

Abstract
High-resolution X-ray crystal structures determined in the past six years dramatically influence our view of ligand induced activation of the epidermal growth factor receptor (EGFR) family of receptor tyrosine kinases. Ligand binding to the extracellular region of EGFR promotes a major domain reorganization, plus local conformational changes, that are required to generate an entirely receptor mediated dimer. In this activated complex the intracellular kinase domains associate to form an asymmetric dimer that supports the allosteric activation of one kinase. These models are discussed with emphasis on recent studies that add details or bolster the generality of this view of activation of this family of receptors. The EGFR family is implicated in several disease states, perhaps most notably in cancers. Activating tumor mutations have been identified in the intracellular and extracellular regions of EGFR. The impact of these on understanding of EGFR activation and its inhibition are discussed.
Keywords: Epidermal growth factor receptor (EGFR), Receptor tyrosine kinase (RTK), Ligand induced receptor dimerization, Mechanisms of activation and inhibition
I. Introduction
The epidermal growth factor receptor (EGFR) is historically the prototypical receptor tyrosine kinase (RTK). It was the first of this large family of transmembrane receptors to be cloned; the first for which the importance of ligand mediated oligomerization in the activation of the enzyme was appreciated; and the first for which a clear connection between aberrant receptor function and cancer could be drawn (58, 67, and references therein).
It is now well accepted that the first step in RTK activation involves ligand-induced receptor dimerization or alteration of a pre-existing dimers (57). This leads to stimulation of the intracellular kinase domain and tyrosine autophosphorylation in trans. Phosphorylated tyrosines act as recruitment sites for down stream signaling molecules containing SH2 and/or PTB domains. The cascade of events that are initiated following activation of EGFR represent some of the most extensively studied sets of signal transduction pathways. Many of the principals governing the regulation of such pathways were elucidated from studies of EGFR signaling (15, 69).
In normal physiological settings EGFR, and the other three homologous members of the EGF receptor family (see next section), regulate key events in coordination of cell growth, differentiation and migration (15). EGFR itself is critical in epithelial development, and other members of the family are essential for cardiac development and/or have well studied roles in mammary glands and the nervous system (7). Aberrant signaling from all 4 receptors, through misregulation of the receptors or of their ligands, has been implicated in diseases including nervous system disorders and many cancers (7, 34, 49). For example, EGFR activation in epithelial tumors has been linked with more aggressive disease and poorer outcomes. Drugs that inactivate EGFR through interaction with either the extracellular or intracellular regions of EGFR are intensively studied in the clinic (49).
This review will consider the impact of high-resolution structural studies upon our current understanding of EGFR regulation. The starting point is the model of ErbB receptor activation that was proposed in 2003 based on structural data available at that time (8, 40). Since 2003, significant new information has been published that increases the sophistication of this model and/or adds new facets that were not previously considered. It is to these studies that I will devote the majority of this review.
II. The Epidermal Growth Factor Receptor family of receptor tyrosine kinases
The EGFR family of RTKs comprises 4 members (collectively referred to as the ErbB or HER family): EGFR itself, ErbB2 (HER2/Neu), ErbB3 (HER3) and ErbB4 (HER4). Like all RTKs, each ErbB receptor comprises a large extracellular region, a single spanning transmembrane (TM) domain, an intracellular juxtamembrane (JM) region, a tyrosine kinase domain and a C-terminal regulatory region (Fig. 1A). The ligands that regulate ErbB receptors can be separated into two main groups (Fig. 2): the ‘EGF agonists’ that activate EGFR, and the neuregulins (NRG) that bind ErbB3 and ErbB4 (69). There are at least 7 different EGF agonists: EGF, transforming growth factor α (TGFα), amphiregulin (AR), betacellulin (BTC), epigen (EPN), epiregulin (EPR) and heparin binding EGF-like growth factor (HB-EGF) (30). Of these, a sub-set can also activate ErbB4, and are known as the bispecific ligands (BTC, EPR and HB-EGF). ErbB3 and ErbB4 are regulated by multiple differently spliced variants of the 4 different NRG gene products (20). Each ErbB ligand contains an EGF-like core domain of about 60 amino acids (Fig. 2) that is sufficient for its biological activity (30). ErbB2 has no known soluble ligand and has been proposed play a role in ErbB receptor activation by forming heterodimers with other liganded ErbB family members (14, 69). ErbB2 is also distinguished from other members of this receptor family in that overexpression of ErbB2 causes ligand independent cell transformation (18). As shown in Fig. 2, and discussed in detail below, ErbB2 also turns out to be an outlier structurally.
FIGURE 1. The domains of EGFR.
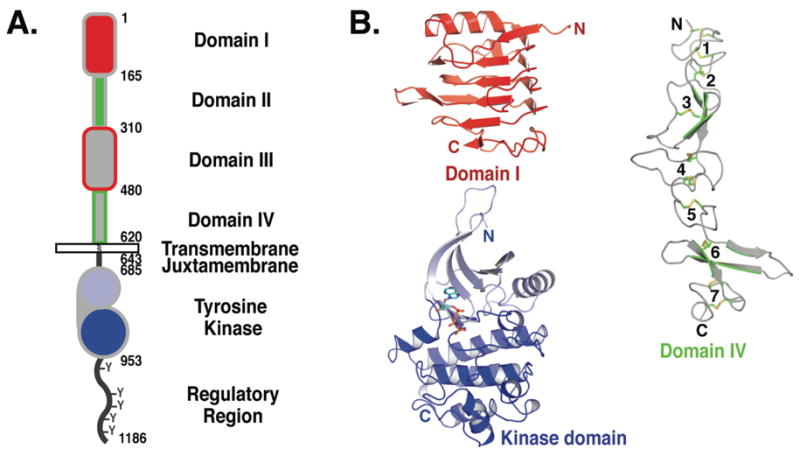
A. The extracellular region comprises 4 domains: I–IV, sometimes referred to as L1, CR1, L2 and CR2 or L1, S1, L2 and S2. Domains I (red) and III (gray with red outline) share about 37% sequence identity, while domains II (green) and IV (gray with green outline) are cystine rich. The N-lobe of the kinase domain is in light blue and the C-lobe in darker blue. This color scheme is used in all figures unless otherwise noted. Amino acid numbers are noted for each domain boundary. The conventional numbering system is used in which amino acid one of EGFR is the assumed first amino acid of the mature protein. In some recent papers, including those defining EGFR cancer mutations, alternative numbering is used where the signal peptide of EGFR is included. To convert to this alternative scheme add 24 to numbers used here.
B. Representative cartoons of the domains of EGFR. Domains I and III adopt a β-helix fold, here domain I from pdb id 1YY9 is shown. Domains II and IV adopt extended structures comprising a series of disulfide-bonded modules. Domain IV from pdb id 1YY9 is shown with the disulfides in stick representation and the disulfide-bonded modules numbered. There are two types of disulfide-bonded module. One has a single disulfide bond and the intervening loops adopt a bow-like arrangement (modules 2, 3, 5 & 6). The second type has two disulfide bonds with consecutive cysteines linked in the pattern Cys1–Cys3 and Cys2–Cys4 (modules 1, 4 & 7). The inactive kinase is shown (pdb id 2GS7) with the ATP analogue (AMP-PNP) in stick representation.
FIGURE 2. The extracellular regions of ErbB receptors and their activating ligands.

Two orthogonal cartoon views of each unliganded ErbB receptor (pdb ids 1NQL, 1N8Z, 1M6B and 2AHX). The coordinates of domain III only were used to align the structures. ErbB2 is an outlier adopting an extended rather than tethered arrangement of domains (see text). Ligands are listed, grouped according to the receptors they activate. Cartoons of TGFα (left; pdb id 1MOX) and NRG1α (right; pdb id 1HRE) are shown in cyan as representative structures of the EGF-like domain of ErbB ligands. The scale for the ligands is twice that used for the receptor extracellular regions.
