

Abstract
Embryogenesis is regulated by genetic programs that are dynamically executed in a stereotypic manner, and deciphering these molecular mechanisms requires the ability to control embryonic gene function with similar spatial and temporal precision. Chemical technologies can enable such genetic manipulations, as exemplified by the use of caged morpholino (cMO) oligonucleotides to inactivate genes in zebrafish and other optically transparent organisms with spatiotemporal control. Here we report optimized methods for the design and synthesis of hairpin cMOs incorporating a dimethoxynitrobenzyl (DMNB)-based bifunctional linker that permits cMO assembly in only three steps from commercially available reagents. Using this simplified procedure, we have systematically prepared cMOs with differing structural configurations and investigated how the in vitro thermodynamic properties of these reagents correlate with their in vivo activities. Through these studies, we have established general principles for cMO design and successfully applied them to several developmental genes. Our optimized synthetic and design methodologies have also enabled us to prepare a next-generation cMO that contains a bromohydroxyquinoline (BHQ)-based linker for two-photon uncaging. Collectively, these advances establish the generality of cMO technologies and will facilitate the application of these chemical probes in vivo for functional genomic studies.
Introduction
Embryonic development relies upon the precise control of genetic programs to create complex tissues and organs. Mutagenesis screens and the sequencing of multiple genomes have revealed an extensive list of patterning genes, many of which are expressed in a tissue-specific manner within the developing embryo. One of the remaining challenges in developmental biology is to understand how these genes act in space and time to modulate cell proliferation, migration, and differentiation in a stereotypic manner. Toward that goal, several genetic approaches for conditional gene regulation have been developed, such as the FLP/FRT, Cre/Lox, and Tet-ON/Tet-OFF systems, and these technologies have provided key insights into the molecular mechanisms that underlie tissue patterning and function.(1)
Chemical technologies are also required for surmounting this challenge, especially in biological systems for which reverse-genetic methods are limited. For example, the zebrafish is ideally suited for visualizing vertebrate ontogeny, since its embryos and larvae are optically transparent and develop rapidly ex utero.(2) However, methods for regulating endogenous gene function in zebrafish are underdeveloped relative to those for other model organisms; targeted gene knockouts by homologous recombination and inducible RNA interference technologies have not yet been achieved.(3) In lieu of these approaches, synthetic oligonucleotides such as morpholinos (MOs) and negatively charged peptide nucleic acids (ncPNAs) have been employed as antisense reagents in zebrafish embryos (Figure 1).4,5 MO nucleoside analogues display DNA bases from a morpholine ring system and are connected by a phosphorodiamidate backbone, while ncPNA monomers are composed of alternating trans-4-hydroxy-l-proline/phosphonate polyamides, each functionalized with a DNA base. Because of these non-natural structures, both MOs and ncPNAs are resistant to nucleases and persist for up to 4 days in zebrafish embryos. Thus, when MO or ncPNA oligomers are injected into zebrafish prior to the eight-cell stage, they become uniformly distributed throughout the embryo and constitutively block either RNA splicing or translation, depending on the targeted sequence. In the case of MOs, oligomers containing 25 bases are typically used, while ncPNA oligomers are limited to 18 bases. Unlike RNA interference technologies, MOs and ncPNAs do not promote RNA degradation.(6)
Figure 1.

Structures of DNA, MO, and ncPNA oligonucleotides.
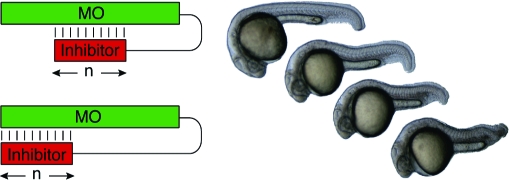
While MOs and ncPNAs have been used to interrogate gene function in zebrafish and other model organisms, the utility of these reagents is restricted by their constitutive activity. Gene expression is inhibited immediately after introduction of the antisense oligomers, and because they are typically injected into early-stage embryos, this blockade persists in most, if not all, cell lineages. Conventional MOs and ncPNAs therefore are less effective for studying genes that are required for embryonic survival and/or have pleiotropic functions at different developmental stages or in distinct tissues. To overcome this limitation, we recently developed caged morpholinos (cMOs) that can be activated by light, taking advantage of the transparency of zebrafish embryos and larvae.(7) This was achieved by tethering a complementary MO-derived inhibitor to the 25-base targeting sequence through a dimethoxynitrobenzyl (DMNB)-based photocleavable linker, resulting in a hairpin structure (Figure 2). The intramolecular duplex suppresses binding of the targeting sequence to its complementary RNA, and the stem−loop configuration is significantly less active in vivo. After linker cleavage with 360-nm light, the inhibitor dissociates from the 25-base MO, allowing the antisense reagent to prevent splicing or translation of its RNA target. Similar caging strategies have previously been described for regulating DNA duplex formation and RNA function in vitro,8−11 and this general approach differs from complementary oligonucleotide-caging technologies that target individual nucleoside bases,12,13 the phosphate backbone,14−16 or oligonucleotide termini.(17)
Figure 2.

Schematic representation of the hairpin cMO strategy.
As a proof of principle, we first used this methodology to conditionally silence a T-box transcription factor called no tail-a (ntla), which is required for the differentiation of axial mesodermal cells into a transient, chordate-specific organ called the notochord.18,19 Embryos lacking ntla function exhibit clear morphological phenotypes in a cell-autonomous manner, providing an ideal system for evaluating the efficacy of cMOs in vivo. In particular, ntla mutants or morphants (as MO-injected embryos are commonly called) lack a notochord, are posteriorly truncated, and exhibit U-shaped rather than V-shaped somites. The latter defect is collateral to notochord ablation, since the notochord secretes morphogens to pattern the flanking myotome.(20) Mutants or morphants lacking ntla function also exhibit an ectopic medial floor plate, a ventral region of the developing spinal cord, and it is believed that ntla acts as a transcriptional switch between notochord and medial floor plate cell fates.18,21 By varying the developmental stage at which we activated the ntla cMO, we found that this transcription factor is required not only for specification of the mesoderm toward notochord cell fates but also for the maturation of notochord progenitors into a highly vacuolated tissue.(7) We also demonstrated our ability to silence ntla expression in a subset of mesodermal cells by activating the ntla cMO in a spatially restricted manner, selectively redirecting these populations to differentiate into medial floor plate cells. A similar caging approach has been applied to ncPNAs targeting the chordin (chd) and dharma (dha) genes, although the use of these photoactivatable ncPNA hairpins to spatially control gene expression has not been reported to date.(22)
While our results established the general principle of using caged oligonucleotides to conditionally regulate in vivo gene expression, there are limitations associated with these initial investigations. First, our previous studies required preparation of the inhibitory oligomer and its appendant DMNB linker through solid-phase chemistries, since conventional MOs amenable to terminal hydroxyl modifications were not commercially available at that time. These procedures are laborious and time-consuming, hindering the synthesis and evaluation of other cMOs. Second, guidelines for the design of other hairpin cMOs were not evident from our findings, as only one inhibitory sequence and structural configuration was tested. Nor could general rules for caged oligonucleotide design be derived from the ncPNA study, which limited its analysis to single chd- and dha-targeting reagents with divergent structures and dosages.(22) Finally, the reliance of both classes of caged oligonucleotides on nitrobenzyl-based chromophores restricts their use to single-photon irradiation with UV light, which can induce DNA lesions and has limited spatial resolution. Photoactivatable MOs and ncPNAs that are compatible with two-photon excitation could be activated with less coincident damage, deeper tissue penetration, and greater spatial precision.(23)
We report herein our resolution of these issues through the development of new hairpin cMOs. We have designed and synthesized a DMNB-based bifunctional linker that can be used to conjugate the targeting MO and its complementary inhibitor in only three steps, starting with appropriately functionalized MO oligomers that are now commercially available. We have utilized this optimized synthetic route to prepare a series of ntla-targeting cMOs with differing structural configurations, thereby allowing us to systematically analyze how the in vivo efficacy of these reagents correlates with their biophysical properties. Through these studies, we have demonstrated that cMO activity in vivo can be modeled in simplified thermodynamic terms, and we have established guidelines for the preparation of other cMOs. These design criteria have enabled us to prepare photoactivatable reagents targeting the zebrafish genes heart of glass (heg),(24)floating head (flh),(25) and endothelial-specific variant gene 2 (etv2).(26) We have further demonstrated the versatility of this approach by replacing the DMNB chromophore with a bromohydroxyquinoline (BHQ) group, which has a significantly greater cross section for two-photon excitation.27,28 These studies represent the first comprehensive analysis of the structure and in vivo function of hairpin-caged oligonucleotides, providing a foundation upon which future chemical technologies for conditional gene regulation can be based.
Results and Discussion
Development of a DMNB-Based Bifunctional Linker for Hairpin cMO Synthesis
Since our current cMO design involves the conjugation of a targeting MO to a complementary inhibitor, modifications of the hydroxyl- and amine-functionalized termini of each oligomer (designated as the 5′ and 3′ ends, respectively) are necessary. At the time of our initial studies, MOs functionalized with 3′ primary amines were commercially available, but 5′-amine oligomers were not. We therefore prepared an inhibitory oligonucleotide conjugated at its 5′ end to a photocleavable linker (1; Chart 1) using solid-phase chemistry. This required the synthesis of all four MO bases, a linker-modified MO monomer, and multiple rounds of solid-phase coupling (Scheme S1 in the Supporting Information).(7) Addition of the linker-functionalized inhibitor to the targeting MO was then achieved through a Cu(I)-catalyzed Huisgen 1,3-dipolar cycloaddition (“click” chemistry(29)). Although this approach is ultimately effective, its utilization of time-consuming and labor-intensive procedures hinders the application of hairpin cMOs to other developmental genes.
Chart 1. Chemical Structures of Linker-Conjugated Inhibitor 1 Obtained through Solid-Phase Chemistry and Bifunctional Linker 2a for Solution-Phase Chemistry.

