sense, this consideration applies even to the GB models considered here25–27, since these GB models were derived from an original GB model which was parameterized to match PB calculations using PARSE radii as a starting point57,58. PARSE radii were themselves derived to fit experimental hydration free energies when using single solute conformations10.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.