The extracellular regions of EGFR family members contain two homologous ligand binding domains (domains I and III) and two cystine rich domains (domains II and IV; Fig. 1). The only other RTKs with a similar extracellular domain arrangement are members of the insulin receptor (IR) family, which share the same domain I/II/III organization, but the membrane proximal cystine rich domain IV of ErbB receptors is replaced by fibronectin type III domains in the IR family. By contrast with the EGFR and IR families most other RTKs have extracellular regions comprised of immunoglobulin or fibronectin type III domains (32). Just as they are distinct in their domain composition, so do the IR and ErbB families differ from other RTKs in their mechanisms of ligand activation (66).
Although high-resolution structural studies of intact RTKs pose technical challenges that have not yet been overcome, there is a wealth of structural data on both the extra- and intra-cellular regions of the EGFR family. X-ray crystal structures have been determined for the extracellular regions of all four ErbB receptors (sErbBs) in their unliganded state (6, 11, 12, 22, 25) (Fig. 2). The structure of the EGFR extracellular region (sEGFR) has also been determined in a dimeric – presumably activated – state induced by binding of EGF or TGFα (26, 54) (Fig. 3). Additional insight into the mechanisms of extracellular control has also been provided by three different structures of sErbB proteins in complex with the Fab fragments of inhibitory therapeutic antibodies (12, 24, 44). The structure of the intracellular kinase domain of EGFR has also been extensively studied in different activation states (62, 68, 71, 74). Structural details of the individual domains of EGFR and their homologues have been extensively reviewed elsewhere (1, 32, 40) and are summarized briefly in the legend to Fig. 1.
FIGURE 3. Ligand induced dimerization of the extracellular region of EGFR.

A. Cartoon of the tethered sEGFR (pdb id 1YY9) oriented as in the upper panel of Fig. 2.
B Cartoon of the TGFα induced dimer of sEGFR501 (pdb id 1MOX). The orientation of domain III is as in the lower panel of sErbB cartoons in Fig. 2. The colors of the left hand molecule have been lightened for contrast.
C. A molecular surface representation of tethered sEGFR in the same orientation as in A with domains I and III in red, II in green and IV in gray. An ≈ 130° rotation about the indicated axis (black dot) plus 20 Å translation into the plane of the page is required to bring domain I from it position in the tethered structure (left) its location in the dimer (right) (22). In this model of the sEGFR dimer, domain IV is included such as to maintain the same domain III/IV relationship as in the tethered structure. Domains I, II and III in the dimer are from pdb id 1IVO and are shown in the same orientation as in B.
Despite this wealth of structural information, there are important regions of EGFR for which relatively little data are available. For example, little is known about the structure of the first ~30 amino acids of the intracellular JM region (Fig. 1A), which may play an important regulatory role (31, 48). Moreover, the most C-terminal ~190 amino acids of EGFR that contains multiple tyrosine phosphorylation sites is poorly characterized, but is clearly implicated in regulation of receptor activation (45).
III. The unexpected receptor mediated dimer of the extracellular region of EGFR
In 2002 two papers published back-to-back in Cell radically changed the mechanistic view of ligand induced EGFR dimerization. They described a dimer in which all contacts between the two molecules were receptor-mediated (26, 54). This contrasts starkly with the direct contribution of the bound ligand to the dimer interface of other cytokine receptor and RTK dimers that have been studied (5, 8, 32, 57, and references therein). For many RTKs, such as those of the platelet derived growth factor (PDGF)/Kit receptor family, the ligands themselves are dimeric and bivalent. In Kit, each protomer in the ligand contacts a different receptor molecule, so that the dimeric ligand effectively crosslinks the receptor into a dimeric complex (46, 72). In other cases, such as the fibroblast growth factor (FGF) receptor, accessory molecules (heparan sulfate proteoglycans) link the two ligands to yield a bivalent complex that effectively crosslinks two receptor molecules (59).
In the dimeric complexes formed when EGF or TGFα bind to the first three domains of sEGFR the growth factor binding sites are distant from the dimer interface, and do not contribute directly to dimer contacts (Fig. 3B). All contacts across the dimer interface are mediated by domain II of the receptor – making these dimers ‘receptor-mediated’ rather than ‘ligand-mediated’ (58). A beta hairpin, referred to as the ‘dimerization arm’, in domain II makes extensive contacts with the domain II of its binding partner, reaching at its tip to interact with the opposite domain I (Fig. 3B & C). EGF/TGFα are bivalent; each ligand binds simultaneously to domains I and III of the same receptor molecule.
Structures of unliganded sErbB receptors (22, 44) indicated that ‘exposure’ of the ‘dimerization arm’ is a key event in regulation of receptor dimerization. Indeed, in the inactive/unliganded form of sEGFR and sErbB3, the dimerization arm is buried by intramolecular interactions with domain IV (11, 22, 44). The intramolecular interactions in this so-called ‘tethered’ conformation of the receptor and the intermolecular interactions across the dimer interface are mutually exclusive. This led to the proposal that the tethered sEGFR and sErbB3 structures represent an autoinhibited conformation (8, 22), since also seen for sErbB4 (6).
The tethered configuration of unliganded sEGFR, sErbB3 and sErbB4 also places the two ligand-binding sites on domains I and III quite distant from one another. For a single EGF molecule to contact these two binding sites simultaneously, a large domain rearrangement is required in sEGFR (Fig. 3C). This domain rearrangement allows domains I and III to dock onto the same EGF molecule, while simultaneously exposing the dimerization arm in an ‘extended’ sEGFR conformation that closely resembles the structure observed in the dimeric complexes with bound EGF or TGFα (going from left to right in Fig. 3C) (22).
IV. A structure based model for ligand induced EGFR dimerization
Exposure of the dimerization arm is the most obvious change induced upon binding of growth factor ligands to the EGFR extracellular region (Fig. 3), but it remains unclear exactly how this is achieved. The ligand-binding domains I and III are identical in structure whether bound to ligand or not (21, 44), arguing against an allosteric mechanism for ‘triggering’ exposure of the dimerization arm. EGF (or TGFα) binds to a truncated sEGFR (sEGFR501) that cannot form the intramolecular domain II/IV tether (with domain IV removed) only ~20–30 fold more strongly than it binds to intact sEGFR (16, 19, 22). This observation suggests that the tether provides only a modest energy barrier to the close apposition of domains I and III – of the order of 2 kcal/mol. If the tether is this weak, it seems reasonable to suggest that the EGFR extracellular region could exist in a dynamic equilibrium, sampling multiple conformations including tethered and various untethered states. Binding of growth factor to domains I and III could then trap untethered receptors in a dimerization-competent extended configuration, driving the system towards the active dimeric form (8). There has been no direct analysis of the structural dynamics of sEGFR invoked by this model, and this remains an important knowledge gap in the field.
Although there is no doubt that exposing the domain II dimerization arm is necessary for EGFR dimerization, it is clearly not sufficient. Indeed, sEGFR501 remains monomeric unless EGF/TGFα is added (19). Recent studies argue that much smaller adjustments in the conformation of domain II at the dimer interface are also critical (16). As can be appreciated in Fig. 4, significant local conformational changes in domain II accompany the dramatic domain rearrangement. These changes result in a different trajectory of the dimerization arm in the extended compared to the tethered receptor, and significantly alter the overall curvature of the long axis of domain II (Fig. 4A). Since domain II forms the dimer interface, this change in curvature upon ligand binding could have substantial implications for dimerization strength.
FIGURE 4. Conformational changes in domain II of sEGFR.