To streamline the synthesis of cMOs, we developed a new strategy that takes advantage of the recent commercial availability of 5′-amine MOs. Our goal was to prepare a bifunctional linker that contains the DMNB chromophore, an N-hydroxysuccinimide ester for amine conjugation, and a terminal alkyne for azide cycloaddition through “click” chemistry. With appropriately functionalized targeting and inhibitory MOs, the cMO could then be assembled in three steps. We first synthesized the bifunctional, photocleavable cross-linker 2a (Chart 1 and Scheme 1). The DMNB amino alcohol 3a(7) was selectively acylated with methyl adipoyl chloride to give amide 4a, which was then activated by 1,1′-carbonyldiimidazole and coupled with 6-amino-N-(prop-2-ynyl)hexanamide(30) to provide the propargyl intermediate 5a. Saponification of 5a and re-esterification of the corresponding acid with disuccinimidyl carbonate then yielded linker 2a, which is orthogonally reactive with azides and amines.
Scheme 1. Synthesis of Bifunctional Linkers 2a and 2b.

Reagents and conditions: (a) methyl adipoyl chloride, DIPEA, CH2Cl2, 86%; (b) 1,1′-carbonyldiimidazole, DMF, 88%; (c) 6-amino-N-(prop-2-ynyl)hexanamide, DIPEA, CH2Cl2, 64%; (d) 2 M NaOH(aq), THF, MeOH, 93%; (e) disuccinimidyl carbonate, pyridine, acetonitrile, 75%. Yields are those obtained for the DMNB derivatives.
To prepare a ntla cMO through this synthetic approach, a targeting MO (5′-GACTTGAGGCAGACATATTTCCGAT-3′) was functionalized with 3-azidopropionic acid succinimidyl ester to yield the azide derivative 6 (Scheme 2). Linker 2a was then reacted with the commercially available 5′-amine- and 3′-fluorescein-functionalized MO (5′-TATGTCTGCC-3′) in 0.1 M sodium borate buffer (pH 8.5) to generate linker-derivatized inhibitory oligomer 7a, and the two MO oligomers were coupled through “click” chemistry in 0.1 M potassium phosphate buffer (pH 8) containing sodium ascorbate, CuI, and tris[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl]amine (TBTA). Purification of the final product by ion-exchange HPLC using NaClO4 as the eluent then yielded the ntla cMO 8a.
Scheme 2. Simplified Strategy for cMO Synthesis.

Reagents and conditions: (a) 2,5-dioxopyrrolidin-1-yl 3-azidopropanoate, 0.1 M sodium borate (pH 8.5)/3:1 (v/v) DMSO, 98%; (b) bifunctional DMNB linker 2a or 2b, 0.1 M sodium borate (pH 8.5)/DMSO 6:1 (v/v), 70−90%; (c) sodium ascorbate, TBTA, CuI, 0.1 M potassium phosphate (pH 8.0), DMSO, 10−25% after HPLC. The ntla cMOs 8a−h, 8a′, and 8e′ were prepared through this synthetic route, using the ntla-targeting MO (5′-GACTTGAGGCAGACATATTTCCGAT-5′) and inhibitory MOs having various lengths and sequences (Table 1).
To evaluate the in vivo efficacy of this reagent, we next injected 8a into wild-type zebrafish embryos at the one-cell stage and globally irradiated approximately half of the embryos with 360-nm light (13 mW/cm2) for 10 s at 3 h post fertilization (hpf). The remaining embryos were cultured in the dark as negative controls, and the resulting phenotypes at 1 day post fertilization (dpf) were then scored. As we have described previously, ntla loss-of-function phenotypes can be categorized into four classes according to their severity: class I = a fully penetrant ntla mutant phenotype characterized by no notochord, U-shaped somites, and a lack of posterior structures; class II = no notochord, U-shaped somites, and some posterior somites; class III = incompletely vacuolated notochord, V-shaped somites, and a shortened anterior−posterior axis; and class IV = wild-type phenotype (Figure 3A).(7) These phenotypes can be recapitulated by injecting early-stage embryos with varying doses of the conventional ntla MO, and classes I−IV correspond respectively to doses of ≥115, 57, 28, and ≤14 fmol/embryo (Figure 3B). Since the class I phenotype can be achieved with ntla MO levels ≥115 fmol/embryo, we utilized 8a at this minimum dose in this experiment. To our surprise, 8a induced class-II and class-III phenotypes in the absence of irradiation (Figure 3C), while our original ntla cMO exhibited little activity prior to uncaging.(7) Since the targeting and inhibitory MOs used in this reagent are identical to those in our previous study, this difference in caging efficiency must reflect changes in the linker structure.
Figure 3.

Conventional and caged MOs targeting the ntla gene and classification of ntla mutant phenotypes. (A) Classification of ntla mutant phenotypes according to morphological cues. Somitic (s), medial floor plate (mfp), notochord (nc), and yolk extension (ye) tissues are labeled. Scale bars: left panels, 200 μm; right panels, 50 μm. Bright-field (left) and differential interference contrast (right) images of 1 dpf embryos are shown. (B) Distribution of phenotypes according to the dose (per embryo) of a conventional ntla MO. (C) Distribution of phenotypes for ntla cMO 8a at a dose of 115 fmol/embryo with and without photoactivation.
Gene-Silencing Activities and Biophysical Properties of Hairpin ntla cMOs
Reasoning that the hairpin cMO efficacy depends on the interplay between inhibitor length, stem−loop configuration, and linker formats, we used linker 2a to prepare ntla cMOs with differing structures (8a−h; Figure 4 and Table 1). In particular, we varied the length of the inhibitory MO (10, 12, 14, and 16 bases) and evaluated both “blunt” and “staggered” stem−loops. Each ntla cMO was synthesized, purified by ion-exchange HPLC, injected into one-cell-stage zebrafish embryos, and photoactivated as before, except that a dose of 230 fmol/embryo was evaluated in each case. This higher cMO concentration was used for these studies to maximize our ability to identify caged configurations with minimum basal activity.
Figure 4.

Activity profiles of ntla cMOs with different structures. The leftmost panels show schematic representations of “staggered” (top) and “blunt” (bottom) cMO configurations (n = number of bases), and the other panels are distributions of phenotypes for each cMO configuration 8a−h (see Table 1 and Figure 3) at a dose of 230 fmol/embryo.
Table 1. Oligomer sequences.
| oligomer | sequence | cMO |
|---|---|---|
| ntla MOa | GACTTGAGGCAGACATATTTCCGAT | 8a-h, 8a′, 8e′ 22a-b |
| heg MOa | GTAATCGTACTTGCAGCAGGTGACA | 9a-c |
| flh MOa | GGGAATCTGCATGGCGTCTGTTTAG | 10 |
| etv2 MOa | CACTGAGTCCTTATTTCACTATATC | 11 |
| spt MOa | GCTTGAGGTCTCTGATAGCCTGCAT | 12 |
| ntla 10-mer | TATGTCTGCC | 8a, 8a′ |
| “Staggered”b | ||
| ntla 12-mer | TATGTCTGCCTC | 8b |
| “Staggered”b | ||
| ntla 14-mer | TATGTCTGCCTCAA | 8c |
| “Staggered”b | ||
| ntla 16-mer | TATGTCTGCCTCAAGT | 8d |
| “Staggered”b | ||
| ntla 10-mer | GCCTCAAGTC | 8e, 8e′ |
| “Blunt”b | ||
| ntla 12-mer | CTGCCTCAAGTC | 8f |
| “Blunt”b | ||
| ntla 14-mer | GTCTGCCTCAAGTC | 8g |
| “Blunt”b | ||
| ntla 16-mer | ATGTCTGCCTCAAGTC | 8h |
| “Blunt”b | ||
| heg 13-mer | CAAGTACGATTAC | 9a |
| “Blunt”b | ||
| heg 11-mer | GCTGCAAGTAC | 9b |
| “Staggered”b | ||
| heg 10-mer | GTACGATTAC | 9c |
| “Blunt”b | ||
| flh 10-mer | GCAGATTCCC | 10 |
| “Blunt”b | ||
| etv2 10-mer | GGACTCAGTG | 11 |
| “Blunt”b | ||
| spt 10-mer | GACCTCAAGC | 12 |
| “Blunt”b | ||
| ntla RNAc | ATCGGAAATATGTCTGCCTCAAGTC | - |
| flh RNAc | CTAAACAGACGCCATGCAGATTCCC | - |
| heg RNAc | TGTCACCTGCTGCAAGTACGATTAC | - |
| etv2 RNAc | GATATAGTGAAATAAGGACTCAGTG | - |
| spt RNAc | ATGCAGGCTATCAGAGACCTCAAGC | - |
MO oligomers for translational inhibition of targeted mRNA.
Inhibitory MO oligomers for modulating cMO activity.
RNA oligomers for thermal denaturation studies.
The resulting phenotypes confirmed that hairpin cMO activity varies significantly with inhibitor length and stem−loop structure. Within a stem−loop configuration, increasing the number of bases in the inhibitory oligomer decreased the cMO activity under both basal and photoactivated conditions. The “blunt” and “staggered” stem−loop configurations also exhibited distinct activity profiles. The “staggered” system failed to achieve an adequate activity differential between the caged and uncaged forms under any of the conditions we tested. As described above, cMO 8a still exhibited gene-silencing activity in its caged form, even though it successfully induced class-I phenotypes upon photoactivation. The other “staggered” cMOs (8b−d) had lower basal activities, but their uncaged forms failed to yield strong ntla mutant phenotypes. In contrast, the “blunt” stem−loop design provided greater caging efficiency, and two cMOs, 8e and 8f, exhibited dynamic ranges appropriate for conditional gene silencing: these two reagents did not induce mutant phenotypes in their caged forms, and photoactivation of the cMOs yielded fully penetrant phenotypes in most embryos. Since our linker-1-based ntla cMO utilized the same inhibitory oligomer as the “staggered” reagent 8a,(7) these results underscore the importance of matching linker and stem−loop structures for optimum cMO activity.
To better understand how cMO structure dictates in vivo activity, we characterized the biophysical properties of each reagent. We first determined the binding energies for the ntla MO/RNA duplex, each ntla MO/inhibitor heterodimer, and various stem−loop structures. Thermal denaturation curves for the ntla MO/RNA duplex and ntla MO/inhibitor heterodimers were acquired by mixing the ntla-targeting MO with the complementary oligomers in a 1:1 ratio and measuring their temperature-dependent changes in hypochromicity at 260 nm (Figure 5 and Tables 1−3). For these studies, we used the commercially available amine-functionalized ntla-targeting and inhibitory MO oligomers prior to their modification for cMO synthesis. The thermal denaturation curves were fit to a two-state oligomer binding model(31) to provide the corresponding ΔG values at 28 °C, the standard temperature for culturing zebrafish embryos. These analyses indicated that the binding free energy for ntla MO and its complementary 25-base RNA is −28.1 kcal/mol, while binding free energies for the various ntla MO/inhibitor duplexes range from −10.7 to −16.2 kcal/mol. The ΔG values for intermolecular ntla MO/inhibitor complexes correlated with the length and sequence content of the inhibitory MOs, independent of the region of complementarity (corresponding to “blunt” vs “staggered” hairpins). To assess binding energies for ntla MO/inhibitor interactions within an intramolecular stem−loop, we next conducted hypochromicity measurements of “blunt” and “staggered” ntla MO hairpins. Since the 260-nm absorbance measurements would photolyze the ntla cMOs (8a−h), we synthesized two ntla MO/inhibitor hairpins (8a′ and 8e′) (Scheme 2) using the bifunctional but nonphotolabile cross-linker 2b, which was prepared from 3-(methylamino)propan-1-ol (3b) in analogy to the DMNB-based reagent 2a (Scheme 2). Derivation of the ΔG values from these thermal denaturation curves using a hairpin-binding model(31) revealed that the intramolecular binding free energies of 8a′ and 8e′ were −4.9 and −6.9 kcal/mol, respectively (Table 4).
Figure 5.

