

Abstract
Autophagy, a highly conserved mechanism of quality control inside cells, is essential for the maintenance of cellular homeostasis and for the orchestration of an efficient cellular response to stress. The decrease in autophagic activity observed in almost all cells and tissues as organisms age was proposed to contribute to different aspects of the aging phenotype and to the aggravation of detrimental age-related diseases. The recent advances in our understanding of the molecular mechanisms underlying autophagy and the identification of the subset of genes involved in this process has enabled the use of genetic manipulations to start testing this hypothesis. Here, I review the recent genetic evidence in support of tight connections between autophagy, health span and aging.
Cellular quality control, autophagy and aging
All cells rely on surveillance mechanisms, chaperones and proteolytic systems to control the quality of their proteins and organelles and to guarantee that any malfunctioning or damaged intracellular components are repaired or eliminated [1,2]. Molecular chaperones interact with unfolded or misfolded proteins and assist in their folding [3]. However, if the extent of protein damage is too great, or the cellular conditions are not adequate for re-folding, the same molecular chaperones often deliver proteins for degradation. Two proteolytic systems contribute to cellular clearance: the ubiquitin-proteasome and the lysosomal systems [4]. Chaperone malfunctions or alterations in the components of the proteolytic systems result in intracellular accumulation of damaged proteins and organelles and underlie the basis of different human pathologies [5]. Accumulation of damage is also characteristic of tissues in all organisms as they age and has been proposed to be responsible for their functional loss in aging. Changes with age in both the ubiquitin-proteasome and the lysosomal system have been described (reviewed in Refs [6,7]), but only recently has the contribution of these changes to the aging phenotype started to be elucidated. The main reason for the recent advances has been the improved molecular characterization of the different cellular degradative pathways. The identification of the genes encoding effectors and regulators of intracellular clearance has now enabled direct analysis of the consequences of downregulation of their activity to levels similar to those observed in old organisms. Likewise, genetic manipulations to prevent the age-related functional decline of some of these systems have confirmed the contribution of the accumulation of intracellular damage to their functional failure in aging. Here, I review the contribution of the lysosomal system to the maintenance of cellular homeostasis - through autophagy (see Glossary) - and the recent findings linking the autophagic system to life-span extension and to different aspects of cellular and organismal aging.
Autophagy: the comeback of an old pathway
Lysosomes are organelles fully devoted to degrading diverse macromolecules both from the extracellular environment and from inside the cells [8]. Lysosomes contain the highest cellular concentration of hydrolases (i.e. proteases, lipases, glycases and nucleotidases) in their lumen, in addition to permeases in their membrane for recycling the essential building blocks of the degraded products (e.g. amino acids, fatty acids and cholesterol, sugars, etc.) to the cytosol [9]. In this respect, lysosomes are real recycling compartments in which cellular structures are broken into their individual components, which can then be reused for synthesis of new cellular structures. Lysosomes degrade both intact (functional) proteins, to guarantee continuous renewal of the cellular proteome, and damaged proteins that are no longer functional, to avoid their accumulation inside cells. Lysosomes not only degrade soluble individual proteins but also particulate structures and complete organelles. This feature makes the lysosomal system particularly relevant under conditions when damaged proteins start to organize into irreversible oligomers and aggregates.
The contribution of the lysosomal system to catabolism and intracellular clearance (autophagy) has been known for more than half a century, since the description of this organelle by deDuve [9]. However, until recently, the understanding of the pathophysiology of autophagy advanced at a very slow pace compared to that of other quality control mechanisms. The most important propellers of the current advances in the autophagy field were three simultaneous yeast genetic studies initiated ∼10 years ago, which identified the first autophagy related genes (ATG) [10] (Box 1). The numbers of ATG genes have escalated to include >30 and the majority are conserved throughout evolution [11]. The possibility of genetically manipulating autophagy - through knockouts, knock-downs and overexpression of the ATG genes - has permitted, for the first time, investigation of the cellular consequences of changes in the activity of this pathway and a link between autophagic malfunction and different human diseases [8,12-14].
Box 1. Identification of autophagy-related genes by genetic screening in yeast.
The identification of the initial subset of genes necessary for autophagy resulted from three genetic screens in Saccharomyces cerevisiae performed almost simultaneously in three different laboratories. [68-70]. The read-outs used for identification of autophagy-defective mutants by the different groups were:
Cellular survival during starvation
Activation of macroautophagy is essential for survival of yeast cells in the absence of food. Autophagy mutants were identified as those mutants unable to grow in nutrient-poor media.
Degradation of intracellular proteins
Starvation-induced macroautophagy results in ‘in bulk’ degradation of the cellular cytosol. Autophagy-defective mutants were identified by comparing the cellular levels of particular cytosolic proteins in cells maintained in nutrient-rich or nutrient-poor media. The inability to increase the degradation of the cytosolic protein (persistent high levels) when changed into a media lacking nutrients is characteristic of the autophagy-defective mutants.
Morphological alteration of the autophagic compartments
Autophagic bodies released into the vacuole are easily identifiable in S. cerevisiae when vacuolar proteases are inhibited. An analysis of these compartments in cells placed in a nutrient-poor media enabled the identification of mutants with alterations in some of the steps of the macroautophagic process (e.g. delivery and fusion of the autophagosomes with the vacuoles, degradation once inside the vacuole, etc.).
Pathophysiology of autophagy
The participation of autophagy in different physiological functions is attributable, in almost all cases, to one of its two main functions: as an energy source or as a mechanism for the removal of unwanted cellular structures [15] (Figure 1). As described earlier, the essential components resulting from macromolecule degradation in lysosomes can be reused not only for the synthesis of new macromolecules but also as cellular fuel. In addition, the unique ability of the lysosomal system to sequester and degrade entire cellular regions gives a prominent role to autophagy in conditions requiring extensive cellular remodeling such as cell differentiation, embryogenesis and even in the complete cellular destruction that occurs in some forms of cell death [8,12-14]. Furthermore, autophagy has been implicated in both innate and acquired immunity through sampling, digestion and presentation of peptides from both invasive cellular pathogens and also from their own cellular milieu [16].
Figure 1.