A. Smoothed backbone representations of domains II from extended (gray) and from tethered (green) sEGFR. The coordinates of domain I and the first three disulfide-bonded modules of domain II (amino acids 1-225) were used to superimpose the two structures. The lines to the right indicate the curvature of the long axis of domain II. Disulfide bonds are shown (A–C & F) and disulfide-bonded modules numbered (A–E, module 1 is not shown in A, D or E).
B. Cartoon of domains I, II and III from extended sEGFR (pdb id 1MOX) oriented as in A. The area of detailed (boxed) shows the domain II/III interactions that contribute to stabilizing module 6. See text for details.
C. Cartoon of domains I, II and III from tethered sEGFR (pdb id 1YY9), oriented as in A. Note the very different trajectory of the end of domain II (in black and marked with an arrowhead) compared to part B.
D. Domain II from the sEGFR501/TGFα dimer is shown with the left hand molecule in gray and the right hand molecule in black. Contact points across the dimer are indicated with asterisks. Using coordinates from disulfide-bonded module 5 only, domain II from tethered sEGFR has been superimposed first on the right hand extended domain II (green) and then on the left hand extended domain II (dark green) to create a model for a “dimer” of two domain II molecules in the tethered conformation.
E. Domain II from extended (gray) and from tethered (green) sEGFR and from sErbB2 (magenta) shown in same orientation as D. See text for details.
F. Cartoon of tethered sEGFR in the same orientation as C and with the positions of somatic mutations in glioblastoma shown in space filling representation. Those mutations that have been show to cause activation of EGFR are underlined.
The domain II conformation remains quite similar in the tethered and extended receptors for the first three disulfide-bonded modules, which are stabilized by interaction with domain I. However, the structures begin to deviate significantly at disulfide-bonded module 4 (marked with an arrow in Fig. 4A), so that domain II has a different overall curvature between disulfide-bonded modules 5 and 8 in the tethered and extended forms. The consequences of the different domain II curvatures can be better appreciated if the two conformations are superimposed using the central disulfide-bonded module 5 as a reference point (Fig. 4D). In dimers constructed from these domain II configurations, disulfide-bonded modules 2 and 6 project further into the dimerization interface in the extended or activated structure (grey) than in the tethered or inactive structure (green). The relative projection of these two modules into the dimer interface allows then to form direct contacts across the dimer interface, as marked with asterisks in Fig. 4D and directly observed in the crystal structures of ligand-bound sEGFR (26, 54). Mutational analysis demonstrates that interfacial interactions from module 6 (involving D279 and H280) contribute substantially to dimer stability, while those from module 2 (involving Q194) contribute to a smaller extent (16). The conformation of domain II in the region of disulfide-bonded module 6 is stabilized in part by direct interactions with domain III (Fig. 4B) (16, 54). Binding of EGF or TGFα to sEGFR drives a dramatic reorientation of domain III (compare Figs. 3B & C), and promotes domain II/III interactions that stabilize the precise conformation of domain II in this region (around module 6) that is required for dimerization.
It has also been suggested that domain IV contributes directly to stabilization of the ligand-induced sEGFR dimer (8, 22), although it was not present in the published dimer structures. If the relationship between domains III and IV remains fixed in tethered (monomeric) and extended (dimeric) sEGFR, the two copies of domain IV are predicted to make contact across the dimer interface (Fig. 3C). However, in studies of soluble sEGFR variants, deletion of a putative domain IV interaction loop or indeed deletion of almost all of domain IV had only minimal effects on dimerization strength (16). It is possible that rather weak domain IV interactions aid in orientating the membrane proximal parts of the EGFR extracellular region in an intact EGFR dimer at the cell surface (4). Indeed, such weak association between the membrane proximal domains of another RTK, Kit, has recently been crystallographically visualized in a ligand-induced dimer (72) and this could be an important theme for interactions in the extracellular region of many RTKs.
V. Constitutively extended ErbB2 and receptor hetero-dimerization
Crystal structures of the orphan ErbB2 extracellular region revealed that it adopts an extended configuration in which the arrangement of the four domains resembles that seen in each molecule of a sEGFR dimer (Fig. 2) (12, 25). Thus, sErbB2 resembles a constitutive, or ligand-independent, activated conformation. This is consistent with the facts that ErbB2 (but not EGFR) overexpression transforms cells (14, 18), and that ErbB2 overexpression is associated with a significant class of human breast cancers (51). Rather than having a growth factor molecule bound between domains I and III – as seen in activated sEGFR – domains I and III of sErbB2 interact directly with one another (Fig. 2), and domain I/III interactions appear to stabilize the extended configuration of sErbB2. This close proximity of sErbB2 domains I and III may explain the failure of countless efforts to identify a high affinity soluble ligand for ErbB2. There is no room for a ligand to bind between domains I and III, so it has been suggested that no such ligand exists (8, 25).
Despite adopting a constitutively extended conformation (Fig. 2), sErbB2 does not homodimerize detectably in solution (23) or even at the very high concentrations present in the crystals used to determine its structure (25, 40). When a symmetric sErbB2 homodimer is modeled by overlaying its dimerization arm with that seen in sEGFR dimers, clashes in the C-terminal portion of domain II (module 8) preclude satisfactory docking. As shown in Fig. 4E, module 6 of domain II of sErbB2 (magenta) adopts the same conformation with respect to the dimerization arm as it does in activated/extended sEGFR (gray). Moreover, the same domain II/III interactions that stabilize this region in the sEGFR dimer are also observed in the sErbB2 structures (25). By contrast the N-terminal part of domain II in sErbB2 does not overlay with the equivalent region in extended sEGFR (Fig. 4E). Thus, although module 6 and the dimerization arm appear to adopt conformations in sErbB2 that are reminiscent of those required for sEGFR homodimerization, disulfide-bonded module 2 is not positioned for optimal interaction across a homodimeric interface, and module 8 will actually disrupt such a symmetric interface. These observations have been interpreted to suggest that the extended sErbB2 structure might be optimal for domain II-mediated heterodimerization of ErbB2 with another ligand-bound (extended) ErbB receptor, in preference to homodimerization (24). For example, X-ray scattering shows that the extracellular region of ErbB3 adopts an extended conformation upon binding to the EGF-like domain of its ligand, neuregulin 1β1 (NRG1-β1) without forming homodimers (17). ErbB2 and ErbB3 are well-known heterodimerization partners (14), and extended ErbB2 may associate with NRG-bound monomeric ErbB3 in a complex reminiscent of an asymmetric version of the dimer depicted in Figs. 3. Some evidence has been presented for formation of an NRG-dependent sErbB2/sErbB3 heterodimer (23), and a high resolution structural view of such a heterodimer remains an important frontier in this field.
VI. sEGFR autoinhibition and the tethered state
The models described above for EGF-induced dimerization of the EGFR extracellular region assume that the crystallographically observed domain II/IV tether represents a set of autoinhibitory interactions. If this is the case, mutations that disrupt the tether should lead to an increased sensitivity of EGFR to activation by ligand. In the extreme case, disrupting such an autoinhibitory tether might lead to constitutive activation of the receptor, and we might anticipate that tether mutations will be found in patients with EGFR-dependent cancers. Contrary to these expectations, mutations designed to disrupt all of the domain II/IV intramolecular interactions in intact EGFR did not substantially alter cell surface activation of the receptor, or the sensitivity of its EGF dependence (16, 47, 64). These results were interpreted to suggest that the tether has a very limited autoinhibitory effect (or even none) on EGFR activity at the cell surface.
Recent studies employing small angle X-ray scattering (SAXS) suggest that a reevaluation of the role played by the tethered configuration of EGFR is required (17). Solution SAXS studies could readily distinguish between the tethered and extended conformations of sEGFR, sErbB3 and sErbB2 at low resolution. However, mutated forms of sEGFR lacking all of the crystallographically-observed domain II/IV interactions thought to stabilize the tethered conformation gave SAXS-derived molecular envelopes that were barely distinguishable from those seen for wild-type tethered sEGFR. Thus, disrupting the domain II/IV tether interactions via mutations is insufficient to drive sEGFR into a significantly extended conformation. The failure of equivalent mutations in full length EGFR to affect receptor activation is therefore likely to reflect their failure to alter the receptor’s conformation, rather than discerning the importance of the tethered state on the cell surface.