Representative thermal denaturation curves for MO duplexes corresponding to ntla cMOs 8a−h (ntla-targeting and inhibitory MOs, 0.5 μM each) and noncleavable hairpins 8a′ and 8e′.
Table 3. Thermodynamic Parameters of MO/RNA Dimers.
| oligomera | Tm (°C) | ΔG (kcal/mol)b |
|---|---|---|
| ntla | 77.7 ± 0.8 | −28.1 ± 2.4 |
| heg | 83.4 ± 0.9 | −28.4 ± 2.2 |
| flh | 83.4 ± 0.8 | −27.5 ± 2.1 |
| etv2 | 73.9 ± 1.0 | −25.1 ± 2.8 |
| spt | 85.2 ± 1.2 | −30.0 ± 3.2 |
Dimers of 25-mer MO and RNA oligomers.
Binding free energy of the MO/RNA dimer at 28 °C.
Table 4. Thermodynamic Parameters of Noncleavable ntla MO Hairpins.
Melting temperature of the noncleavable MO hairpin.
Binding free energy of the hairpin at 28 °C.
Tm and ΔGhairpin values were determined from sigmoidal fits of the thermal denaturation curves using the hairpin algorithm in MeltWin 3.0b software.
Taken together, these in vitro binding energies provide qualitative insights into how cMOs perform in vivo. Intermolecular ntla MO/inhibitor interactions with ΔG values lower than −12 kcal/mol are required for low basal activities, and “blunt” hairpins exhibit higher caging efficiencies than their “staggered” counterparts because of their greater stabilization of the intramolecular MO/inhibitor duplex (Table 2 and Figure 4). Surprisingly, the ΔG values for the “blunt” and “staggered” ntla MO/inhibitor duplexes correlate with the inhibitor length and sequence in a similar manner, yet the activities of their corresponding ntla cMOs upon photoactivation diverge. For example, the intermolecular ntla MO/inhibitor duplexes derived from “blunt” ntla cMOs 8e and 8f have ΔG values of −12.3 and −13.5 kcal/mol, respectively, and activated forms of these cMOs produce strong ntla mutant phenotypes. The other two “blunt” ntla cMOs (8g and 8h) have ΔG values of −15.5 and −16.2 kcal/mol, respectively, and both induce only partial loss-of-function phenotypes upon linker photolysis. An in vitro ntla MO/inhibitor ΔG value between −12 and −14 kcal/mol therefore represents the optimal balance of basal and induced activities for the “blunt” ntla cMOs. However, the “staggered” ntla MOs (8a and 8b) fail to induce full ntla mutant phenotypes after uncaging, even though their corresponding ntla MO/inhibitor duplexes have ΔG values of −10.7 and −13.3 kcal/mol.
Table 2. Thermodynamic Parameters of MO/Inhibitor Dimers.
|
Tm (°C)b,e |
|||
|---|---|---|---|
| oligomera | observed | predictedc | ΔG (kcal/mol)d,e |
| 8a | 36.3 ± 0.4 | 38 | −10.7 ± 0.2 |
| 8b | 49.2 ± 1.3 | 46 | −13.3 ± 0.3 |
| 8c | 52.5 ± 1.5 | 49 | −14.1 ± 0.2 |
| 8d | 54.5 ± 0.9 | 57 | −14.7 ± 0.1 |
| 8e | 45.7 ± 0.6 | 42 | −12.3 ± 0.3 |
| 8f | 49.0 ± 0.8 | 49 | −13.5 ± 0.3 |
| 8g | 56.1 ± 0.6 | 57 | −15.5 ± 0.2 |
| 8h | 59.0 ± 0.7 | 61 | −16.2 ± 0.4 |
| 9a | 41.4 ± 1.6 | 44 | −11.4 ± 0.2 |
| 9b | 41.7 ± 2.3 | 44 | −11.4 ± 0.5 |
| 9c | 32.2 ± 1.1 | 34 | −9.7 ± 0.1 |
| 10 | 43.7 ± 1.1 | 42 | −12.5 ± 0.5 |
| 11 | 40.5 ± 1.1 | 42 | −11.7 ± 0.3 |
| 12 | 42.9 ± 1.2 | 42 | −11.9 ± 0.2 |
Dimers of MO and inhibitory MO oligomers with 3′ and 5′ amine modifications, respectively.
Melting temperature of the MO/inhibitor dimer.
Predicted melting temperature according to eq 10, which is based upon the Tm and ΔG values for 8a−h.
Binding free energy of the dimer at 28 °C.
Tm and ΔG values were determined from sigmoidal fits of the thermal denaturation curves using the non-self-complementary algorithm in MeltWin 3.0b software.
The molecular basis for these differing activities is unclear. Our original solid-phase-chemistry-derived ntla MO(7) and the “staggered” cMO 8a have identical targeting and inhibitory MO sequences, suggesting that linker elements may contribute to cMO activity after uncaging. In addition, the photolysis products of ntla cMOs 8a and 8e are functionalized with linker substituents that are not present in the oligomers used in our binding energy measurements; the targeting MO liberated upon cMO activation is 3′-functionalized with a 1,2,3-triazole and an aliphatic amine, while the inhibitory oligomer is 5′-functionalized with an aliphatic chain and the DMNB-derived chromophore. We therefore irradiated 8a and 8e with 360-nm light and obtained thermal denaturation curves for the resulting ntla MO/inhibitor heterodimers. Analysis of the photolysis products by HPLC confirmed that the two hairpin oligonucleotides are uncaged with comparable efficacies (∼75% conversion), and the ΔG values for the resulting linker-functionalized ntla MO/inhibitor complexes are similar to those observed for their amine-functionalized counterparts (Figure S1 and Table S1 in the Supporting Information). Since we were unable to discern any thermodynamic differences between the intermolecular MO/inhibitor duplexes corresponding to 8a and 8e, we next interrogated whether RNA strand exchange rates might account for their divergent activities in vivo. We incubated each ntla MO/inhibitor duplex with complementary 25-base RNA for different lengths of time and resolved the resulting MO/RNA duplexes by polyacrylamide gel electrophoresis (Figure S2 in the Supporting Information). The RNA exchange rates for the “staggered” and “blunt” MO/inhibitor duplexes were indistinguishable in this assay; MO/RNA hybridization was complete in both cases within the time frame of RNA addition. Thus, the activity differences between 8a and 8e cannot be explained by in vitro MO/inhibitor thermodynamics or kinetics alone, and the two cMOs might exhibit divergent photolysis, inhibitor dissociation, or RNA exchange rates in vivo.
Modeling of cMO Activity in Vivo
While the correlation between MO/inhibitor interaction strength and cMO activity provides qualitative guidelines for reagent design, a quantitative thermodynamic analysis is necessary to predict cMO efficacy with greater precision (Figure 6). Prior to photolysis, cMOs adopt “open” and “closed” states according to the equilibrium constant Khairpin (eq 1):
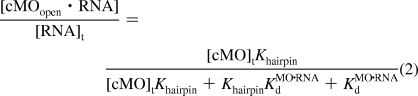
With the assumption that the total concentration of cMO prior to uncaging ([cMO]t) significantly exceeds that of its RNA target ([RNA]t), the basal level of RNA bound by the nonphotolyzed cMO, given by ([cMOopen·RNA]/[RNA]t), can be described as a function of [cMO]t, Khairpin, and the dissociation constant for the MO/RNA duplex (KMO·RNAd) (eq 2):
 |
Upon photoactivation, the fraction of RNA complexed with the released targeting MO is a function of [cMO]t, KMO·RNAd, and the dissociation constant for the MO/inhibitor complex (KMO·INHd) (eqs 3 and 4):
Setting [cMO]t to a value of 4.6 μM, which approximates the embryonic concentration of the ntla cMO (230 fmol/embryo, 50 nL embryonic volume at 5 hpf), and using our ntla cMO 8e data to establish ΔG values of −28.1, −12.3, and −6.9 kcal/mol for the MO/RNA duplex, intermolecular MO/inhibitor duplex, and intramolecular MO/inhibitor hairpin, respectively, led to the prediction that essentially all of the ntla RNA is MO-complexed before and after cMO photolysis. This conclusion clearly deviates from the phenotypes we observed with ntla cMO 8e-injected embryos.
Figure 6.