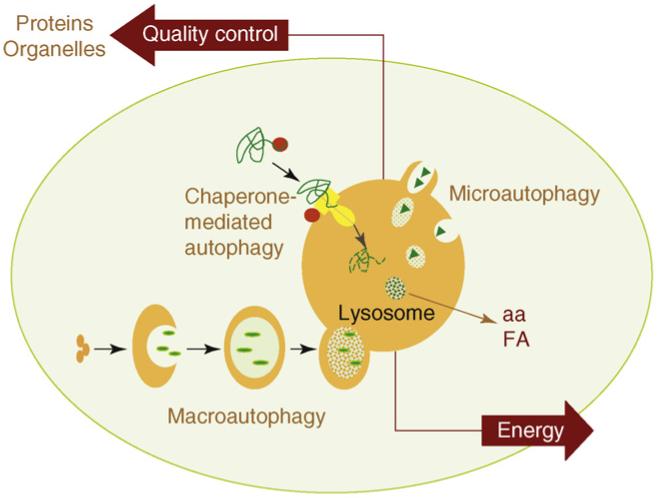
Schematic model of the different types of autophagy in mammalian cells. Three different main types of autophagy have been described in mammalian cells. In macroautophagy, entire regions of the cytosol, including proteins and organelles, are sequestered in a double membrane vesicle that then fuses with lysosomes to enable cargo degradation. During microautophagy, the lysosomal membrane invaginates or protrudes to internalize cytosolic components in single membrane vesicles that are then rapidly degraded in the lysosomal lumen. Selective degradation of soluble cytosolic proteins can be attained by chaperone-mediated autophagy, in which candidate substrate proteins (green) are brought to a translocation complex (yellow) at the lysosomal membrane by cytosolic chaperones (red). Substrate proteins can only reach the lysosomal lumen by this pathway after undergoing complete unfolding in the cytosolic side of the lysosomal membrane. The two most important functions of autophagy are also depicted in this model: (i) as an alternative source of energy, by providing essential components - such as amino acids (aa) or free fatty acids (FA) - that can be used for cellular fueling; and (ii) as an essential component of the cellular mechanisms for quality control, by guaranteeing removal of altered proteins and organelles.
Genetic manipulation of autophagy in different organisms has also revealed the contribution of autophagic malfunction to disease. In recent years, autophagy has been shown to underlie the basis of certain types of cancer, muscular disorders, metabolic alterations, including some complications of the diabetic syndrome, infectious diseases and protein conformational disorders, among which neurodegenerative diseases stand by themselves in their well established connections to the autophagic process [8,12-14]. In later sections, I comment on recent studies supporting a causal role for the failure of autophagy in some common age-related diseases, but a comprehensive analysis of the changes in the autophagic system in disease is beyond the scope of the present review (readers are referred to some of the published work on this topic [8,12-14]).
Basic mechanisms of autophagy
The detailed molecular characterization of autophagy and the improved understanding of the physiological roles for this pathway have also resulted in a growing number of terms to refer to this process. The differences among the autophagic variations reside in the type of cargo, the mechanism for its delivery to lysosomes and the conditions in which that particular process is activated. However, despite the variety of autophagic processes described, they all fall into one of three main types: macroautophagy, microautophagy or chaperone-mediated autophagy [8,14,17] (Figure 1).
Macroautophagy is the most extensively analyzed and quantitatively more important type of autophagy in which entire regions of the cytosol are sequestered by a de novo-formed limiting membrane that seals into an autophagosome [11] (Figure 1). These double-membrane vesicles acquire the proteases required for degradation of the sequestered material by fusion with secondary lysosomes. Two conjugation events - a protein to protein and a protein to lipid conjugation - and two intracellular kinase complexes - the beclin-VPS34 (named for vacuolar protein sorting) complex and the mTOR (mammalian target of rapamycin) complex - act coordinately to modulate formation, sealing and fusion of the autophagic compartments with lysosomes [11,18]. Although macroautophagy was considered an inducible type of autophagy, growing evidence supports the existence of basal macroautophagy, essential for maintenance of cellular homeostasis in different organs [19-22]. In addition to this ‘in bulk’ degradation, selective sequestration of cellular structures into autophagosomes is also a common mechanism for removal of organelles or particulate protein complexes (aggregates) [23].
The process of microautophagy is poorly characterized, at least in mammalian cells. Like in macroautophagy, entire cytosolic regions constitute the microautophagy cargo, but it is the lysosomal membrane itself that invaginates or protrudes to seclude the cytosolic components [24] (Figure 1). Variations of microautophagy have been described in yeast for the selective degradation of peroxisomes (micropexophagy) or even nuclear regions (piecemeal microautophagy) [23].
Selective autophagy of soluble cytosolic proteins can occur via chaperone-mediated autophagy (CMA), the only type of autophagy that requires unfolding of the protein cargo before internalization into lysosomes [13,25]. Selectivity is determined via the recognition by a cytosolic chaperone of a pentapeptide in the amino acid sequence of the substrate proteins that are, thus, delivered to the surface of the lysosomes (Figure 1). To reach the lysosomal lumen, substrate proteins interact with a receptor protein, the lysosome-associated protein type 2A (LAMP-2A), and a complex of lysosomal chaperones at both sides of the lysosomal membrane [25]. Although CMA is maximally activated under stress, different levels of basal activity can be detected in many cell types [13].
Autophagy and aging: before the genetic dissection
A decrease in proteolytic activity has been considered responsible, at least in part, for the accumulation of damaged cellular components in almost all tissues of aging organisms (reviewed in Refs [6,7,26]). Indeed, age-dependent alterations in the lysosomal system and declined autophagic activity were described long before the molecular basis for this process was fully understood. Most of the early functional studies were performed in the liver, as this was also the organ in which the most extensive characterization of the different autophagic pathways was available [26]. However, the morphological features of the aging lysosomal system (e.g. expansion of lysosomal compartments, accumulation of autophagic vacuoles and deposition of undigested material inside lysosomes in the form of an autofluorescent pigment termed lipofuscin) were also described in non-dividing cells of many other tissues (e.g. brain, heart, muscle and kidney). Failure of the quality control mechanisms are particularly detrimental in post-mitotic cells, in which distribution of damaged components to daughter cells is not possible because their cells no longer undergo cell division [27,28].