If direct domain II/IV interactions do not hold sEGFR in a tethered or compact conformation, what does? As pointed out by Dawson et al., (17) the relative orientation of domains II and III are very similar in tethered sEGFR and in domain I/II/III fragments from the IR family (1). This comparison suggests that the region of polypeptide linking disulfide-bonded module 8 of domain II with domain III (marked with an arrowhead in Fig. 4C) may be unexpectedly rigid. Rigidity of this linkage could play a significant part in maintaining the EGFR extracellular region in a tethered-like conformation. Consistent with this notion, mutation of a cysteine in the domain II module 8 disulfide bond causes a partial gain-of-function in the C. elegans EGFR ortholog Let-23 (37). Conserved prolines in this region may also contribute to local mainchain stability, consistent with the relatively low crystallographic temperature factors (B-factors) observed in this region in each tethered sErbB receptor. By contrast the B-factors in disulfide-bonded module 8 in the ligand bound dimers are higher than average, suggesting that in the extended configuration this region is under strain. Additional stabilization of the tethered configuration could come from oligosaccharides, not fully visualized in the X-ray crystal structures.
VII. Extracellular EGFR mutations in cancer
Somatic mutations were recently identified in glioblastomas that map to the extracellular region of EGFR, and a subset of these shown to enhance receptor activation (Fig. 4F) (41). Several of these mutations fall in the vicinity of the intramolecular tether (e.g. P572L and G574V) and could destabilize domain II/IV interactions – although these mutations are not likely to be sufficient for EGFR activation given the discussion presented above. Other mutations cluster in different parts of the extracellular region. One group falls close to disulfide-bonded module 8 of domain II, and could affect the mainchain rigidity in this region – although the effects of these mutations on EGFR activation was not reported. Another cluster of mutations at the domain I/II interface is interesting. In the tethered receptor, A265 from disulfide-bonded module 5 of domain II packs against the aliphatic portion of R84 from domain I (Fig. 4F). Mutations at either of these positions leads to EGFR activation (41), which could possibly result from alterations in the conformation of domain II.
Further studies of the effects of these mutations (and their combinations) on the conformational properties of the EGFR extracellular region should provide important insight into the structural restraints that keep sEGFR in a tethered-like conformation, and into the energetic barriers to its extension and dimerization.
VIII. Activation of the intracellular tyrosine kinase domain of EGFR
The unique mechanism of ligand-induced dimerization is not the only feature that sets EGFR apart from other RTKs. Recent structural studies of the intracellular EGFR tyrosine kinase domain (EGFR-TK) also suggest that it is regulated through an unexpected set of interactions (74), with the formation of an asymmetric kinase domain dimer being critical.
The first reported crystal structures of EGFR-TK revealed a conformation with characteristics of an activated kinase (62), based on the structural features of its activation loop and the orientation of the C-helix (in the N-terminal lobe). Although the apparently constitutive adoption of such an active conformation was surprising, it was consistent with the fact that EGFR is unusual in not requiring activation-loop phosphorylation to promote its activity (27). These structures suggested a notable absence of autoinhibitory interactions in the EGFR kinase domain, by contrast with the well-defined interactions that maintain the kinase domains from the insulin receptor, FGF receptor, and other RTKs in their inactive states (33). From among the possible interpretations of this structural view (reviewed by Burgess et al., 8), elegant studies from the Kuriyan laboratory (74) argue that crystal packing mimics interactions found in an active receptor dimer, which lead to activation of EGFR-TK through an allosteric mechanism.
Structures of EGFR-TK bound to the therapeutic inhibitor lapatinib (68) and of an EGFR kinase mutant bound to AMP-PNP (74) revealed that EGFR-TK can also adopt a characteristic inactive structure (Fig. 5A) with clear intramolecular autoinhibitory interactions. The two inactive EGFR-TK structures are virtually identical to one another, and resemble inactive forms of cyclin-dependent kinases (CDKs) and Src-family kinases (33). In each case, a short helical region in the activation loop packs against the catalytically-critical C-helix, which contains a conserved glutamate that must form an ion pair with a lysine that coordinates ATP’s α- and β-phosphates. The C-helix is displaced (and this ion pair is disrupted) by interaction with the activation loop in inactive EGFR-TK, Src-family, or CDK kinases.
FIGURE 5. Activation of the EGFR kinase domain.

A. Cartoon of the EGFR kinase domain in the inactive conformation with AMP-PNP in stick representation (pdb id 2GS7). The activation loop (A-loop) is in magenta and the catalytically important C-helix is in yellow.
B. Cartoon of the asymmetric EGFR kinase domain dimer (pdb id 2GS6) with the ATP moiety of the bound ATP-peptide conjugate in stick representation. The conformation of the activation loop (magenta) and position of the C-helix (yellow) are consistent with an active kinase (33).
C. A cartoon view of the inactive kinase domain in the same orientation and colors as in A. The locations of somatic mutations identified in NSCLC are indicated.
Mutations that disrupt the interactions between the C-helix and activation loop (Fig. 5) activate EGFR-TK (13, 56, 74), and are clinically very important (see below). Interestingly, the C-helix/activation loop interactions are also incompatible with the packing of EGFR-TK molecules in crystals of the active state. In numerous crystals of active EGFR-TK, an asymmetric dimer can be identified in which the helical C-lobe of one EGFR-TK molecule abuts the N-lobe of its neighbor in the crystal, in an interaction that is highly reminiscent of the activating interaction between helical cyclins and the cyclin dependent kinases (CDKs) (35, 74). This asymmetric dimer occurs in the crystal lattice of all cases where the active conformation is observed for EGFR-TK, and interactions of the C-helix of one EGFR-TK molecule (in the N-lobe) with the C-lobe of its neighbor (Fig. 5B) appear to disrupt the autoinhibitory interactions described above (and stabilized by the inhibitor lapatinib). In fact, EGFR-TK could only be crystallized in its inactive conformation in the presence of lapatinib (68) or with a mutation that disrupts the CDK/cyclin-like dimer (74). Moreover, mutations designed to disrupt the asymmetric dimer interface shown in Fig. 5B prevented EGF-activation of the intact EGFR (74).
Thus, contrary to initial suggestions, EGFR-TK does adopt an autoinhibited conformation in the absence of ligand-induced dimerization, as observed for most other RTKs. However, the mechanism of EGFR-TK activation is quite unique. Whereas most RTKs reach full activity following trans-autophosphorylation of their kinase domains within a dimer, EGFR-TK forms an asymmetric dimer that allosterically activates the kinase domain.
IX. Intracellular EGFR mutations in cancer
The importance of EGFR-TK autoinhibition is underscored by the growing numbers of somatic EGFR mutations reported in certain cancers, particularly in non-small cell lung cancer (NSCLC). In clinical NSCLC trials with EGFR-targeted tyrosine kinase inhibitors (TKIs), a small subset of patients showed dramatic initial responses, and this response correlated with the occurrence of somatic mutations in exons 18 to 21 in the EGFR kinase domain (56, 61). Point mutations in the nucleotide binding loop (the P-loop; exon 18) or in the activation loop (exon 21), and deletions immediately preceding the catalytically important C-helix all lead to enhanced sensitivity to TKIs. In vitro cellular and biochemical studies have shown that these alterations activate the EGFR kinase domain, leading to ligand-independent signaling that is effectively inhibited by the TKIs (56). The initial patient response to TKIs therefore appears to reflect inhibition of constitutive, oncogenic, signaling by EGFR in their tumors.