Schematic representation of cMO/RNA, MO/inhibitor (INH), and MO/RNA equilibria.
Since these predictions do not take into account the complexity of MO and RNA interactions in live organisms, we sought to empirically derive the activity profiles of MOs in vivo. We first investigated whether the relationship between ntla RNA activity and total ntla MO concentration can still be described as a two-state equilibrium, even though the apparent equilibrium constant for in vivo MO/RNA interactions (KMO·RNAapp, analogous to KMO·RNAd in Figure 6) would include contributions from oligonucleotide-binding proteins, RNA secondary structure, and other embryonic factors. We injected the targeting MO into one-cell-stage zebrafish at doses of 0, 14, 28, 57, and 115 fmol/embryo (approximate final concentrations of 0, 0.28, 0.56, 1.1, and 2.3 μM, respectively), lysed the embryos at 10 hpf, and then detected the Ntla protein by quantitative immunoblotting (Figure 7A). The ntla MO reduced the Ntla protein levels in a dose-dependent manner that can be modeled as two-state thermodynamic interaction, with the fraction of wild-type RNA activity remaining in MO-injected embryos (RNAActMO/RNAActWT) described as a function of total MO concentration ([MO]t) and KMO·RNAapp (Figure 7B and eq 5):
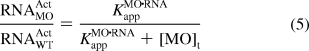 |
Through this analysis, an apparent free-energy value (ΔGMO·RNAapp) of −8.7 kcal/mol for embryonic ntla MO/RNA interactions was obtained.
Figure 7.

ntla MO/RNA and ntla MO/inhibitor interactions in vivo. (A) Ntla protein knockdown in zebrafish embryos injected at the one-cell stage with various ntla MO doses. Ntla and β-actin levels at 10 hpf were detected by immunoblotting and quantified. (B) These data can be modeled as a two-state equilibrium (solid line), yielding an apparent free energy value (ΔGMO·RNAapp = −8.7 kcal/mol) that describes MO/RNA interactions in live embryos. (C) Ntla protein knockdown in embryos injected at the one-cell stage with ntla MO (115 fmol/embryo) and various doses of the 14-base inhibitory oligomer corresponding to ntla cMO 8g. Ntla and β-actin levels at 10 hpf were detected by immunoblotting and quantified as above. (D) Modeling of the 8g data in (C) as a three-state, competitive equilibrium (solid line) yields an apparent free-energy value (ΔGMO·INHapp = −7.3 kcal/mol) that describes 8g MO/inhibitor interactions in vivo. The graphical data are means of triplicate samples with error bars representing the standard deviations.
It is important to note that this ΔGMO·RNAapp value does not reflect the actual binding constant for the ntla MO/RNA duplex in zebrafish embryos but instead is an aggregate descriptor of MO/RNA affinity, RNA accessibility, MO/protein interactions, and the influence of other embryonic factors on MO efficacy. Indeed, the 19.4 kcal/mol difference between ΔGMO·RNAapp and the corresponding in vitro ΔG value underscores how significant these other variables can be. Our empirical data, however, suggest that this thermodynamic description can have predictive value, even though it does not explicitly consider MO and RNA interactions with other cellular components or possible kinetic contributions to in vivo function. Thus, MO-induced gene silencing can be modeled in these simplified thermodynamic terms.
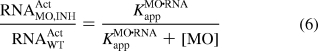
To determine whether cMO activity can also be modeled in simple thermodynamic terms, we next analyzed MO/inhibitor interactions in vivo. We injected zebrafish embryos with the ntla MO (115 fmol/embryo; ∼2.3 μM) and various doses of the 14-base inhibitor corresponding to ntla cMO 8g (0, 150, 450, and 1350 fmol/embryo; ∼0, 3, 9, and 27 μM, respectively). The resulting Ntla protein levels at 10 hpf were then quantified as before (Figure 7C). The inhibitory oligomer repressed the ntla MO activity in a concentration-dependent manner that can be modeled as a three-state, competitive equilibrium involving MO, inhibitor, and RNA interactions (Figure 7D). The fraction of wild-type RNA activity associated with each MO and inhibitor dose, RNAActMO,INH/RNAActWT, can be expressed as a function of the apparent equilibrium constant for MO/inhibitor interactions (KMO·INHapp; analogous to KMO·INHd in Figure 6), the total concentration of the inhibitory oligomer ([INH]t), and [MO]t (eqs 6 and 7):
 |
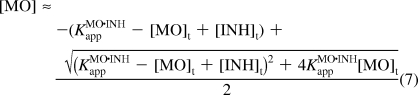 |
Through this analysis, we derived an apparent ΔGMO·INHapp value of −7.3 kcal/mol for 8g MO/inhibitor interactions. As with the ΔGMO·RNAapp value we determined for MO-dependent ntla silencing, this apparent free-energy value does not reflect the actual binding constant for the ntla MO/inhibitor duplex in vivo but instead integrates other interactions between these synthetic oligonucleotides and cellular components. Since the 8.2 kcal/mol difference between ΔGMO·INHapp and the corresponding in vitro ΔG value is significantly smaller than the 19.4 kcal/mol difference we observed for MO/RNA interactions (see Table 3 and Figure 7B), cellular factors appear to impact RNA activity to a greater extent than MO function.
To assess the validity of modeling in vivo MO, RNA, and inhibitor interactions in these simplified terms, we investigated whether the apparent ΔGMO·RNAapp and ΔGMO·INHapp values can be used to predict how cMO gene-silencing activity will be influenced by changes in inhibitor structure. As discussed above, the fraction of total RNA activity inhibited by a complementary MO and the mitigating influence of an inhibitory oligomer can be described by eqs 5−7. In the case of a cMO, [MO]t and [INH]t will be equal after photoactivation, and the fraction of wild-type RNA activity remaining in the presence of photoactivated cMO (RNAActcMO/RNAActWT) is therefore a function of KMO·RNAapp, KMO·INHapp, and [cMO]t (eqs 8 and 9):
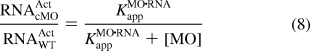 |
In the case of our ntla cMO experiments shown in Figure 4, KMO·RNAapp and [cMO]t can be approximated as 0.48 μM (ΔGMO·RNAapp = −8.7 kcal/mol) and 4.6 μM, respectively. If it is assumed that “blunt” MO/inhibitor interactions generally exhibit a 8.2 kcal/mol difference between ΔGMO·INHapp and the corresponding in vitro ΔG, the KMO·INHapp values for MO/inhibitor interactions associated with ntla cMOs 8e, 8f, 8g, and 8h can be estimated as 1100, 140, 50, and 1.6 μM, respectively. Using these parameters in the model described by eqs 8 and 9 leads to the prediction that ntla cMOs 8e and 8f will be similar in efficacy to the conventional ntla MO, achieving at least a 90% knockdown of Ntla protein expression levels (red lines in Figure 8A,B). In contrast, ntla cMOs 8g and 8h are predicted to exhibit weaker efficacies at the same embryonic dose, since the inhibitory oligomer interacts more strongly with the targeting MO (red lines in Figure 8C,D).
Figure 8.