Measurement of autophagic activity (the rate of degradation of long-lived proteins) in rodent livers revealed an age-dependent decrease in lysosomal-mediated degradation that correlated with an increase in damaged proteins in this organ [29]. The reasons for this reduced macroautophagic activity with age, at least in the liver, seem to be twofold: a defect in the clearance of autophagic vacuoles and problems in the hormonal regulation of this type of autophagy (Figure 2). The half-life of newly formed autophagosomes most likely increases because of the inability of the secondary lysosomes to fuse and/or degrade the autophagosome cargo [30]. Failure of the lysosomal hydrolases, as a result of the gradual increase of lipofuscin in lysosomes and of primary damage to this compartment by toxic protein products, has been proposed to underlie the slow clearance of autophagosomes in the aging liver [31]. Other studies in rodent liver have identified alterations in the response of macroautophagy to changes in circulating levels of insulin and glucagon with age [32,33]. In young organisms, the increase in glucagon levels during starvation upregulates macroautophagy, whereas insulin, secreted as nutrients are absorbed in the gut, enhances the mTOR-mediated repression of this pathway (Figure 2). The inability of glucagon to upregulate macroautophagy in aging livers has been proposed to result from the basal signaling activity of the insulin receptor with age, which is also enhanced by oxidative stress in the absence of insulin (reviewed in Ref. [34]). Macroautophagy deregulation in old organisms could also be a consequence of its persistent activation because of the gradual resistance to insulin, typical of aging cells. Although increased autophagy could be initially beneficial, its maintained activation could lead to chronic depletion of essential autophagic components and failure to upregulate this pathway when needed [35].
Figure 2.
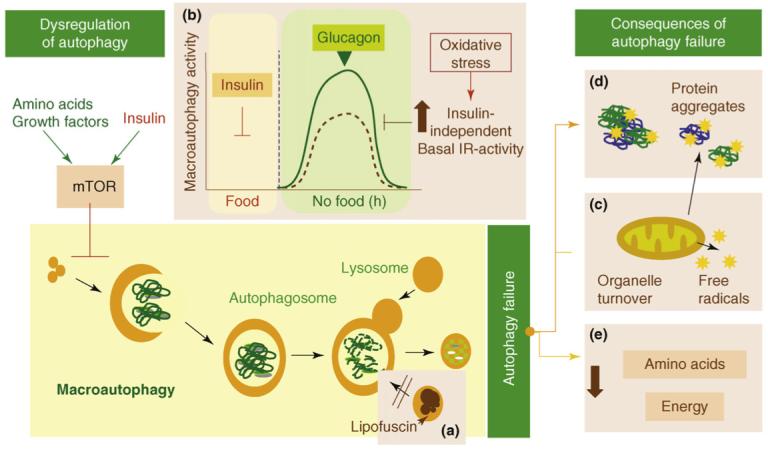
Failure of macroautophagy in aging. Possible causes and consequences of the failure of macroautophagy in old organisms are depicted in this schematic model (brown boxes). (a) The accumulation of autophagic vacuoles with age could result from the inability of lipofuscin-loaded lysosomes to fuse with autophagic vacuoles and degrade the sequestered content. (b) In addition, the formation of autophagosomes in old cells might be reduced because of the inability of macroautophagy enhancers (such as glucagon) to induce full activation of this pathway. The stimulatory effect of glucagon is compromised in old cells because of maintained negative signaling through the insulin receptor (IR) even under basal conditions (i.e. in the absence of insulin). Maintained insulin signaling would activate mTOR, a known repressor of macroautophagy. (c) Inadequate turnover of organelles, such as mitochondria, in aging cells could increase levels of free radicals that generate protein damage and (d) could also potentiate the inhibitory signaling through the insulin receptor. (e) An age-dependent decline in macroautophagy can also result in energetic compromise of the aging cells.
Studies in rodent livers also provided the first evidence that caloric restriction, the only intervention known to effectively decelerate aging, prevented the decline of macroautophagy activity with age [32,33,36]. Limited dietary intake decreases the incidence of age-related disorders and increases life span in numerous experimental models from yeast and invertebrates to mammals including primates (reviewed in Refs [37,38]). The logical connection between caloric restriction and autophagy stems from the findings that that autophagy is a catabolic process upregulated during nutrient deprivation and that tissues from calorically restricted animals contain markedly lower levels of damaged components when compared to age matched ad libitum fed animals. Both roles of autophagy, as an energy source when nutrients are scarce and as a process to remove cellular damage, fit well with the beneficial effect of caloric restriction.
The age-related decrease in the other nutritionally regulated autophagy pathway - CMA - was also described before its molecular components were identified. Early studies in cultured primary human fibroblasts revealed that the degradation of a selective pool of cytosolic proteins in lysosomes (through what is now known as CMA) was gradually impaired during passage in culture [39]. This decrease in CMA in aging human fibroblasts was later confirmed in different tissues of old rodents [40].
The new connections between autophagy, aging and life span
Genetic screens in yeast (in the case of macroautophagy) and novel biochemical approaches (in the case of CMA) have helped to identify the subset of genes and protein products that participate as effectors and modulators of these autophagic pathways. Manipulations in these genes have confirmed the tight connection between autophagy, life span and aging (Table 1).
Table 1. Genes shown to affect both autophagy and life span in C. elegans.
| Gene namea | Modification | Autophagy | Life span | Refs |
|---|---|---|---|---|
| cep-1/p53 | Mutant | Increased | Increased | [46] |
| daf-2 | Mutant | Increased | Increased | [43,45,49] |
| daf-15/Raptor | Mutant | Increased | Increased | [45] |
| daf-16/FOXO | Mutant | No change (in daf-2 mutants) | Decreased | [45] |
| TOR/mTOR | Inhibition | Increased | Increased | [45] |
| RNAi | Increased | Increased | [45,49] | |
| eat-2 (DR)d | Mutant | Increased | Increased | [45,52] |
| eat-3 (DR) | Mutant | Increased | Increased | [52] |
| pha-2 (DR) | Mutant | Increased | Increased | [52] |
| pha-3 (DR) | Mutant | Increased | Increased | [52] |
| pha-4/FOXA | RNAi | Decreased | Decreased in daf-2 mutants | [45] |
| rab-10 | Mutant, RNAi | Increased | Increased in DR | [45] |
| No change in fed | ||||
| vps-34c | RNAi | Decreased | Decreased in daf-2, eat-2 mutants | [45] |
| No change in wild type | [45] | |||
| unc-51/ATG-1 | Mutant | Decreased | Decreased in eat-2 mutants | [43,49] |
| Decreased in wild type?b | [43,49] | |||
| bec-1/beclin-1 | Mutant, RNAi | Decreased | Decreased in: daf-2, daf-15, eat-2 mutants | [43-46,49,50] |
| No change/decreased in wild type?b | [43-46,49,50] | |||
| M7.5/ATG-7 | RNAi | Decreased | Decreased in daf-2, eat-2 mutants | [44,50] |
| No change/decreased in wild type?b | [44,50] | |||
| lgg-1/LC3 | RNAi | Decreased | Decreased in daf-2 mutants | [43] |
| No change/decreased in wild type?b | [43,49] | |||
| atg-9 | RNAi | Decreased | Decreased in wild type | [49] |
| atg-12 | RNAi | Decreased | Decreased in daf-2 mutants | [44] |
| Decreased in wild type | [44] | |||
| atg-18 | Mutant | Decreased | Decreased in wild type and daf-2 | [49] |
Gene name denotes C. elegans gene nomenclature. When double nomenclature is used, the second name is the mammalian ortholog.