Each class of EGFR-TK mutations found in NSCLC (Fig. 5C) is likely to destabilize the inactive conformation of the EGFR kinase domain (71, 74). For example the L834R substitution (L858R in kinase mutation literature) disrupts interactions between the helical turn in the activation loop and the C-helix in the inactive conformation (Fig. 5A,C). L834 is relatively surface exposed in the active state. Similarly, deletions in the region preceding the C-helix also remove interactions likely to stabilize the inactive conformation of the activation loop (74). The occurrence and properties of these cancer mutations thus strongly argue that in inactive EGFR, as for most other RTKs (32), the kinase domain adopts an autoinhibited state. Normal activation requires ligand-induced dimerization that promotes allosteric activation of EGFR-TK. The cancer mutations circumvent the need for ligand activation by disrupting interactions that maintain the kinase in its autoinhibited inactive state.
X. Mechanism of EGFR activation at the cell membrane
In Fig. 6 an overall model is presented that combines structural information for EGFR on the outside and on the inside the membrane. In the resting state, EGFR is shown with its extracellular region in the tethered configuration and its kinase domain in the inactive form. Ligand binding to the extracellular region induces receptor-mediated dimerization that brings the intracellular domains into close proximity, and promotes the association of the kinase domains in an asymmetric dimer. In the asymmetric EGFR-TK dimer, one molecule is activated through interaction of its N-lobe with the C-lobe of the cyclin-like activator (shown in the inactive conformation). It is thought that the activated kinase phosphorylates the C-terminal tail of the activator (cyclin-like) receptor. In a subsequent step (not shown) it is proposed that the roles of the two receptors switch, such that both intracellular domains can become trans-autophosphorylated.
FIGURE 6. Mechanism of EGFR activation.

The crystal structures from EGFR are placed so as to provide a framework to consider the mechanism of activation of EGFR at the cell membrane. The same scale is used for the cartoon representations (plus transparent molecular surface) of the extracellular region and kinase domain. The TM domain is shown as an α-helix (gray), also to this same scale. Regions that have not been crystallographically defined are shown with a dashed or solid lines. The missing stretches of the inactive kinase are shown in brown, while those of the active kinase are in black. See text for details.
There is structural information for all but the most C-terminal half dozen amino acids of the extracellular region, which link to the presumably helical TM domain. The TM helices of ErbB receptors have been shown to self (and hetero)-associate in membranes (50). Although TM interactions of this sort may aid in stabilizing the dimer, or in orienting its components, mutations that disrupt TM domain association do not influence receptor signaling (10, 36; J.M. Mendrola & M.A. Lemmon, unpublished data).
On the intracellular side of the membrane, several key pieces of information remain missing – as implied in Fig. 6. There is no reliable structural information for the first ~30 amino acids of the intracellular JM region. By analogy with other RTKs, this region may play a regulatory role – possibly contributing to autoinhibition (31). A basic stretch at the beginning of this JM region has been reported to associate with acidic lipids in the inner leaflet of the membrane and this could promote association of the kinase domain itself with the membrane through electrostatic interactions (48). Membrane tethering of this type is proposed to have an autoinhibitory influence on EGFR, which is reversed when calcium/calmodulin (Ca/CaM) binds to the JM region and dissociates it from the membrane. Once released from the membrane the intracellular domains are proposed to come together to form the asymmetric dimer outlined above. If the asymmetric dimer depicted in Fig. 6 forms with its TM domains in contact, the JM region will have to adopt an extended structure in order to link the N-termini of the two kinase domains, which are separated in this model by ~50Å.
The C-terminal ~190 amino acids of EGFR have not yet been resolved in any crystal structure. Circular dichroism studies indicate a significant amount of secondary structure in this region (42). Hydrodynamic studies show that the relationship between the large C-terminal regulatory region and the tyrosine kinase domain is altered upon autophosphorylation (9). FRET studies indicate that the C-terminal and TK domains become separated – to give a more extended molecule – following activation and phosphorylation (43), possibly reflecting loss of intramolecular interactions. It is intriguing that the side-chains of Y974 and Y992 in the C-terminal domain, two of the EGFR autophosphorylation sites (60), make well-defined interactions with the tyrosine kinase domain (68, 74). Disruption of interactions like this could be responsible for the changes in overall intracellular region structure upon EGFR autophosphorylation, and could in turn influence enzymatic activity of the tyrosine kinase domain. The C-terminal region of EGFR may thus play an autoinhibitory role (65), and autophosphorylation could reverse this through the types of conformational changes seen in hydrodynamic and FRET studies. A structural view of this region remains one of the key challenges in this field.
XI. EGF receptor family as targets for anticancer therapy
Therapeutic agents that target both the extracellular and intracellular regions of ErbB receptors are in clinical use and/or development. There are many excellent reviews covering this area, see for example (2, 49, 53, 73). Here, comments are restricted to the impact of structural studies on understanding the mechanisms of receptor inhibition by these drugs. Several monoclonal antibodies with clinical efficacy that bind the EGFR or ErbB2 extracellular regions have been studied. An EGFR-targeted antibody (cetuximab/Erbitux™) binds directly to the ligand-binding site on domain III (44), and will also sterically block the transition from the tethered to extended states depicted in Fig. 3. An ErbB2-targeted antibody (pertuzumab, Omnitarg™) impedes receptor dimerization by binding directly to the domain II (presumed) heterodimerization site in ErbB2 (24). Perhaps the most well-known of the ErbB-targeted antibody therapeutics (trastuzumab/Herceptin™) binds to domain IV of the ErbB2 extracellular domain (12). This appears to block ErbB2 ectodomain shedding that occurs when ErbB2 is expressed at very high levels and that is linked to oncogenesis (52). Although these direct effects on ErbB receptor signaling contribute to the inhibitory effects of the antibodies, antibody-dependent cellular cytotoxicity also plays a significant role in each case. Based on the structural models for ligand-induced ErbB receptor activation described above, it is highly likely that antibodies with other modes of binding to ErbB receptor extracellular regions will prove to be very valuable. One obvious example would be antibodies that stabilize the tethered conformation.
The small molecule tyrosine kinase inhibitors (TKIs) all bind to the ATP-binding site of the kinase domain and structural details of a number of these inhibitors bound to EGFR-TK have been reported (62, 68, 71, 74). Some of the TKIs, notably erlotinib/Tarceva™ and gefitinib/Iressa™, bind to the active configuration of EGFR-TK (62, 71). By contrast, lapatinib/Tykerb™ binds to (and appears to stabilize) the inactive, autoinhibited form of EGFR-TK (68). As described above, numerous EGFR mutations in NSCLC and head-and-neck cancer have been reported that appear to destabilize the autoinhibited conformation of EGFR-TK (56, 61), and screening patients for these mutations can aid in treatment decisions. It should be noted that these mutations destabilize the EGFR-TK conformation preferred by lapatinib (68), but stabilize the conformation bound by erlotinib (62), getitinib and other TKIs (71). The presence of these mutations may therefore indicate the use of erlotinib/gefitinib-type drugs rather than the more bulky lapatinib. This example illustrates how a structural understanding of the effects of both mutations and therapeutic inhibitors on autoinhibitory interactions can have important clinical implications.
Finally, a sobering aspect of the use of TKIs in EGFR-dependent NSCLC is the occurrence of resistance mutations, in particular the T790M mutation (61). This mutation is predicted to clash with inhibitors such as erlotinib or gefitinib (71). Irreversible inhibitors of similar chemical structure (anilinoquinazolines) have been reported to inhibit T790M EGFR-TK (39), and may represent vialble treatment alternatives. Further structural studies are needed to define the optimal inhibitors.