Modeling of in vivo cMO activity. Photoactivated cMOs and MOs inhibit their RNA targets with different efficacies, which diverge as the MO/RNA interaction strength decreases. This divergent activity profile is exacerbated when the MO/inhibitor interaction strength increases. RNA activity curves for MOs and photoactivated cMOs associated with in vivo MO/RNA interaction energies of (red) −8.7 and (blue) −7.3 kcal/mol are shown for intermolecular MO/inhibitor interaction energies estimated for the “blunt” ntla cMOs (A) 8e, (B) 8f, (C) 8g, and (D) 8h, assuming complete uncaging upon irradiation. Actual photoactivation yields in vivo are likely 50−75% (see text). Photoactivated cMO and conventional MO activity profiles are drawn as solid and dashed colored lines, respectively. The embryonic ntla cMO concentration used in our structure−activity studies (see Figure 4) is indicated in each panel by the vertical black line.
Our observations in vivo match these predictions. Photoactivated 8e and 8f yielded the strongest mutant phenotypes of the ntla cMO configurations we tested. Moreover, the predicted efficacies of 8g and 8h are consistent with their photoinduced phenotypes and the relationship between ntla RNA activity and phenotypic class (compare Figures 3B, 4, 7A,B, and 8C,D), especially considering that UV light penetrance and therefore cMO uncaging efficiencies in vivo will be attenuated to some degree. On the basis of our in vitro uncaging results (see Figure S1 in the Supporting Information) and the observed phenotypes for irradiated ntla cMO-injected embryos, we estimate that our whole-organism irradiation procedure achieves cMO photoactivation yields of 50−75%. Our model also predicts that cMO efficacy will be increasingly compromised as ΔGMO·RNAapp increases (compare the blue and red lines in Figure 8, which represent a 10-fold change in MO/RNA interaction strength), since the concentration of photoactivated cMO required to achieve a given level of RNA silencing increases in a manner disproportionate to that of a conventional MO. This latter issue is of particular importance because the MO doses required to induce mutant phenotypes vary significantly between genes, reflecting different RNA activity thresholds for wild-type physiology and variable RNA sequence accessibilities. In addition, MO doses greater than 1000 fmol/embryo (∼20 μM) are generally avoided to minimize cytotoxicity.
In contrast, the MO/inhibitor interaction strength associated with optimum cMO efficacy should not vary significantly between genes. The 25-base targeting MOs are typically designed to avoid secondary structures, and they are not known to interact in a sequence-specific manner with DNA- or RNA-binding proteins. Rather, the binding free energies for the intra- and intermolecular MO/inhibitor duplexes dictate the fraction of targeting MO that is free to anneal to its RNA target, thereby establishing the basal and photoinduced activities of a given cMO. Our findings suggest that cMOs should have ΔGMO·INHapp values of approximately −5 kcal/mol or greater for their corresponding intermolecular MO/inhibitor duplexes, as this interaction strength in vivo would allow efficient gene-silencing upon cMO photoactivation for a broad range of targeting MO potencies. Maximizing the cMO activity in this manner, however, is counterbalanced by the need to maintain the “closed” cMO hairpin state prior to photoactivation. In fact, the ΔGMO·INHapp value of −4.1 kcal/mol predicted for ntla cMO 8e may represent the optimum thermodynamic parameter for cMO efficacy. Further attenuation of the MO/inhibitor interaction will eventually yield undesirable levels of basal activity, and a small fraction of embryos injected with the ntla cMO 8e exhibited weak mutant phenotypes without irradiation (see Figure 4).
Guidelines for Hairpin cMO Design
Taken together, our results demonstrate that cMO activity can be modeled as a competitive three-state equilibrium, even though this approach does not explicitly consider how MO activity and RNA accessibility are influenced by cellular proteins, RNA structure, and other embryonic factors. Moreover, this analysis does not require the determination of actual MO/RNA or MO/inhibitor binding affinities in whole embryos. We therefore sought to establish simple guidelines for the preparation of cMOs targeting other genes and to empirically test their validity. Such design criteria would significantly advance the use of cMOs in chemical and developmental biology research, especially considering the financial and time investments associated with these studies. The disparate efficacies of the two stem−loop structures indicate that “blunt” cMO hairpins are preferable to “staggered” configurations, and within our series of “blunt” ntla cMOs 8e−h, in vitro MO/inhibitor ΔG values between −12 and −14 kcal/mol yield an optimum balance between caged and uncaged activities. Our modeling of cMO activity in vivo further suggests that the higher MO/inhibitor ΔG value associated with ntla cMO 8e should be preferred, since it would maximize the inducible gene-silencing activity over a broader range of targeting MO efficacies. This binding energy corresponds to a duplex melting temperature of ∼45 °C (see Figure 5 and Table 2).
To facilitate the design of thermodynamically equivalent cMOs against other genes, we first determined the relationship between MO duplex sequence and thermal stability. By multiple-regression analysis of the ntla MO/inhibitor pairs listed in Table 2, we determined that the melting temperature of MO duplexes (TMOm, in °C) is correlated with sequence content according to eq 10 and can be empirically related to its in vitro ΔG value (in kcal/mol at 28 °C) by eq 11:
In eq 10, each nj (j = A, T, G, C) is the number of times the corresponding base appears in the MO sequence. Using this equation, one can identify an appropriate inhibitor for a given targeting MO, which ideally would generate a “blunt” hairpin and have a predicted duplex melting temperature between 40 and 45 °C. This empirically derived algorithm should be most accurate with MO duplexes similar in length to those in this study, since it does not take into account the contribution of nearest-neighbor effects.
We next tested the validity of our design criteria by targeting four other zebrafish genes: heg, flh, etv2, and spadetail/tbx16 (spt). The transmembrane protein heg is expressed in the endocardium during embryogenesis, mediating a signal that is required for concentric growth of the heart.(24) Embryos lacking heg function exhibit abnormally large heart chambers with walls that are only one cell thick and therefore incapable of sustaining blood circulation. These defects are apparent by 2 dpf and can be recapitulated with the heg-targeting MO (5′-GTAATCGTACTTGCAGCAGGTGACA-3′) at doses of 230 fmol/embryo (∼4.6 μM) or higher (79%, n = 48). By 4 dpf, heg mutants and morphants exhibit severe cardiac edema (Figure 9A). To generate a heg cMO (9a), we conjugated the targeting MO to an inhibitory oligomer 5′-CAAGTACGATTAC-3′ using linker 2a (Tables 1−3). We then injected wild-type zebrafish embryos with 9a at the one-cell stage (460 fmol/embryo; ∼9.2 μM) and either irradiated the embryos with 360-nm light at 2 hpf or cultured them in the dark. By 4 dpf, the irradiated embryos had no blood circulation and exhibited cardiac edema (86%, n = 21), while the nonirradiated zebrafish had normal cardiac patterning and function (98%, n = 44) (Figure 9B,C and Figure S3 in the Supporting Information). To further test the predictive value of our cMO design criteria, we evaluated two additional heg cMOs: one contained a “staggered” inhibitor (9b; inhibitor sequence 5′-GCTGCAAGTAC-3) with an intermolecular MO/inhibitor ΔG value comparable to that for 9a, and the other contained a shorter “blunt” inhibitor (9c; inhibitor sequence 5′-GTACGATTAC-3′) with a weaker MO/inhibitor interaction strength (Tables 1 and 2). In comparison to 9a, these alternative heg cMOs were less effective in vivo. The majority of zebrafish embryos injected with an equivalent dose of either 9b or 9c exhibited heg mutant phenotypes in the absence of irradiation (9b: 66%, n = 53; 9c: 62%, n = 45), indicating that these structural configurations do not adequately cage the MO activity (Figure S3). Thus, the design parameters established through our ntla cMO structure−activity relationship studies are transferable to other genes.
Figure 9.

Evaluation of heg, flh, and spt cMO activities in vivo. (A) Embryos injected with a conventional heg MO recapitulate heg mutant phenotypes, including enlarged heart chambers, no blood circulation, and cardiac edema. (B, C) Embryos injected with heg cMO 9a exhibit heg mutant phenotypes upon photoactivation. (D) Embryos injected with a conventional flh MO recapitulate flh mutant phenotypes, including notochord ablation and somite fusion through the trunk midline. (E, F) Embryos injected with flh cMO 10 exhibit flh mutant phenotypes upon photoactivation. (G) Embryos injected with a conventional spt MO recapitulate spt mutant phenotypes, including a loss of trunk somitic tissue and mislocalized mesodermal progenitors in the posterior (“spadetail” morphology). (H, I) Embryos injected with spt cMO 12 exhibit partial spt mutant phenotypes upon photoactivation. Nonspecific toxicity is also observed (data not shown). Developmental stages: (A−C) 4 dpf; (D−F, G−I) 1 dpf. Scale bars: (A−C) 400 μm; (D−F, G−I) 200 μm.
The flh homeobox transcription factor is coexpressed with ntla in the zebrafish mesoderm, and loss of flh expression also causes ablation of the notochord.25,32 However, mesodermal progenitors normally fated to become the notochord do not transform into medial floor plate cells in flh mutants; rather, these populations differentiate into ectopic muscle to create fused somites. Since no flh-targeting MO had yet been described, we first identified a flh-blocking oligonucleotide (5′-GGGAATCTGCATGGCGTCTGTTTAG-3′) and demonstrated its efficacy in zebrafish embryos (Figure 9D). This antisense reagent is a potent inhibitor of flh function, and a dose of 60 fmol/embryo (∼1.2 μM) is sufficient to cause replacement of the notochord with axial muscle cells in 1 dpf zebrafish (89%, n = 19). On the basis of our cMO design criteria, we synthesized a hairpin flh cMO (10) using linker 2a and a 5′-GCAGATTCCC-3′ oligomer as the MO inhibitor (Tables 1−3). Wild-type zebrafish were injected at the one-cell stage with 10 at a dose of 100 fmol/embryo (∼2 μM) and either globally irradiated with 360-nm light for 10 s at 2 hpf or cultured in the dark. As expected, the majority of nonirradiated flh cMO-injected embryos developed normally (89%, n = 28), while the irradiated embryos lacked a notochord and had fused somites (100%, n = 11), matching the mutant phenotype (Figure 9E,F).
The etv2 transcription factor is expressed in the lateral mesoderm during early somitogenesis and then in vascular endothelial cells of the axial, head, and intersomitic vessels.(26) It is believed that these early etv2-expressing cells are endothelial precursors, and etv2 mutants or embryos injected with an etv2-blocking MO (5′-CACTGAGTCCTTATTTCACTATATC-3′) exhibit disrupted blood-vessel formation and lack circulation.(26) MO doses of 115 fmol/embryo (∼2.3 μM) or higher are sufficient to induce a fully penetrant mutant phenotype (data not shown). We therefore synthesized a hairpin etv2 cMO (11) containing a 5′-GGACTCAGTG-3′ inhibitory oligomer and injected it into wild-type zebrafish at the one-cell stage (230 fmol/embryo; ∼4.6 μM) (Tables 1−3). Embryos then irradiated with 360 nm light for 10 s at 3 hpf had limited or no blood circulation by 2.5 dpf (100%, n = 12; see Supplementary Movie 1 in the Supporting Information), but most of the etv2 cMO-injected embryos cultured in the dark exhibited normal blood flow (87%, n = 15; see Supplementary Movie 2 in the Supporting Information).
Finally, we designed and synthesized a cMO targeting spt, which is another mesodermal T-box transcription factor.21,33 The ntla and spt genes are expressed in overlapping domains during early embryogenesis and then become restricted to the axial and paraxial mesoderm, respectively. Cells with ntla function become the notochord, while the spt-expressing cells contribute to the skeletal muscle in the flanking somites. A loss of spt function therefore causes a severe deficit in trunk somitic mesoderm as well as a gross mislocalization of the corresponding progenitor cells to posterior regions (hence the “spadetail” name). These phenotypes can be recapitulated with a spt-targeting MO 12 (5′-GCTTGAGGTCTCTGATAGCCTGCAT-3′)(34) at doses of 345 fmol/embryo (∼6.9 μM) or higher (61%, n = 23) (Figure 9G). As with the other cMOs described above, we prepared the corresponding spt cMO hairpin using linker 2a and the inhibitory oligomer 5′-GACCTCAAGC-3′ (Tables 1−3). Wild-type embryos at the one-cell stage were injected with 12, and a subset was globally irradiated with 360 nm light for 10 s at 2 hpf. The majority of embryos cultured in the dark developed normally (87%, n = 24). Zebrafish injected with a dose of 700 fmol/embryo (∼14 μM) exhibited a loss of trunk mesoderm but not posteriorly mislocalized progenitor cells upon cMO photoactivation (31%, n = 16) and also showed nonspecific developmental defects due to general MO toxicity (62%, n = 16) (Figure 9H,I). These phenotypes indicate that the photoactivated spt cMO 12 is not able to fully recapitulate the activity of the conventional MO and that this reagent also exhibits greater nonspecific toxicity. Thus, although our design criteria were successful for cMOs targeting ntla, heg, flh, and etv2, there can be unforeseen MO or inhibitor interactions with embryonic factors that reduce cMO efficacy in certain situations. In the case of the spt cMO, one possibility is that a 700 fmol/embryo dose of the 35-base reagent approaches the toxicity level of a 1000 fmol/embryo dose of the conventional 25-base MO.
Development of a BHQ-Based cMO for Two-Photon Activation
To conclude our studies, we investigated the ability of our hairpin cMO design to incorporate other photocleavable groups. While nitrobenzyl-based chromophores such as the DMNB group in our ntla, flh, heg, etv2, and spt cMOs have been widely used in biological applications, the UV light required for their photolysis is potentially damaging and difficult to restrict in three-dimensional space. Two-photon excitation typically uses wavelengths greater than 700 nm and affords greater spatial resolution, but the DMNB group and most other caging moieties have small two-photon cross sections and are therefore inefficiently cleaved under these conditions. Two notable exceptions are the bromohydroxycoumarin (BHC)(23) and bromohydroxyquinoline (BHQ)(27) groups. Since the low fluorescence of BHQ chromophores makes them particularly useful for biological applications, we designed a BHQ-based bifunctional cross-linker (13, Scheme 3) for the preparation of cMOs. As with our DMNB-based linker 2a, this two-photon-sensitive linker incorporates an N-hydroxysuccinimide ester and a terminal alkyne to enable orthogonal coupling to appropriately modified MO oligomers.
Scheme 3. Synthesis of Bifunctional BHQ Linker 13.