‘?’ denotes controversial findings.
Shadowed boxes denote essential autophagy genes.
Abbreviations: DR, dietary restriction.
The first genetic connection between autophagy and aging was established in the worm Caenorhabditis elegans, an organism extensively used to analyze changes in protein degradation with age [41], in addition to the identification of genes that mediate life-span extension [42]. Mutations in different genes that reduce signaling through the equivalent of the insulin-signaling pathway in worms, such as daf-2, can extend longevity [42]. Knock-down of essential autophagy proteins in daf-2 mutants drastically reduced their life-span extension [43,44] (Figure 3). Functional autophagy has also proven necessary to attain the maximal life-span extension mediated by deletion of other genes such as those encoding mTOR [45] or the tumor suppressor p53 [46], which curiously represses macroautophagy in mammals [47]. Likewise, factors that promote longevity in invertebrates, such as the Foxo family of Forkhead transcription factors, upregulate macroautophagy [48]. Indeed, macroautophagy activation is a common feature of all the long-lived mutant worms [43-45,49]. In contrast to the well established role of autophagy in the ‘superlongevity’ of these mutant worms, the requirement of functional autophagy for normal life span remains a controversial matter in this model. Thus, whereas mutation of essential autophagic genes shortened the life span of wild-type worms in some of these studies [44,49], it had no effect in others [43,46,50]. The reasons for these discrepant results remain to be elucidated. Regardless of this controversy, accumulation of intracellular damage and signs of cellular aging have been observed in both worms and flies defective for essential autophagy genes [49,51].
Figure 3.
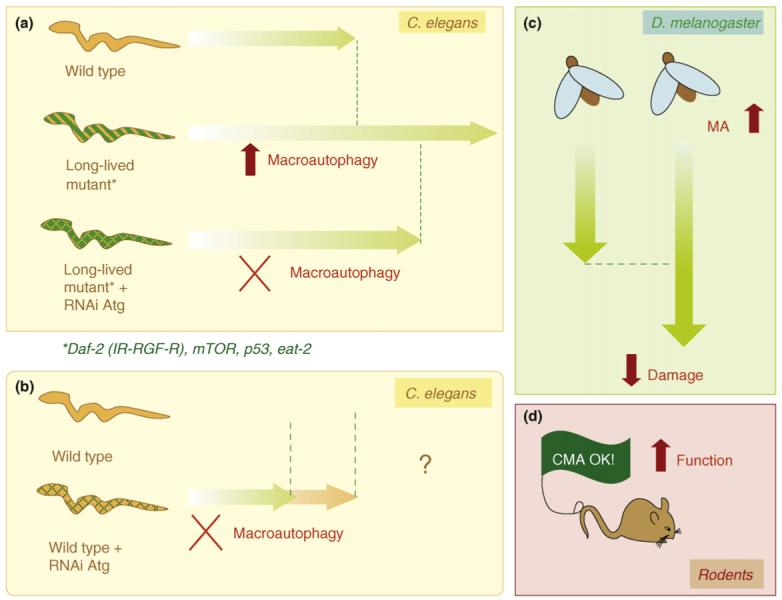
Effects of genetic manipulations on autophagy in cellular aging and longevity. Studies in worms (a) have revealed that macroautophagy is upregulated in different long-lived mutants (life span is indicated by green arrows). Blockage of macroautophagy by siRNA against essential autophagy proteins (Atg) in these mutant worms reduces their life-span extension, supporting the view that functional autophagy is required to attain maximal life extension in these worms. (b) Whether or not macroautophagy blockage by similar procedures reduces normal life span in wild-type worms remains controversial (denoted by?). (c) Macroautophagy upregulation in neurons decreases age-dependent neuronal damage and increases life span in flies. (d) Recently, the prevention of the age-related decline in CMA activity in the liver of a transgenic mouse model improved overall hepatic function.
The role of macroautophagy in life-span extension mediated by caloric restriction was recently genetically confirmed in C. elegans. Macroautophagy is upregulated in feeding-defective worm mutants used to model dietary restriction [52] and disruption of essential autophagy genes shortened their life span [50]. This is an interesting finding because caloric restriction further increases life span in the long-lived insulin-signaling mutants, indicating that both interventions influence aging by different mechanisms. Macroautophagy, thus, reveals itself as a possible common effector of both pathways, a role probably attributable to the dual regulation of macroautophagy by insulin-signaling and mTOR (Figure 3). Mutations in the TOR pathway increase life span in invertebrates, but these mutants no longer respond to caloric restriction, thereby supporting the idea that attenuation of TOR signaling is part of the downstream mechanism involved in the beneficial effects of caloric restriction [53]. TOR is a negative regulator of macroautophagy that signals as a downstream kinase of the insulin-signaling pathway. However, insulin-independent signaling through TOR is possible because this kinase is also activated by nutrients and growth factors [54]. Blockage of this last signaling mechanism is probably responsible for maintained activation of macroautophagy during caloric restriction. Interestingly, although macroautophagy is required for life-span extension in all these long-lived worm mutants, at least in worms, it is not sufficient and it probably acts in parallel with other downstream pathways [45].
The identification of the different molecular components of CMA has facilitated a better understanding of the reasons for the declined activity of this type of autophagy with age. Indeed, a step-by-step comparative analysis of CMA in livers of young and old rodents revealed that substrate recognition by the cytosolic chaperones, targeting to the lysosomal membrane and degradation in the lysosomal lumen are preserved until late in life [40]. The problem, however, lies in the binding and lysosomal translocation of the substrate proteins because of progressively lower levels of the CMA receptor at the lysosomal membrane with age [40]. This defect - also confirmed in other tissues of old rodents and in human aging fibroblasts - is not because of transcriptional downregulation of this receptor, but instead it is a consequence of instability of this protein once it reaches the lysosomal membrane. Changes with age, probably in the lipid composition of the lysosomal membrane, are behind the instability of the CMA receptor, LAMP-2A, in old organisms [55].