XII. Outstanding questions
A major outstanding issue is precisely how to correlate the structural and biophysical studies that have been the focus of this review with EGFR activation at the cell surface. Since early studies of EGFR it has been known that there are two affinity “classes” for EGF binding to its cell surface receptor (28, 29). These classes are evident in curvilinear Scatchard plots for EGF binding that have been interpreted as indicating a high-affinity (2% - 5%) class and a low-affinity (the remainder) class of sites (3). Several studies attempt to correlate these states with the structure-based model for dimerization of the extracellular region of EGFR with conflicting conclusions (38, 47, 64). It is likely that the low affinity states correspond to the binding of EGF to form a symmetric dimer akin to that in the crystal structures (55). The nature of the high affinity states is less clear, yet of critically importance since this class of receptors is likely the mediator of normal EGFR activation (63). One suggestion is that these high affinity receptors may be preformed oligomers, for which there is some evidence (70). An intriguing and untested possibility is that the extracellular region of EGFR in the high affinity ligand bound state is not symmetrical, but rather mirrors the asymmetry of the intracellular dimer that is proposed in the allostreric activation of the EGFR-TK.
SUMMARY POINTS
Ligand binding to the extracellular region of EGFR induces an entirely receptor mediated symmetric dimer.
The arrangement of the domains of the extracellular region of EGFR is dramatically different in the unliganded and ligand bound states.
Ligand binding induces local conformational changes in domain II of sEGFR that are critical for dimerization.
The EGFR-TK is activated allosterically in an asymmetric homodimer.
EGFR is autoinhibited by interactions in the extracellular region, tyrosine kinase domain and possibly also from the JM and C-terminal regions.
Cancer mutations in both the extracellular region and kinase domain can activate EGFR most likely by destabilizing the inactive, autoinhibited state.
FUTURE ISSUES
A structure of an intact ErbB receptor, or of the entire intracellular region, would clearly be highly informative.
Additional high-resolution structures of ligand induced homo- and hetero-dimers of sErbB receptors will confirm (or not) current models proposed based on available data.
Determination of the features that distinguish the population of cell surface EGFR that binds ligand with high affinity is key to gaining a complete understanding of normal receptor activation.
A molecular understanding of differences in cellular responses to stimulation by different ErbB ligands.
The role of ligand independent EGFR dimerization/clustering needs to be clarified.
Full characterization of the effects of cancer mutations on EGFR (and other ErbB) activity and application of this information to development of optimal therapeutic strategies for patients bearing such mutations.
Greater appreciation of similarities and differences in the mechanisms of activation of the EGF and IR receptor family.
Acknowledgments
We thank Mark Lemmon, and members of the Ferguson laboratory for valuable discussions and critical comments on the manuscript. K.F. is the recipient of a Career Award in the Biomedical Sciences from the Burroughs Wellcome Fund and is the Dennis and Marsha Dammerman Scholar supported by the Damon Runyon Cancer Research Foundation (DRS-52-06).
MINI-GLOSSARY
- ErbB receptors
The members of the EGFR family of RTKs
- EGFR/ErbB1/HER1
ErbB2/HER2/neu, ErbB2/HER3, ErbB4/HER4
- EGF-agonists
The growth factor ligands that activate EGFR
- Tethered sEGFR
The configuration of the 4 domains of the extracellular region of EGFR that is observed in crystal structures of all unliganded/inactive sEGFR, sErbB3 and sErbB4
- Extended sEGFR
The configuration of the 4 domains of the extracellular region of EGFR in the ligand bound form. This is a model based on the crystal structure of the EGF and TGFα complexes with sEGFR in which the domains I, II and III plus the first disulfide-bonded module of domain IV are observed. The remainder of domain IV is added based on the domain III/IV relationship observed in tethered structures of sEGFR. This configuration is also observed the crystal structures of the entire extracellular region of unliganded sErbB2
ACRONYMS
- EGFR
epidermal growth factor receptor
- EGF
epidermal growth factor
- JM
juxtamembrane
- RTK
receptor tyrosine kinase
- sEGFR/sErbBs
the soluble extracellular region of EGFR or of the ErbB receptors
- sEGFR501
a truncated sEGFR that terminates at amino acid 501, after the first disulfide-bonded module of domain IV
- TGFα
transforming growth factor alpha
- TM
transmembrane
- TKI
tyrosine kinase inhibitor
LITERATURE CITED
- 1.Adams TE, Epa VC, Garrett TP, Ward CW. Structure and function of the type 1 insulin-like growth factor receptor. Cell Mol Life Sci. 2000;57:1050–93. doi: 10.1007/PL00000744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baselga J, Arteaga CL. Critical update and emerging trends in epidermal growth factor receptor targeting in cancer. J Clin Oncol. 2005;23:2445–59. doi: 10.1200/JCO.2005.11.890. [DOI] [PubMed] [Google Scholar]
- 3.Bellot F, Moolenaar W, Kris R, Mirakhur B, Verlaan I, et al. High-affinity epidermal growth factor binding is specifically reduced by a monoclonal antibody, and appears necessary for early responses. J Cell Biol. 1990;110:491–502. doi: 10.1083/jcb.110.2.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Berezov A, Chen J, Liu Q, Zhang HT, Greene MI, Murali R. Disabling receptor ensembles with rationally designed interface peptidomimetics. J Biol Chem. 2002;277:28330–39. doi: 10.1074/jbc.M202880200. [DOI] [PubMed] [Google Scholar]
- 5.Boulanger MJ, Garcia KC. Shared Cytokine Signaling Receptors: Structural Insights from the Gp130 System. Adv Protein Chem. 2004;68:107–46. doi: 10.1016/S0065-3233(04)68004-1. [DOI] [PubMed] [Google Scholar]
- 6.Bouyain S, Longo PA, Li S, Ferguson KM, Leahy DJ. The extracellular region of ErbB4 adopts a tethered conformation in the absence of ligand. Proc Natl Acad Sci USA. 2005;102:15024–29. doi: 10.1073/pnas.0507591102. Unliganded sErbB4 is also in the tethered configuration, supporting a general model for all ErbB receptor dimerization. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bublil EM, Yarden Y. The EGF receptor family: spearheading a merger of signaling and therapeutics. Curr Opin Cell Biol. 2007;19:124–34. doi: 10.1016/j.ceb.2007.02.008. [DOI] [PubMed] [Google Scholar]
- 8.Burgess AW, Cho HS, Eigenbrot C, Ferguson KM, Garrett TP, et al. An open-and-shut case? Recent insights into the activation of EGF/ErbB receptors. Mol Cell. 2003;12:541–52. doi: 10.1016/s1097-2765(03)00350-2. Summary of the view of EGFR dimerization and activation that emerged from the structures of the extracellular region of ErbB receptors and early studies of the EGFR kinase domain. [DOI] [PubMed] [Google Scholar]