Reagents and conditions: (a) benzenesulfonyl chloride, DIPEA, CH2Cl2, 97%; (b) SeO2, dioxane, 80 °C, 91%; (c) In powder, allyl bromide, NH4Cl(aq), THF, 96%; (d) K2OsO4·2H2O, lutidine, dioxane, H2O, then NaIO4, 75%; (e) methylamine(aq), NaBH(OAc)3, MeOH, H2O, 81%; (f) methyl adipoyl chloride, DIPEA, CH2Cl2, 59%; (g) carbonyldiimidazole, DMF then 6-amino-N-(prop-2-ynyl)hexanamide, DIPEA, DMF, 81%; (h) 0.2 M NaOH(aq), THF, MeOH, 82%; (i) N-hydroxysuccinimide, EDCI, DMF, 48%.
To synthesize linker 13, we first prepared BHQ derivative 14 as previously described(27) and then protected its phenolic hydroxyl group with a benzenesulfonyl moiety to give 15. The 2-methyl group of 15 was then oxidized with selenium dioxide, and the resulting aldehyde 16 was allylated to give alcohol 17. Oxidative cleavage of 17 yielded aldehyde 18,(35) which was reductively aminated with aqueous methylamine in the presence of NaBH(OAc)3 to give the amino alcohol intermediate 19. In analogy to Scheme 1, compound 19 was acylated to form the amide 20, and 1,1′-carbonyldiimidazole-mediated coupling of this product with 6-amino-N-(prop-2-ynyl)hexanamide afforded carbamate 21. Phenol deprotection and ester saponification of 21 were simultaneously accomplished with 0.2 M NaOH to give an acid intermediate, which was re-esterified with N-hydroxysuccinimide in the presence of 1-ethyl-3-(3′-dimethylaminopropyl)carbodiimide to give the two-photon-sensitive linker 13. The bifunctional linker was then used to assemble fluorescein-conjugated and nonfluorescent ntla cMOs (22a and 22b, respectively; Chart 2) using the corresponding inhibitory oligomers (23a and 23b), which are analogous to that in the optimum DMNB-containing ntla cMO 8e (Figure 4 and Table 1). Since the BHQ group has not been used previously with carbamates and other BHQ-caged compounds have not been tested in cultured cells or live organisms, we first determined whether the BHQ carbamate is stable in vivo and can be efficiently photolyzed. We injected ntla cMO 22a into wild-type zebrafish embryos at the one-cell stage (230 fmol/embryo) and irradiated a subset of them with 360-nm light for 10 s at 3 hpf. The embryos were then cultured for another day and scored according to the four phenotypic classes described above. As we hoped, the BHQ-based ntla cMO exhibited activity essentially identical to that of the DMNB-based reagent 8e; embryos injected with this reagent developed normally when cultured in the dark but displayed a ntla phenotype upon irradiation (Figure S4 in the Supporting Information).
Chart 2. Schematic Representation of BHQ-Based ntla cMOs 22a and 22b and Their Corresponding Precursors 23a and 23b.

We then examined whether the BHQ-based ntla cMO 22b could be activated in targeted regions of the zebrafish embryo using two-photon excitation. These experiments followed studies conducted previously in our laboratory, in which we uncaged a DMNB-based ntla cMO in a subset of mesodermal progenitor cells, selectively inducing them to differentiate into medial floor plate cells rather than the notochord.(7) To identify the irradiated cells in this earlier investigation, we coinjected the zebrafish embryos with mRNA encoding Kaede fluorescent protein, which undergoes a green-to-red photoconversion upon one-photon excitation. Because Kaede is inefficiently converted by two-photon excitation, we utilized a caged coumarin fluorophore conjugated to dextran (dextran−HCC-NPE) as a cell-autonomous photoactivatable tracer (Figure S5 in the Supporting Information). This new class of caged coumarins is highly sensitive to two-photon irradiation,36,37 and dextran−HCC-NPE has demonstrated efficacy in vivo.(38) We injected dextran−HCC-NPE into one-cell-stage embryos with either the BHQ-based ntla cMO 22b or the BHQ-conjugated inhibitory oligomer 23b. An 80 μm × 60 μm × 50 μm region of the posterior, axial mesoderm at the end of gastrulation was then subjected to two-photon irradiation for 2 min (Figure 10A−D). These cells normally differentiate into vacuolated notochord cells by 1 dpf, and the embryos injected with the coumarin tracer and BHQ-functionalized ntla MO inhibitor 23b contained a cluster of blue-fluorescent notochord cells toward the end of the yolk extension (Figure 10A,E). In contrast, embryos injected with both the photoactivatable tracer and ntla cMO 22b exhibited medial floor plate cells with blue fluorescence and few, if any, fluorescently labeled notochord cells (Figure 10F). These observations confirm the sensitivity of the BHQ-based reagent to two-photon photolysis and illustrate the versatility of the hairpin cMO architecture.
Figure 10.