Consequences of autophagic failure in aging
In light of the myriad of physiological functions of autophagy, it is easy to infer that the described age-related decline in autophagic activity will affect normal cell functioning and contribute to different aspects of the aging phenotype. Recent studies using tissue-specific conditional autophagy-knockout mice have confirmed the important role of this catabolic process in the maintenance of cellular homeostasis and proper response to stress [19-22]. Thus far, changes in life span and health span have not been analyzed in these rodents with impaired autophagy. However, worms and flies with defective autophagy have decreased life span and earlier features of cellular aging [49,51,56]. An important limitation of the studies both in invertebrates and in mammals is that autophagic blockage is induced early in life, whereas the age-dependent decrease in this pathway does not begin in most organisms until middle age. It is, thus, possible that compensatory mechanisms that can be activated early in life in response to the autophagic failure are no longer possible late in life, resulting in even more dramatic consequences. In support of this possibility, recent studies in mouse models with temporally controlled knockout of an essential autophagy gene in the heart have revealed that autophagic impairment early in development does not result in altered heart function, unless these animals are exposed to additional stress. By contrast, autophagic blockage late in adulthood results in marked heart failure, even under normal resting conditions [22]. Future studies on the consequences of macroautophagic decline with age should take into account these temporal differences.
The use of LAMP-2A knockdown in cultured fibroblasts to reproduce the age-related decline in CMA activity has confirmed the importance of this pathway in the removal of altered cytosolic proteins and as part of the cellular response to stress [57]. However, selective blockage of CMA in a whole organism, to analyze the functional consequences of the accumulation of damaged products observed in the cultured cells, has not been possible yet. The main limitation in this respect is the fact that LAMP-2A is one of the three spliced variants of the lamp2 gene and, consequently, knockout of the whole gene would present a more complex phenotype than that resulting only from defective CMA. The use of RNAi against LAMP-2A in animals could help overcome this limitation in the future.
Recently, particular attention has been dedicated to the consequences of reduced macroautophagy and CMA with age in the progression of the so called age-related disorders (Box 2). The fact that alterations in cellular homeostasis or in the cellular response to stress are common to many of these late-onset disorders explains why alterations in autophagy are often found to underlie the basis of these pathologies and/or act as aggravating factors in the course of the disease. Interested readers are referred to recent comprehensive reviews on this topic [8,12,14,58].
Box 2. Autophagy and age-related disorders.
Age-related disorders are a broad group of diseases of higher prevalence in the elder population for which aging is considered a risk factor (e.g. cancer, neurodegenerative diseases, metabolic disorders such as diabetes, etc.). Here, I provide some examples of those disorders for which the aggravating effect of aging in the progression of the disease has been proposed to be, at least in part because of age-related changes in autophagy (reviewed in Refs [8,12,14,58]).
Neurodegenerative disorders
Alzheimer’s disease (AD) and Parkinson’s disease (PD), the two neurodegenerative disorders of highest prevalence in our society, are both late-onset diseases. Alterations in macroautophagy have been identified as one of the early changes in AD affected neurons, whereas a primary defect in CMA has been described in PD. Neurons respond to autophagic failure by up-regulating other proteolytic mechanisms (including autophagic pathways not affected by the disease). It has been proposed that this compensatory stage could be compromised with age because of the functional decline of the different autophagic pathways. This defect would eventually result in accumulation of pathogenic products, cellular death and on-set of symptoms related to loss of the affected neurons.
Cancer
Autophagy acts in tumor suppression by removing damaged organelles and reducing chromosome instability. As autophagic activity decreases with age, accumulation of intracellular damage, dysfunctional organelles and chromosome aberrations increase the chances of oncogenic transformation of somatic cells.
Immunosenescence
Different aspects of the gradual deterioration of the immune system with age could be related to autophagic dysfunction. Decreased autophagy in professional antigen presenting cells (i.e. dendritic cells, macrophages and B-cells) could lead to inefficient presentation of exogenous antigens, poor T-cell activation and failure to orchestrate a proper immune response to pathogens. Furthermore, the inability of non-professional cells to present their own antigens through sampling of the cellular milieu by macroautophagy could underlie the abnormal autoimmune response sometimes observed in aging organisms.
Myopathies
Defective autophagosome clearance in the aging muscle could contribute to muscle wasting (sarcopenia) characteristic of old organisms. As autophagic vacuoles accumulate with age, they interfere with the contractile properties of muscle fibers weakening them and favoring small ruptures by traction, which eventually could lead to muscle atrophy.
Enhancing autophagy as an anti-aging intervention
Autophagy might prevent or decelerate aging by guaranteeing the stability of the cellular proteome and proper organelle turnover. For example, mitochondrial dysfunction, extensively reported to contribute to cellular aging, has been proposed to occur, at least in part, because of the inability of old cells to remove their non-functional mitochondria (reviewed in Refs [59,60]). The recent advances in our understanding of autophagy have motivated a growing interest in developing strategies to enhance autophagic activity to slow-down aging progression.
Pharmacological upregulation of macroautophagy has been successfully applied to different fly and mouse models of neurodegeneration to ameliorate symptoms and slow-down disease progression [61]. Activation of macroautophagy has also been proposed to underlie the anti-aging effect of antilipolytic drugs, although genetic confirmation of its contribution is still pending [62].
Persistent activation of macroautophagy, however, might not be the ideal anti-aging intervention. In fact, recent studies in different mouse models of premature aging (progeria) have reported extensive basal activation of autophagy as a consequence of changes in lipid and glucose metabolism triggered by the nuclear damage. In these cases, chronic activation of this pathway seems to contribute to systemic degeneration and weakening [63]. Instead, a better approach could be to repair the autophagic defect to preserve proper activity until late in life. One current limitation of this approach, however, is the lack of information regarding the macroautophagy component(s) at fault in old organisms. Multiple factors could contribute to the loss of autophagic activity with age and the reason for failure could be tissue-specific. In fact, comparative studies of changes in basal macroautophagy with age in heart and liver have revealed different responses of both tissues to caloric restriction [64]. However, the recent studies in flies are encouraging, in which a decrease in the expression of several autophagy genes in neurons has been observed with age [51] (Figure 3). Enhanced expression of the defective genes has proven effective in prolonging life span and providing resistance to oxidative stress [51]. Decreased expression of some autophagy genes in aging human brain has been recently reported [65], but restorative manipulations like the ones described in flies have not yet been performed in aging mammals.
We have been able to prevent the age-dependent decrease of CMA in liver through genetic manipulation in mice [66] (Figure 3). Activation of an exogenously added copy of the CMA receptor in these dual transgenic mice when the levels of the endogenous protein start to decrease (around 10 months of age) prevents the decline in CMA activity in old animals. The livers of the mice with preserved CMA function show better cellular homeostasis, improved ability to respond to the cell damage induced by different stressors and an overall liver function comparable to that in young mice [66]. This is the first evidence in mammals that preventing the age-related decline in autophagic activity slows-down cellular aging and preserves organ function. Interestingly, the activity of other quality control mechanisms (e.g. macroautophagy and the ubiquitin-proteasome system) was also improved in these animals [66]. It is possible that a cleaner cellular milieu has a beneficial effect on the function of different cellular processes, including other clearance mechanisms, or that the previously described cross-talk among these mechanisms could promote their functional improvement [57].