- 9.Cadena DL, Chan CL, Gill GN. The intracellular tyrosine kinase domain of the epidermal growth factor receptor undergoes a conformational change upon autophosphorylation. J Biol Chem. 1994;269:260–5. [PubMed] [Google Scholar]
- 10.Carpenter CD, Ingraham HA, Cochet C, Walton GM, Lazar CS, et al. Structural analysis of the transmembrane domain of the epidermal growth factor receptor. J Biol Chem. 1991;266:5750–55. [PubMed] [Google Scholar]
- 11.Cho HS, Leahy DJ. Structure of the extracellular region of HER3 reveals an interdomain tether. Science. 2002;297:1330–33. doi: 10.1126/science.1074611. [DOI] [PubMed] [Google Scholar]
- 12.Cho HS, Mason K, Ramyar KX, Stanley AM, Gabelli SB, et al. Structure of the extracellular region of HER2 alone and in complex with the Herceptin Fab. Nature. 2003;421:756–60. doi: 10.1038/nature01392. [DOI] [PubMed] [Google Scholar]
- 13.Choi SH, Mendrola JM, Lemmon MA. EGF-independent activation of cell-surface EGF receptors harboring mutations found in gefitinib-sensitive lung cancer. Oncogene. 2007;26:1567–76. doi: 10.1038/sj.onc.1209957. [DOI] [PubMed] [Google Scholar]
- 14.Citri A, Skaria KB, Yarden Y. The deaf and the dumb: the biology of ErbB-2 and ErbB-3. Exp Cell Res. 2003;284:54–65. doi: 10.1016/s0014-4827(02)00101-5. [DOI] [PubMed] [Google Scholar]
- 15.Citri A, Yarden Y. EGF-ERBB signalling: towards the systems level. Nat Rev Mol Cell Biol. 2006;7:505–16. doi: 10.1038/nrm1962. [DOI] [PubMed] [Google Scholar]
- 16.Dawson JP, Berger MB, Lin CC, Schlessinger J, Lemmon MA, Ferguson KM. Epidermal growth factor receptor dimerization and activation require ligand-induced conformational changes in the dimer interface. Mol Cell Biol. 2005;25:7734–42. doi: 10.1128/MCB.25.17.7734-7742.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dawson JP, Bu Z, Lemmon MA. Ligand-induced structural transitions in ErbB receptor extracellular domains. Structure. 2007;15:942–54. doi: 10.1016/j.str.2007.06.013. First direct evidence for the ligand-induced transition from the tethered to the extended states for sEGFR and sErbB3. Also highlights the relatively minor role played by the intramolecular domain II/IV tether in maintaining unliganded sEGFR in a stable tethered configuration. [DOI] [PubMed] [Google Scholar]
- 18.Di Fiore PP, Pierce JH, Kraus MH, Segatto O, King CR, Aaronson SA. erbB-2 is a potent oncogene when overexpressed in NIH/3T3 cells. Science. 1987;237:178–82. doi: 10.1126/science.2885917. [DOI] [PubMed] [Google Scholar]
- 19.Elleman TC, Domagala T, McKern NM, Nerrie M, Lonnqvist B, et al. Identification of a determinant of epidermal growth factor receptor ligand-binding specificity using a truncated, high-affinity form of the ectodomain. Biochemistry. 2001;40:8930–39. doi: 10.1021/bi010037b. [DOI] [PubMed] [Google Scholar]
- 20.Falls DL. Neuregulins: functions, forms, and signaling strategies. Exp Cell Res. 2003;284:14–30. doi: 10.1016/s0014-4827(02)00102-7. [DOI] [PubMed] [Google Scholar]
- 21.Ferguson KM. Active and inactive conformations of the epidermal growth factor receptor. Biochem Soc Trans. 2004;32:742–45. doi: 10.1042/BST0320742. [DOI] [PubMed] [Google Scholar]
- 22.Ferguson KM, Berger MB, Mendrola JM, Cho HS, Leahy DJ, Lemmon MA. EGF activates its receptor by removing interactions that autoinhibit ectodomain dimerization. Mol Cell. 2003;11:507–17. doi: 10.1016/s1097-2765(03)00047-9. [DOI] [PubMed] [Google Scholar]
- 23.Ferguson KM, Darling PJ, Mohan MJ, Macatee TL, Lemmon MA. Extracellular domains drive homo- but not hetero-dimerization of erbB receptors. EMBO J. 2000;19:4632–43. doi: 10.1093/emboj/19.17.4632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Franklin MC, Carey KD, Vajdos FF, Leahy DJ, de Vos AM, Sliwkowski MX. Insights into ErbB signaling from the structure of the ErbB2-pertuzumab complex. Cancer Cell. 2004;5:317–28. doi: 10.1016/s1535-6108(04)00083-2. [DOI] [PubMed] [Google Scholar]
- 25.Garrett TP, McKern NM, Lou M, Elleman TC, Adams TE, et al. The crystal structure of a truncated ErbB2 ectodomain reveals an active conformation, poised to interact with other ErbB receptors. Mol Cell. 2003;11:495–505. doi: 10.1016/s1097-2765(03)00048-0. [DOI] [PubMed] [Google Scholar]
- 26.Garrett TP, McKern NM, Lou M, Elleman TC, Adams TE, et al. Crystal structure of a truncated epidermal growth factor receptor extracellular domain bound to transforming growth factor alpha. Cell. 2002;110:763–73. doi: 10.1016/s0092-8674(02)00940-6. [DOI] [PubMed] [Google Scholar]
- 27.Gotoh N, Tojo A, Hino M, Yazaki Y, Shibuya M. A highly conserved tyrosine residue at codon 845 within the kinase domain is not required for the transforming activity of human epidermal growth factor receptor. Biochem Biophys Res Commun. 1992;186:768–74. doi: 10.1016/0006-291x(92)90812-y. [DOI] [PubMed] [Google Scholar]
- 28.Greenebaum E, Nicolaides M, Eisinger M, Vogel RH, Weinstein IB. Binding of phorbol dibutyrate and epidermal growth factor to cultured human epidermal cells. J Natl Cancer Inst. 1983;70:435–41. [PubMed] [Google Scholar]
- 29.Gullick WJ, Downward DJ, Marsden JJ, Waterfield MD. A radioimmunoassay for human epidermal growth factor receptor. Anal Biochem. 1984;141:253–61. doi: 10.1016/0003-2697(84)90454-8. [DOI] [PubMed] [Google Scholar]
- 30.Harris RC, Chung E, Coffey RJ. EGF receptor ligands. Exp Cell Res. 2003;284:2–13. doi: 10.1016/s0014-4827(02)00105-2. [DOI] [PubMed] [Google Scholar]
- 31.Hubbard SR. Juxtamembrane autoinhibition in receptor tyrosine kinases. Nat Rev Mol Cell Biol. 2004;5:464–71. doi: 10.1038/nrm1399. [DOI] [PubMed] [Google Scholar]
- 32.Hubbard SR, Till JH. Protein tyrosine kinase structure and function. Annu Rev Biochem. 2000;69:373–98. doi: 10.1146/annurev.biochem.69.1.373. [DOI] [PubMed] [Google Scholar]
- 33.Huse M, Kuriyan J. The conformational plasticity of protein kinases. Cell. 2002;109:275–82. doi: 10.1016/s0092-8674(02)00741-9. [DOI] [PubMed] [Google Scholar]
- 34.Hynes NE, Lane HA. ERBB receptors and cancer: the complexity of targeted inhibitors. Nat Rev Cancer. 2005;5:341–54. doi: 10.1038/nrc1609. [DOI] [PubMed] [Google Scholar]
- 35.Jeffrey PD, Russo AA, Polyak K, Gibbs E, Hurwitz J, et al. Mechanism of CDK activation revealed by the structure of a cyclinA-CDK2 complex. Nature. 1995;376:313–20. doi: 10.1038/376313a0. [DOI] [PubMed] [Google Scholar]
- 36.Kashles O, Szapary D, Bellot F, Ullrich A, Schlessinger J, Schmidt A. Ligand-induced stimulation of epidermal growth factor receptor mutants with altered transmembrane regions. Proc Natl Acad Sci USA. 1988;85:9567–71. doi: 10.1073/pnas.85.24.9567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Katz WS, Lesa GM, Yannoukakos D, Clandinin TR, Schlessinger J, Sternberg PW. A point mutation in the extracellular domain activates LET-23, the Caenorhabditis elegans epidermal growth factor receptor homolog. Mol Cell Bio. 1996;16:529–37. doi: 10.1128/mcb.16.2.529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Klein P, Mattoon D, Lemmon MA, Schlessinger J. A structure-based model for ligand binding and dimerization of EGF receptors. Proc Natl Acad Sci USA. 2004;101:929–34. doi: 10.1073/pnas.0307285101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kwak EL, Sordella R, Bell DW, Godin-Heymann N, Okimoto RA, et al. Irreversible inhibitors of the EGF receptor may circumvent acquired resistance to gefitinib. Proc Natl Acad Sci USA. 2005;102:7665–70. doi: 10.1073/pnas.0502860102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Leahy DJ. Structure and function of the epidermal growth factor (EGF/ErbB) family of receptors. Adv Protein Chem. 2004;68:1–27. doi: 10.1016/S0065-3233(04)68001-6. [DOI] [PubMed] [Google Scholar]