Two-photon uncaging of the BHQ-based ntla cMO in vivo. (A) Schematic representation of embryos injected with the dextran−HCC-NPE photoactivatable tracer and then two-photon-irradiated within the posterior axial mesoderm at the 10 hpf stage (red square, dorsal view). By 1 dpf, these irradiated cells normally differentiate into vacuolated notochord cells (red rectangle, lateral view) toward the end of the yolk extension. (B) IR gradient contrast image of the posterior axial mesoderm with the targeted irradiation area indicated by the dashed red box (dorsal view). (C) Fluorescent image of the same region prior to irradiation (excitation, 820 nm; emission, 525/70 nm). (D) Fluorescent image post-irradiation (same conditions as in C). (E) Embryos injected with the dextran−HCC-NPE and oligomer 23b contained a cluster of blue-fluorescent notochord (nc) cells by 1 dpf (lateral view; excitation, 436/20 nm; emission, 480/40 nm). (F) Embryos injected with dextran−HCC-NPE and ntla cMO 22b exhibited medial floor plate (mfp) cells with blue fluorescence and few fluorescently labeled notochord cells (same conditions as in E). Scale bars: 50 μm.
Conclusion
Photoactivatable MOs are valuable probes of embryonic patterning mechanisms, and their implementation by the developmental biology community will require efficient synthetic procedures and general principles for cMO design. To advance these goals, we have devised a bifunctional linker for the synthesis of hairpin cMOs that enables these probes to be prepared in three steps from commercially available reagents. This optimized synthetic procedure has enabled us to prepare several ntla-targeting cMOs of differing configurations, and we have used these structural variants to determine how the thermodynamic properties of cMOs correlate with their in vivo functions. Our studies reveal that hairpin cMO function is highly dependent on the sequence and location of the complementary inhibitor as well as the linker structure.
We further observed that MO/MO and MO/RNA binding free energies in vitro differ dramatically from the apparent free energies of their functional interactions in vivo, undoubtedly reflecting the influence of oligonucleotide-binding proteins, RNA secondary structure, and other embryonic factors. Although the apparent free-energy values obtained through our in vivo studies cannot accurately describe the true MO and RNA binding affinities in zebrafish embryos, our findings demonstrate that these thermodynamic descriptors can be used to predict MO efficacy. Thus, cMO activity in vivo can be modeled as a competitive three-state equilibrium, and the complex interactions between MOs and embryonic factors can be treated as an aggregate phenomenon. Our findings establish a metric for cMO design, which we have successfully applied to cMOs that target the flh, heg, and etv2 genes, and these empirically derived principles will help guide future applications of cMOs in functional genomic studies. Our insights should also be applicable to other caged hairpin reagents, including those based upon ncPNAs and 2′-O-methyl RNAs.(22) Although these oligonucleotide analogues interact with complementary oligomers with distinct in vitro and in vivo free energy values, their design and biological application are subject to the same principles outlined in this study. As illustrated by the partial phenotypes we observed with our spt cMO, however, the activity of caged hairpin oligonucleotides can be attenuated in vivo through mechanisms that are not yet understood. We anticipate that our guidelines for cMO design will become more refined as this chemical technology is more broadly utilized in embryological research.
Finally, the modularity of our hairpin cMOs provides an opportunity for future improvements to this technology, as illustrated by our development of a BHQ-based cMO that is compatible with two-photon excitation. For example, linkers that minimize the entropic loss associated with stem−loop formation could increase the energetic difference between inter- and intramolecular MO/inhibitor duplexes, thereby improving the dynamic range of basal and photoinduced activities. Alternative approaches might involve incorporating the photocleavable bond within the inhibitory oligomer or caging the MO with two short complementary oligomers rather than one long inhibitor. Investigating these possibilities through organic synthesis will expand the utility of cMO technologies and help shed new light on the molecular mechanisms of embryonic development.
Experimental Section
General Synthetic Procedures
All of the reactions were carried out in flame-dried glassware under an argon atmosphere using commercial reagents without further purification, unless otherwise indicated. Reactions were magnetically stirred and monitored by thin-layer chromatography (TLC) using glass-backed silica gel 60F254 (Merck, 250 μm thickness). Yields refer to chromatographically and spectroscopically pure compounds, unless otherwise stated. SiO2 chromatography was carried out with EM Science silica gel (60 Å, 70−230 mesh) as a stationary phase. 1H NMR and 13C NMR spectra were acquired on Varian 300, 400, and 500 MHz spectrometers and standardized to the NMR solvent peak. Electrospray mass spectrometry (ESI-MS) was performed using a Micromass ZQ single-quadrupole liquid chromatograph−mass spectrometer (LC−MS) and a Micromass Q-TOF hybrid quadrupole LC−MS. Detailed synthetic procedures and structural characterization data are provided in the Supporting Information.
Representative Procedure for MO Inhibitor Synthesis (7e)
An MO oligomer (5′-GCCTCAAGTC-3′) with 5′-amine and 3′-fluorescein modifications was purchased from Gene-Tools, LLC and used without further purification. This oligomer (100 nmol) was dissolved in borate buffer [0.1 M Na2B4O7 (pH 8.5), 100 μL] and combined with linker 2a (0.76 mg, 1.5 μmol) in DMSO (15 μL). The reaction was shaken overnight in the dark and then lyophilized to dryness. The resulting yellow gum was dissolved in water (0.5 mL), washed three times with CHCl3 (0.5 mL), and diluted to 1.5 mL with water. The yellow solution was loaded onto Toyopearl Super-Q resin (400 μL), washed three times with wash solution [aqueous 2.5 mM Na2B4O7 (pH 8.5), 50% acetonitrile (ACN)] and two times with water. Fluoresceinated oligomers were eluted from the resin with 600 μL of aqueous 5% HOAc/50% ACN, washed three times with CHCl3 (0.6 mL), and neutralized with 10% NH4OH(aq) (0.3 mL). Solvent was removed in vacuo, and the remaining NH4OAc was removed by repeated aqueous solubilization and lyophilization, affording 7e as a yellow solid (70 nmol, 70%). MS-ESI for 7e [M + H]+ (m/z): calcd for C184H264N69O61P10, 4728; found, 4728.
Representative Procedure for cMO Synthesis (8e)
The inhibitory oligomer 7e (50 nmol) and azide-functionalized ntla MO(7)6 (50 nmol) were dissolved in phosphate buffer [KH2PO4 (pH 8.0), 230 μL]. To this mixture was added sodium ascorbate (99.0 μg, 500 nmol) in 25 μL of water followed by TBTA (265 μg, 500 nmol) and CuI (95.2 μg, 500 nmol) in 50 μL of DMSO. The reaction mixture was briefly sonicated and then stirred overnight at room temperature in the dark. Precipitate was removed from the reaction mixture by centrifugation, and the supernatant was split and desalted over two Zeba Desalt size-exclusion columns (Pierce) according to the manufacturer’s instructions. The desired product was purified from the reaction mixture by adjusting the solution pH to 11.5 with 1 M NaOH(aq) and loading it onto a DNAPac PA-100 ion-exchange HPLC column (Dionex, 9 mm × 250 mm). Aqueous running buffers were (A) 0.02 M NaOH, 1% ACN and (B) 0.375 M NaClO4 in 0.02 M NaOH and 1% ACN. A stepwise gradient was used to separate the product and starting materials, with specific conditions determined by column capacity. A representative purification gradient was the following: 7 to 19% B in 5 min, 19 to 22% B in 10 min, 22 to 50% B in 1 min, and 50% B for 9 min (flow rate 4 mL/min). Elution fractions were collected with the UV−vis flow-cell lamp turned off to prevent photolysis. Fractions (1 mL) were collected every 15 s and buffered with 1 M NH4OAc(aq) (pH 5.0) (40 μL). The fractions containing fluoresceinated product were combined and desalted over a Zeba size-exclusion column. The eluent volume was reduced in vacuo to 50 μL, and the cMOs were precipitated with acetone (400 μL). After centrifugation, the supernatant was discarded, and the cMO pellet was washed with ACN (100 μL) and briefly lyophilized, affording 8e as a yellow solid (7 nmol, 14%). MS-ESI for 8e [M + H]+ (m/z): calcd for C488H737N219O164P35, 13379; found, 13380.
Zebrafish Aquaculture and Husbandry
Adult zebrafish (wild-type AB strain) were acquired from the Zebrafish International Resource Center. Embryos used in these studies were obtained by natural matings and cultured in E3 embryo medium at 28.5 °C according to standard procedures.(39)
MO Microinjections
Various MO, MO/inhibitor duplex, and cMO solutions containing 0.1% (w/v) phenol red were prepared and microinjected at 1 or 2 nL/embryo. For example, to inject 115 fmol of MO, 2 nL of a 57.5 μM solution containing 0.1% (w/v) phenol red was injected into each zebrafish embryo at the one-cell stage. All embryo injections were done according to standard procedures, and the embryos were subsequently cultured in E3 medium at 28.5 °C. For two-photon experiments, solutions containing 1.25 mM dextran−HCC-NPE and 0.1% (w/v) phenol red with or without 57.5 μM cMO 22b were injected at 2 nL/embryo.
Photolysis of cMOs in Vitro
Photolysis reactions were performed by dissolving 1 nmol of cMO hairpin in water (2 μL) and irradiating for 1 min using a Leica DM4500B compound microscope equipped with an A4 filter cube (ex: 360 nm, 40 nm bandpass) and a 20× water-immersion objective (0.50 NA, 13 mW/cm2 intensity at 360 nm). Longer irradiation times did not improve reaction yields. The solutions were then adjusted to pH 11.5 with 0.02 M NaOH and analyzed by HPLC using a DNAPac PA-200 ion-exchange column (Dionex, 4 mm × 250 mm). Aqueous running buffers were (A) 0.02 M NaOH, 1% ACN and (B) 0.375 M NaClO4 in 0.02 M NaOH and 1% ACN. The HPLC gradient was 7 to 50% B in 27 min at 1.2 mL/min.
Photoactivation of cMOs in Vivo
Zebrafish embryos between the 64- and 256-cell stages were arrayed in an agarose microinjection template. Mercury lamp light was focused onto the individual embryos for 10 s using a Leica DM4500B compound microscope equipped with an A4 filtercube and a 20× water-immersion objective. Embryos were oriented with the animal pole facing the light source. Following photoactivation, embryos were cultured in E3 medium at 28.5 °C.
Two-Photon Irradiation of cMOs
Two-photon cMO photoactivation in zebrafish embryos was performed on an upright two-photon confocal microscope (Ultima XY, Prairie Technologies, Inc., Middleton, WI) equipped with two Ti:sapphire lasers (Mai Tai HP, Spectra Physics, Mountain View, CA) and a 40× (0.8 NA) water-immersion objective (LUMPlanFl/IR, Olympus America, Center Valley, PA). The 820-nm illumination from the first laser (10 mW at the back focal plane of the objective) was used to collect two initial images for each embryo: an epifluorescence image (bandpass: 525 nm center, 70 nm fwhm) and an IR gradient contrast image (820-nm illumination).(40) Using the gradient contrast image, an 80 μm × 60 μm × 50 μm region of interest (ROI) was selected for photoactivation. The ROI was then illuminated for 2 min at 750 nm (65 mW at the back focal plane of the objective) with the second laser. Following photoactivation, the embryo was reimaged with 820-nm illumination. Following two-photon irradiation, embryos were cultured in E3 medium at 28.5 °C.