Concluding remarks and future perspectives
Genetic studies both in invertebrate and in mouse models have recently provided strong evidence of interplay between autophagy, health span and aging. The essential role of autophagy in the maintenance of cellular homeostasis and in the forefront of the response to stress positions it as a crucial player in the fight against cellular aging. Nevertheless, the mechanistic studies of the anti-aging effect of autophagy have just taken off. Generalizations have frequently been made in light of particular findings; however, more detailed studies are required to answer several key questions: (i) Is autophagy required in all interventions known to prolong life span (e.g. contribution of autophagy to the anti-aging effect of methionine restriction has not been experimentally tested yet)? (ii) Is autophagy upregulation a common feature of all models of accelerated aging (e.g. although morphological changes of the autophagic system have been described in the short-lived Klotho mutant mice [67], the analysis of their autophagic function is still missing)? (iii) Will similar manipulations to those used in the liver enable the prevention of the age-related decline of CMA in other tissues?
Likewise, a detailed dissection of the macroautophagy step or steps affected in aging organisms is essential before any restorative attempt can be implemented. Future studies should also concentrate on resolving some of the current controversies regarding the effect of autophagy on life span in different worm models and on comparing them to the effect(s) in other organisms. Despite the large number of ‘pending tasks’, the role of autophagy as an aging-fighter is now widely accepted and it is only a matter of time until the knowledge gained through these types of studies can be applied for the preservation of a healthy life.
Acknowledgements
I thank my numerous colleagues both in the fields of autophagy and of biology of aging who, through their animated discussions, have helped shape this review. I am in particular debt to Ashish Massey, Fernando Macian and Susmita Kaushik for critically reading the manuscript. Work in my laboratory is supported by NIH/NIA (AG021904, AG031782), NIH/NIDDK (DK041918) and a Glenn Foundation Award. I sincerely apologize to colleagues whose work has not been cited owing to space limitations.
Footnotes
- Autophagy
- degradation of any type of intracellular components including protein, organelles or any type of particulate structures (e.g. protein aggregates, cellular inclusions, etc.) in lysosomes.
- Catabolism
- breakdown of macromolecules into their essential components as a result of the coordinate function of different intracellular metabolic pathways. Catabolic processes provide cells with chemical energy and building blocks for the synthesis of new macromolecules.
- Chaperone-mediated autophagy
- autophagic pathway responsible for the selective degradation of a subset of cytosolic proteins in lysosomes upon unfolding and translocation across the membrane.
- Health span
- total number of healthy years of life of an organism.
- Life span
- average length of survival of an organism.
- Long-lived worm mutants
- worms bearing a mutation that increases in their life span in comparison to their wild-type counterparts.
- Lysosomes
- single membrane vesicles with acid luminal pH that contain higher concentrations of different types of hydrolases able to degrade all type of macromolecules.
- Macroautophagy
- type of autophagy by which entire regions of the cytosol sequestered inside a double membrane structure (autophagosome) are delivered as a whole into lysosomes through vesicular fusion.
- Microautophagy
- type of autophagy in which the lysosomal membrane invaginates or protrudes to seclude cytosolic regions that are internalized into the lumen as single membrane vesicles.
- mTOR
- nutrient-regulated mammalian kinase identified by its sensitivity to rapamycin (target of rapamycin) that acts as negative regulator of macroautophagy.
References
- 1.Morimoto RI. Proteotoxic stress and inducible chaperone networks in neurodegenerative disease and aging. Genes Dev. 2008;22:1427–1438. doi: 10.1101/gad.1657108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Goldberg AL. Protein degradation and protection against misfolded or damaged proteins. Nature. 2003;426:895–899. doi: 10.1038/nature02263. [DOI] [PubMed] [Google Scholar]
- 3.Feldman DE, Frydman J. Protein folding in vivo: the importance of molecular chaperones. Curr. Opin. Struct. Biol. 2000;10:26–33. doi: 10.1016/s0959-440x(99)00044-5. [DOI] [PubMed] [Google Scholar]
- 4.Ciechanover A. Proteolysis: from the lysosome to ubiquitin and the proteasome. Nat. Rev. Mol. Cell Biol. 2005;6:79–87. doi: 10.1038/nrm1552. [DOI] [PubMed] [Google Scholar]
- 5.Finkbeiner S, et al. Disease-modifying pathways in neurodegeneration. J. Neurosci. 2006;26:10349–10357. doi: 10.1523/JNEUROSCI.3829-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ward WF. Protein degradation in the aging organism. Prog. Mol. Subcell. Biol. 2002;29:35–42. doi: 10.1007/978-3-642-56373-7_3. [DOI] [PubMed] [Google Scholar]
- 7.Martinez-Vicente M, et al. Protein degradation and aging. Exp. Gerontol. 2005;40:622–633. doi: 10.1016/j.exger.2005.07.005. [DOI] [PubMed] [Google Scholar]
- 8.Mizushima N, et al. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–1075. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.De Duve C, Wattiaux R. Functions of lysosomes. Annu. Rev. Physiol. 1966;28:435–492. doi: 10.1146/annurev.ph.28.030166.002251. [DOI] [PubMed] [Google Scholar]
- 10.Klionsky DJ, et al. A unified nomenclature for yeast autophagy-related genes. Dev. Cell. 2003;5:539–545. doi: 10.1016/s1534-5807(03)00296-x. [DOI] [PubMed] [Google Scholar]
- 11.Yorimitsu T, Klionsky DJ. Autophagy: molecular machinery for self-eating. Cell Death Differ. 2005;12:1542–1552. doi: 10.1038/sj.cdd.4401765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shintani T, Klionsky D. Autophagy in health and disease: a double-edged sword. Science. 2004;306:990–995. doi: 10.1126/science.1099993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Massey AC, et al. Chaperone-mediated autophagy in aging and disease. Curr. Top. Dev. Biol. 2006;73:205–235. doi: 10.1016/S0070-2153(05)73007-6. [DOI] [PubMed] [Google Scholar]
- 14.Cuervo AM. Autophagy: in sickness and in health. Trends Cell Biol. 2004;14:70–77. doi: 10.1016/j.tcb.2003.12.002. [DOI] [PubMed] [Google Scholar]
- 15.Mizushima N, Klionsky D. Protein turnover via autophagy: implications for metabolism. Annu. Rev. Nutr. 2007;27:19–40. doi: 10.1146/annurev.nutr.27.061406.093749. [DOI] [PubMed] [Google Scholar]
- 16.Levine B, Deretic V. Unveiling the roles of autophagy in innate and adaptive immunity. Nat. Rev. Immunol. 2007;7:767–777. doi: 10.1038/nri2161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cuervo AM. Autophagy: many pathways to the same end. Mol. Cell. Biochem. 2004;263:55–72. doi: 10.1023/B:MCBI.0000041848.57020.57. [DOI] [PubMed] [Google Scholar]
- 18.Ohsumi Y. Molecular dissection of autophagy: two ubiquitin-like systems. Nat. Rev. Mol. Cell Biol. 2001;2:211–216. doi: 10.1038/35056522. [DOI] [PubMed] [Google Scholar]
- 19.Hara T, et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441:885–889. doi: 10.1038/nature04724. [DOI] [PubMed] [Google Scholar]
- 20.Komatsu M, et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J. Cell Biol. 2005;169:425–434. doi: 10.1083/jcb.200412022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Komatsu M, et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441:880–884. doi: 10.1038/nature04723. [DOI] [PubMed] [Google Scholar]
- 22.Nakai A, et al. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat. Med. 2007;13:619–624. doi: 10.1038/nm1574. [DOI] [PubMed] [Google Scholar]
- 23.Klionsky DJ, et al. How shall I eat thee? Autophagy. 2007;3:413–416. doi: 10.4161/auto.4377. [DOI] [PubMed] [Google Scholar]
- 24.Marzella L, et al. Autophagy, heterophagy, microautophagy and crinophagy as the means for intracellular degradation. Virchows Arch. B Cell Pathol. Incl. Mol. Pathol. 1981;36:219–234. doi: 10.1007/BF02912068. [DOI] [PubMed] [Google Scholar]
- 25.Dice JF. Chaperone-mediated autophagy. Autophagy. 2007;3:295–299. doi: 10.4161/auto.4144. [DOI] [PubMed] [Google Scholar]
- 26.Cuervo AM, et al. Autophagy and aging: the importance of maintaining “clean” cells. Autophagy. 2005;1:131–140. doi: 10.4161/auto.1.3.2017. [DOI] [PubMed] [Google Scholar]
- 27.Stroikin Y, et al. Testing the ‘garbage’ accumulation theory of ageing: mitotic activity protects cells from death induced by inhibition of autophagy. Biogerontology. 2005;6:39–47. doi: 10.1007/s10522-004-7382-y. [DOI] [PubMed] [Google Scholar]
- 28.Navratil M, et al. Giant mitochondria do not fuse and exchange their contents with normal mitochondria. Exp. Cell Res. 2008;314:164–172. doi: 10.1016/j.yexcr.2007.09.013. [DOI] [PubMed] [Google Scholar]
- 29.Vittorini S, et al. The age-related accumulation of protein carbonyl in rat liver correlates with the age-related decline in liver proteolytic activities. J. Gerontol. A Biol. Sci. Med Sci. 1999;54:B318–B323. doi: 10.1093/gerona/54.8.b318. [DOI] [PubMed] [Google Scholar]
- 30.Terman A. The effect of age on formation and elimination of autophagic vacuoles in mouse hepatocytes. Gerontology. 1995;41:319–325. doi: 10.1159/000213753. [DOI] [PubMed] [Google Scholar]
- 31.Brunk UT, Terman A. Lipofuscin: mechanisms of age-related accumulation and influence on cell function. Free Radic. Biol. Med. 2002;33:611–619. doi: 10.1016/s0891-5849(02)00959-0. [DOI] [PubMed] [Google Scholar]
- 32.Donati A, et al. Age-related changes in the regulation of autophagic proteolysis in rat isolated hepatocytes. J. Gerontol. A Biol. Sci. Med. Sci. 2001;56:B288–B293. doi: 10.1093/gerona/56.7.b288. [DOI] [PubMed] [Google Scholar]
- 33.Donati A, et al. Effect of aging and anti-aging caloric restriction on the endocrine regulation of rat liver autophagy. J. Gerontol. A Biol. Sci. Med. Sci. 2008;63:550–555. doi: 10.1093/gerona/63.6.550. [DOI] [PubMed] [Google Scholar]
- 34.Droge W, Kinscherf R. Aberrant insulin receptor signaling and amino acid homeostasis as a major cause of oxidative stress in aging. Antioxid. Redox Signal. 2008;10:661–678. doi: 10.1089/ars.2007.1953. [DOI] [PubMed] [Google Scholar]
- 35.Meijer AJ, Codogno P. Macroautophagy: protector in the diabetes drama? Autophagy. 2007;3:523–526. doi: 10.4161/auto.4449. [DOI] [PubMed] [Google Scholar]
- 36.Cavallini G, et al. The protection of rat liver autophagic proteolysis from the age-related decline co-varies with the duration of anti-ageing food restriction. Exp. Gerontol. 2001;36:497–506. doi: 10.1016/s0531-5565(00)00224-2. [DOI] [PubMed] [Google Scholar]
- 37.Lane MA, et al. Caloric restriction mimetics: a novel approach for biogerontology. Methods Mol. Biol. 2007;371:143–149. doi: 10.1007/978-1-59745-361-5_11. [DOI] [PubMed] [Google Scholar]
- 38.Masoro EJ. Caloric restriction and aging: controversial issues. J. Gerontol. A Biol. Sci. Med. Sci. 2006;61:14–19. doi: 10.1093/gerona/61.1.14. [DOI] [PubMed] [Google Scholar]
- 39.Okada AA, Dice JF. Altered degradation of intracellular proteins in aging human fibroblasts. Mech. Ageing Dev. 1984;26:341–356. doi: 10.1016/0047-6374(84)90105-2. [DOI] [PubMed] [Google Scholar]
- 40.Cuervo AM, Dice JF. Age-related decline in chaperone-mediated autophagy. J. Biol. Chem. 2000;275:31505–31513. doi: 10.1074/jbc.M002102200. [DOI] [PubMed] [Google Scholar]
- 41.Reznick AZ, Gershon D. The effect of age on the protein degradation system in the nematode Turbatrix aceti. Mech. Ageing Dev. 1979;11:403–415. doi: 10.1016/0047-6374(79)90016-2. [DOI] [PubMed] [Google Scholar]