Bright-Field and Fluorescence Microscopy
Chorions were manually removed from 1 dpf embryos, which were immobilized in E3 medium containing 0.7% (w/v) low-melt agarose and 0.05% (w/v) tricaine. Bright-field images were obtained at 5× with a Leica MZFLIII fluorescence stereoscope equipped with a Leica DC300F digital camera. Differential interference contrast images and time-lapse movies were obtained with a Leica DM4500B fluorescence microscope equipped with a 10× (0.25 NA) objective and a QImaging Retiga-SRV digital camera. Fluorescence images were also obtained with this equipment and a CFP filterset (excitation, 436/20 nm; emission, 480/40 nm).
Determination of MO Duplex Binding Energies
For intermolecular MO duplexes, the complementary oligomers (0.5 μM, 1:1 molar ratio) in buffer [100 mM KCl, 20 mM HEPES, 10 mM MgCl2, 0.1 mM EDTA (pH 7.0), 1 mL] were denatured at 95 °C for 5 min. Thermal denaturation curves were obtained by monitoring temperature-dependent changes in the absorbance of 260-nm light using a Varian Cary 300 spectrophotometer (annealing at 0.5 °C/min). The hypochromicity curves were fitted to a sigmoidal function, and thermodynamic parameters were calculated using the non-self-complementary algorithm in MeltWin 3.0b software. Binding free energies were calculated at 28 °C. For intermolecular MO/RNA duplexes, the complementary MO and RNA oligomers were used in a 2:1 molar ratio to minimize hypochromicity changes due to RNA self-annealing. For intramolecular duplexes, thermodynamic parameters were calculated using the hairpin algorithm in MeltWin 3.0b software.
Western Blot Analysis
At bud stage (10 hpf), wild-type and MO-injected embryos were dechorinated with Pronase (1 mg/mL) for 10 min at 28 °C. The embryos were transferred to microcentrifuge tubes and homogenized with a pipet in TM1 buffer [180 μL/sample; 100 mM NaCl, 5 mM KCl, 5 mM HEPES (pH 7.0), 1% (w/v) PEG-20000] containing protease inhibitors [1 mM PMSF, 5 mg/mL complete Mini protease inhibitor cocktail, EDTA-free (Roche)] to remove yolks. Following centrifugation (500g, 5 min, 4 °C), the TM1 solution was replaced, and the pelleted cells were homogenized again with a pipet and recentrifuged. Eighteen deyolked embryos from each set of experimental conditions were vortexed in SDS-PAGE loading buffer [50 μL/sample; 100 mM Tris-HCl (pH 6.8), 330 mM 2-mercaptoethanol, 4% (w/v) SDS, 20% (v/v) glycerol, 100 mM DTT], sonicated for 1 min, and heated to 100 °C for 5 min. The lysates were resolved on a 4−12% Bis-Tris gradient acrylamide gel (five embryos/lane) and blotted onto nitrocellulose according to standard protocols. Anti-Ntla antibody was used at a 1:2000 dilution in 1× phosphate-buffered saline containing 0.1% (v/v) Tween 20 and 0.2% (w/v) I-Block (Roche). The anti-Ntla antibody was then detected using a horseradish peroxidase-conjugated anti-rabbit IgG antibodies (GE) at 1:10000 dilution and the SuperSignal West Dura Extended Duration Substrate kit (Pierce) according to the manufacturer’s instructions. The chemiluminescence from the membrane was digitally imaged (ChemiDoc XRS, Biorad), and the band intensity was measured with Quantity One 4.5 software. The nitrocellulose membranes were then reprobed with mouse anti-β-actin [sc-8432 (Santa Cruz Biotechnology) at a 1:250 dilution or clone AC-15 (Sigma) at a 1:10000 dilution] and horseradish peroxidase-conjugated anti-mouse IgG antibodies (GE) at 1:10000 dilution to normalize for loading differences between lanes.
Gel-Shift Analysis of MO/Inhibitor Exchange with RNA
Targeting and inhibitory oligomers corresponding to ntla cMOs 8a and 8e (5 μM, 1:1 molar ratio) in buffer [100 mM KCl, 20 mM HEPES, 10 mM MgCl2, 0.1 mM EDTA (pH 7.0), 28 μL] were denatured at 95 °C for 2 min and annealed by cooling to 28 °C over 15 min. The complementary, 3′-fluoresceinated 25-base RNA (3 μM in the above buffer, 2 μL) was then added to the annealed MO duplex solution to achieve a final RNA concentration of 0.2 μM. The mixture of oligomers was incubated at 28 °C for either 1 or 10 min and then chilled to 4 °C on ice. The “0 min” sample was prepared by adding the RNA to the annealed MO duplex solution at 4 °C, and the MO/RNA duplex was prepared by heat denaturation and annealing, as was done for the MO/inhibitor duplexes. All of the samples were then immediately mixed with chilled loading dye [6× stock: 60% glycerol, 0.1 M Tris-HCl, 90 mM boric acid, 1 mM EDTA, 0.9 mM xylene cyanol (pH 8.4)] and resolved on a 15% Tris-borate−EDTA acrylamide gel at 200 V for 20 min at 4 °C. After electrophoresis, the acrylamide gel was analyzed with a GE Typhoon imager (488 nm excitation, 580 nm emission).
Acknowledgments
We thank Profs. Dan Herschlag (Stanford), Eric Kool (Stanford), and Timothy Dore (University of Georgia) for thoughtful discussions, Prof. Kool and the Stanford Center on Polymer Interfaces and Macromolar Assemblies for access to instrumentation, and Profs. John Mably (Harvard) and Nathan Lawson (University of Massachusetts-Worcester) for providing us with MO reagents that we used to synthesize the heg and etv2 cMOs, respectively. I.A.S. and X.O. acknowledge training support from the California Institute for Regenerative Medicine (T1-0001) and a Bio-X Stanford Interdisciplinary Graduate Fellowship, respectively. This work was supported by the National Institutes of Health (R01 GM072600 to J.K.C. and R01 GM077593 to W.-H.L.), the March of Dimes Foundation (Basil O’Connor Research Starter Awards 5-FY04-205 and 1-FY-08-433 to J.K.C.), and the Welch Foundation (I-1510 to W.-H. L.).
Supporting Information Available
Synthetic procedures, NMR spectra, and mass spectrometry data for all compounds; supporting figures, charts, and schemes; supplementary movies; and derivations of eqs 1−9. This material is available free of charge via the Internet at http://pubs.acs.org.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Shestopalov I. A.; Chen J. K. Chem. Soc. Rev. 2008, 37, 1294–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grunwald D. J.; Eisen J. S. Nat. Rev. Genet. 2002, 3, 717–724. [DOI] [PubMed] [Google Scholar]
- Skromne I.; Prince V. E. Dev. Dyn. 2008, 237, 861–882. [DOI] [PubMed] [Google Scholar]
- Nasevicius A.; Ekker S. C. Nat. Genet. 2000, 26, 216–220. [DOI] [PubMed] [Google Scholar]
- Urtishak K. A.; Choob M.; Tian X.; Sternheim N.; Talbot W. S.; Wickstrom E.; Farber S. A. Dev. Dyn. 2003, 228, 405–413. [DOI] [PubMed] [Google Scholar]
- Summerton J. Biochim. Biophys. Acta 1999, 1489, 141–158. [DOI] [PubMed] [Google Scholar]
- Shestopalov I. A.; Sinha S.; Chen J. K. Nat. Chem. Biol. 2007, 3, 650–651. [DOI] [PubMed] [Google Scholar]
- Ordoukhanian P.; Taylor J. S. J. Am. Chem. Soc. 1995, 117, 9570–9571. [Google Scholar]
- Richards J. L.; Tang X.; Turetsky A.; Dmochowski I. J. Bioorg. Med. Chem. Lett. 2008, 18, 6255–6258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang X.; Dmochowski I. J. Mol. Biosyst. 2007, 3, 100–110. [DOI] [PubMed] [Google Scholar]
- Tang X.; Swaminathan J.; Gewirtz A. M.; Dmochowski I. J. Nucleic Acids Res. 2008, 36, 559–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer G.; Krock L.; Mikat V.; Engeser M.; Heckel A. ChemBioChem 2005, 6, 1966–1970. [DOI] [PubMed] [Google Scholar]
- Young D. D.; Lusic H.; Lively M. O.; Yoder J. A.; Deiters A. ChemBioChem 2008, 9, 2937–2940. [DOI] [PubMed] [Google Scholar]
- Ando H.; Furuta T.; Tsien R. Y.; Okamoto H. Nat. Genet. 2001, 28, 317–325. [DOI] [PubMed] [Google Scholar]
- Shah S.; Rangarajan S.; Friedman S. H. Angew. Chem., Int. Ed. 2005, 44, 1328–1332. [DOI] [PubMed] [Google Scholar]
- Monroe W. T.; McQuain M. M.; Chang M. S.; Alexander J. S.; Haselton F. R. J. Biol. Chem. 1999, 274, 20895–20900. [DOI] [PubMed] [Google Scholar]
- Nguyen Q. N.; Chavli R. V.; Marques J. T.; Conrad P. G. Jr.; Wang D.; He W.; Belisle B. E.; Zhang A.; Pastor L. M.; Witney F. R.; Morris M.; Heitz F.; Divita G.; Williams B. R.; McMaster G. K. Biochim. Biophys. Acta 2006, 1758, 394–403. [DOI] [PubMed] [Google Scholar]
- Halpern M. E.; Ho R. K.; Walker C.; Kimmel C. B. Cell 1993, 75, 99–111. [PubMed] [Google Scholar]
- Schulte-Merker S.; van Eeden F. J.; Halpern M. E.; Kimmel C. B.; Nusslein-Volhard C. Development 1994, 120, 1009–1015. [DOI] [PubMed] [Google Scholar]
- Stemple D. L. Development 2005, 132, 2503–2512. [DOI] [PubMed] [Google Scholar]
- Amacher S. L.; Draper B. W.; Summers B. R.; Kimmel C. B. Development 2002, 129, 3311–3323. [DOI] [PubMed] [Google Scholar]
- Tang X.; Maegawa S.; Weinberg E. S.; Dmochowski I. J. J. Am. Chem. Soc. 2007, 129, 11000–11001. [DOI] [PubMed] [Google Scholar]
- Furuta T.; Wang S. S.; Dantzker J. L.; Dore T. M.; Bybee W. J.; Callaway E. M.; Denk W.; Tsien R. Y. Proc. Natl. Acad. Sci. U.S.A. 1999, 96, 1193–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mably J. D.; Mohideen M. A.; Burns C. G.; Chen J. N.; Fishman M. C. Curr. Biol. 2003, 13, 2138–2147. [DOI] [PubMed] [Google Scholar]
- Talbot W. S.; Trevarrow B.; Halpern M. E.; Melby A. E.; Farr G.; Postlethwait J. H.; Jowett T.; Kimmel C. B.; Kimelman D. Nature 1995, 378, 150–157. [DOI] [PubMed] [Google Scholar]
- Sumanas S.; Lin S. PLoS Biol. 2006, 4, e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedoryak O. D.; Dore T. M. Org. Lett. 2002, 4, 3419–3422. [DOI] [PubMed] [Google Scholar]
- Zhu Y.; Pavlos C. M.; Toscano J. P.; Dore T. M. J. Am. Chem. Soc. 2006, 128, 4267–4276. [DOI] [PubMed] [Google Scholar]
- Rostovtsev V. V.; Green L. G.; Fokin V. V.; Sharpless K. B. Angew. Chem., Int. Ed. 2002, 41, 2596–2599. [DOI] [PubMed] [Google Scholar]
- Bonnet D.; Ilien B.; Galzi J. L.; Riche S.; Antheaune C.; Hibert M. Bioconjugate Chem. 2006, 17, 1618–1623. [DOI] [PubMed] [Google Scholar]
- Marky L. A.; Breslauer K. J. Biopolymers 1987, 26, 1601–1620. [DOI] [PubMed] [Google Scholar]
- Melby A. E.; Kimelman D.; Kimmel C. B. Dev. Dyn. 1997, 209, 156–165. [DOI] [PubMed] [Google Scholar]
- Griffin K. J.; Amacher S. L.; Kimmel C. B.; Kimelman D. Development 1998, 125, 3379–3388. [DOI] [PubMed] [Google Scholar]
- Bisgrove B. W.; Snarr B. S.; Emrazian A.; Yost H. J. Dev. Biol. 2005, 287, 274–288. [DOI] [PubMed] [Google Scholar]
- Yu W.; Mei Y.; Kang Y.; Hua Z.; Jin Z. Org. Lett. 2004, 6, 3217–3219. [DOI] [PubMed] [Google Scholar]
- Dakin K.; Li W. H. Nat. Methods 2006, 3, 959. [DOI] [PubMed] [Google Scholar]
- Zhao Y.; Zheng Q.; Dakin K.; Xu K.; Martinez M. L.; Li W. H. J. Am. Chem. Soc. 2004, 126, 4653–4663. [DOI] [PubMed] [Google Scholar]
- Guo Y. M.; Chen S.; Shetty P.; Zheng G.; Lin R.; Li W. H. Nat. Methods 2008, 8, 835–841. [DOI] [PubMed] [Google Scholar]
- Zebrafish: A Practical Approach; Nusslein-Volhard C., Dahm R., Eds.; Oxford University Press: New York, 2002; Vol. 261. [Google Scholar]
- Dodt H.; Eder M.; Frick A.; Zieglgansberger W. Science 1999, 286, 110–113. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.