- 42.Johnson TE. Caenorhabditis elegans 2007: the premier model for the study of aging. Exp. Gerontol. 2008;43:1–4. doi: 10.1016/j.exger.2007.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Melendez A, et al. Autophagy genes are essential for dauer development and life-span extension in C. elegans. Science. 2003;301:1387–1391. doi: 10.1126/science.1087782. [DOI] [PubMed] [Google Scholar]
- 44.Hars ES, et al. Autophagy regulates ageing in C. elegans. Autophagy. 2007;3:93–95. doi: 10.4161/auto.3636. [DOI] [PubMed] [Google Scholar]
- 45.Hansen M, et al. A role for autophagy in the extension of lifespan by dietary restriction in C. elegans. PLoS Genet. 2008;4:e24. doi: 10.1371/journal.pgen.0040024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tavernarakis N, et al. The effects of p53 on whole organism longevity are mediated by autophagy. Autophagy. 2008;4:870–873. doi: 10.4161/auto.6730. [DOI] [PubMed] [Google Scholar]
- 47.Crighton D, et al. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell. 2006;126:121–134. doi: 10.1016/j.cell.2006.05.034. [DOI] [PubMed] [Google Scholar]
- 48.Salih DA, Brunet A. FoxO transcription factors in the maintenance of cellular homeostasis during aging. Curr. Opin. Cell Biol. 2008;20:126–136. doi: 10.1016/j.ceb.2008.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tóth ML, et al. Longevity pathways converge on autophagy genes to regulate life span in Caenorhabditis elegans. Autophagy. 2008;4:330–338. doi: 10.4161/auto.5618. [DOI] [PubMed] [Google Scholar]
- 50.Jia K, Levine B. Autophagy is required for dietary restriction-mediated life span extension in C. elegans. Autophagy. 2007;3:597–599. doi: 10.4161/auto.4989. [DOI] [PubMed] [Google Scholar]
- 51.Simonsen A, et al. Promoting basal levels of autophagy in the nervous system enhances longevity and oxidant resistance in adult Drosophila. Autophagy. 2008;4:176–184. doi: 10.4161/auto.5269. [DOI] [PubMed] [Google Scholar]
- 52.Mörck C, Pilon M. C. elegans feeding defective mutants have shorter body lengths and increased autophagy. BMC Dev. Biol. 2006;6:39. doi: 10.1186/1471-213X-6-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kaeberlein M, et al. Regulation of yeast replicative life span by TOR and Sch9 in response to nutrients. Science. 2005;310:1193–1196. doi: 10.1126/science.1115535. [DOI] [PubMed] [Google Scholar]
- 54.Sarbassov DD, et al. Growing roles for the mTOR pathway. Curr. Opin. Cell Biol. 2005;17:596–603. doi: 10.1016/j.ceb.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 55.Kiffin R, et al. Altered dynamics of the lysosomal receptor for chaperone-mediated autophagy with age. J. Cell Sci. 2007;120:782–791. doi: 10.1242/jcs.001073. [DOI] [PubMed] [Google Scholar]
- 56.Juhasz G, et al. Atg7-dependent autophagy promotes neuronal health, stress tolerance, and longevity but is dispensable for metamorphosis in Drosophila. Genes Dev. 2007;21:3061–3066. doi: 10.1101/gad.1600707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Massey AC, et al. Consequences of the selective blockage of chaperone-mediated autophagy. Proc. Natl. Acad. Sci. U. S. A. 2006;103:5905–5910. doi: 10.1073/pnas.0507436103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Martinez-Vicente M, Cuervo AM. Autophagy and neurodegeneration: when the cleaning crew goes on strike. Lancet Neurol. 2007;6:352–361. doi: 10.1016/S1474-4422(07)70076-5. [DOI] [PubMed] [Google Scholar]
- 59.Jin S. Autophagy, mitochondrial quality control, and oncogenesis. Autophagy. 2006;2:80–84. doi: 10.4161/auto.2.2.2460. [DOI] [PubMed] [Google Scholar]
- 60.Lemasters JJ. Selective mitochondrial autophagy, or mitophagy, as a targeted defense against oxidative stress, mitochondrial dysfunction, and aging. Rejuvenation Res. 2005;8:3–5. doi: 10.1089/rej.2005.8.3. [DOI] [PubMed] [Google Scholar]
- 61.Ravikumar B, et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat. Genet. 2004;36:585–595. doi: 10.1038/ng1362. [DOI] [PubMed] [Google Scholar]
- 62.Donati A, et al. In vivo effect of an antilipolytic drug (3,5′-dimethylpyrazole) on autophagic proteolysis and autophagy-related gene expression in rat liver. Biochem. Biophys. Res. Commun. 2008;366:786–792. doi: 10.1016/j.bbrc.2007.12.023. [DOI] [PubMed] [Google Scholar]
- 63.Marino G, et al. Premature aging in mice activates a systemic metabolic response involving autophagy induction. Hum. Mol. Genet. 2008;17:2196–2211. doi: 10.1093/hmg/ddn120. [DOI] [PubMed] [Google Scholar]
- 64.Wohlgemuth SE, et al. Autophagy in the heart and liver during normal aging and calorie restriction. Rejuvenation Res. 2007;10:281–292. doi: 10.1089/rej.2006.0535. [DOI] [PubMed] [Google Scholar]
- 65.Shibata M, et al. Regulation of intracellular accumulation of mutant Huntingtin by Beclin 1. J. Biol. Chem. 2006;281:14474–14485. doi: 10.1074/jbc.M600364200. [DOI] [PubMed] [Google Scholar]
- 66.Zhang C, Cuervo AM. Restoration of chaperone-mediated autophagy in aging liver improves cellular maintenance and hepatic function. Nat. Med. 2008;14:959–965. doi: 10.1038/nm.1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Shiozaki M, et al. Morphological and biochemical signs of age-related neurodegenerative changes in klotho mutant mice. Neuroscience. 2008;152:924–941. doi: 10.1016/j.neuroscience.2008.01.032. [DOI] [PubMed] [Google Scholar]
- 68.Tsukada M, Ohsumi M. Isolation and characterization of autophagy-defective mutants of Saccharomyces cerevisiae. FEBS Lett. 1993;333:169–174. doi: 10.1016/0014-5793(93)80398-e. [DOI] [PubMed] [Google Scholar]
- 69.Thumm M. Isolation of autophagocytosis mutants of Saccharomyces cerevisiae. FEBS Lett. 1994;349:275–280. doi: 10.1016/0014-5793(94)00672-5. [DOI] [PubMed] [Google Scholar]
- 70.Harding TM. Isolation and characterization of yeast mutants in the cytoplasm to vacuoles protein targeting pathway. J. Cell Biol. 1995;131:591–602. doi: 10.1083/jcb.131.3.591. [DOI] [PMC free article] [PubMed] [Google Scholar]