- 41.Lee JC, Vivanco I, Beroukhim R, Huang JH, Feng WL, et al. Epidermal Growth Factor Receptor Activation in Glioblastoma through Novel Missense Mutations in the Extracellular Domain. PLoS Med. 2006;3:e485. doi: 10.1371/journal.pmed.0030485. The first report of activating mutations in the full-length extracellular region of EGFR from tumor samples. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee NY, Hazlett TL, Koland JG. Structure and dynamics of the epidermal growth factor receptor C-terminal phosphorylation domain. Protein Sci. 2006;15:1142–52. doi: 10.1110/ps.052045306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lee NY, Koland JG. Conformational changes accompany phosphorylation of the epidermal growth factor receptor C-terminal domain. Protein Sci. 2005;14:2793–803. doi: 10.1110/ps.051630305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li S, Schmitz KR, Jeffrey PD, Wiltzius JJ, Kussie P, Ferguson KM. Structural basis for inhibition of the epidermal growth factor receptor by cetuximab. Cancer Cell. 2005;7:301–11. doi: 10.1016/j.ccr.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 45.Linggia B, Carpenter G. ErbB receptors: new insights on mechanisms and biology. Trends Cell Biol. 2006;16:649–56. doi: 10.1016/j.tcb.2006.10.008. [DOI] [PubMed] [Google Scholar]
- 46.Liu H, Chen X, Focia PJ, He X. Structural basis for stem cell factor-KIT signaling and activation of class III receptor tyrosine kinases. EMBO J. 2007;26:891–901. doi: 10.1038/sj.emboj.7601545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mattoon D, Klein P, Lemmon MA, Lax I, Schlessinger J. The tethered configuration of the EGF receptor extracellular domain exerts only a limited control of receptor function. Proc Natl Acad Sci USA. 2004;101:923–28. doi: 10.1073/pnas.0307286101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.McLaughlin S, Smith SO, Hayman MJ, Murray D. An electrostatic engine model for autoinhibition and activation of the epidermal growth factor receptor (EGFR/ErbB) family. J Gen Physiol. 2005;126:41–53. doi: 10.1085/jgp.200509274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mendelsohn J, Baselga J. Epidermal growth factor receptor targeting in cancer. Semin Oncol. 2006;33:369–85. doi: 10.1053/j.seminoncol.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 50.Mendrola JM, Berger MB, King MC, Lemmon MA. The single transmembrane domains of ErbB receptors self-associate in cell membranes. J Biol Chem. 2002;277:4704–12. doi: 10.1074/jbc.M108681200. [DOI] [PubMed] [Google Scholar]
- 51.Meric-Bernstam F, Hung MC. Advances in Targeting Human Epidermal Growth Factor Receptor-2 Signaling for Cancer Therapy. Clin Can Res. 2006;12:6326–30. doi: 10.1158/1078-0432.CCR-06-1732. [DOI] [PubMed] [Google Scholar]
- 52.Molina MA, Codony-Servat J, Albanell J, Rojo F, Arribas J, Baselga J. Trastuzumab (herceptin), a humanized anti-Her2 receptor monoclonal antibody, inhibits basal and activated Her2 ectodomain cleavage in breast cancer cells. Cancer Res. 2001;61:4744–9. [PubMed] [Google Scholar]
- 53.Normanno N, De Luca A, Bianco C, Strizzi L, Mancino M, et al. Epidermal growth factor receptor (EGFR) signaling in cancer. Gene. 2006;366:2–16. doi: 10.1016/j.gene.2005.10.018. [DOI] [PubMed] [Google Scholar]
- 54.Ogiso H, Ishitani R, Nureki O, Fukai S, Yamanaka M, et al. Crystal structure of the complex of human epidermal growth factor and receptor extracellular domains. Cell. 2002;110:775–87. doi: 10.1016/s0092-8674(02)00963-7. [DOI] [PubMed] [Google Scholar]
- 55.Ozcan F, Klein P, Lemmon MA, Lax I, Schlessinger J. On the nature of low- and high-affinity EGF receptors on living cells. Proc Natl Acad Sci USA. 2006;103:5735–40. doi: 10.1073/pnas.0601469103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Riese DJ, Gallo RM, Settleman J. Mutational activation of ErbB family receptor tyrosine kinases: insights into mechanisms of signal transduction and tumorigenesis. Bioessays. 2007;29:558–65. doi: 10.1002/bies.20582. An excellent survey of mutations in ErbB receptors and their interpretation in context of models for ErbB receptor activation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schlessinger J. Cell Signaling by Receptor Tyrosine Kinases. Cell. 2000;103:211–25. doi: 10.1016/s0092-8674(00)00114-8. [DOI] [PubMed] [Google Scholar]
- 58.Schlessinger J. Ligand-induced, receptor-mediated dimerization and activation of EGF receptor. Cell. 2002;110:669–72. doi: 10.1016/s0092-8674(02)00966-2. [DOI] [PubMed] [Google Scholar]
- 59.Schlessinger J, Plotnikov AN, Ibrahimi OA, Eliseenkova AV, Yeh BK, et al. Crystal structure of a ternary FGF-FGFR-heparin complex reveals a dual role for heparin in FGFR binding and dimerization. Mol Cell. 2000;6:743–50. doi: 10.1016/s1097-2765(00)00073-3. [DOI] [PubMed] [Google Scholar]
- 60.Schulze WX, Deng L, Mann M. Phosphotyrosine interactome of the ErbB-receptor kinase family. Mol Syst Biol. 2005;1:5–13. doi: 10.1038/msb4100012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sharma SV, Bell DW, Settleman J, Haber DA. Epidermal growth factor receptor mutations in lung cancer. Nat Rev Cancer. 2007;7:169–81. doi: 10.1038/nrc2088. [DOI] [PubMed] [Google Scholar]
- 62.Stamos J, Sliwkowski MX, Eigenbrot C. Structure of the epidermal growth factor receptor kinase domain alone and in complex with a 4-anilinoquinazoline inhibitor. J Biol Chem. 2002;277:46265–72. doi: 10.1074/jbc.M207135200. [DOI] [PubMed] [Google Scholar]
- 63.Ullrich A, Schlessinger J. Signal transduction by receptors with tyrosine kinase activity. Cell. 1990;61:203–12. doi: 10.1016/0092-8674(90)90801-k. [DOI] [PubMed] [Google Scholar]
- 64.Walker F, Orchard SG, Jorissen RN, Hall NE, Zhang HH, et al. CR1/CR2 interactions modulate the functions of the cell surface epidermal growth factor receptor. J Biol Chem. 2004;279:22387–98. doi: 10.1074/jbc.M401244200. [DOI] [PubMed] [Google Scholar]
- 65.Walton GM, Chen WS, Rosenfeld MG, Gill GN. Analysis of deletions of the carboxyl terminus of the epidermal growth factor receptor reveals self-phosphorylation at tyrosine 992 and enhanced in vivo tyrosine phosphorylation of cell substrates. J Biol Chem. 1990;265:1750–54. [PubMed] [Google Scholar]
- 66.Ward CW, Lawrence MC, Streltsov VA, Adams TE, McKern NM. The insulin and EGF receptor structures: new insights into ligand-induced receptor activation. Trends Biochem Sci. 2007;32:129–37. doi: 10.1016/j.tibs.2007.01.001. The similarities and differences between the EGF and insulin receptor structures are well summarized. [DOI] [PubMed] [Google Scholar]
- 67.Waterfield M. Cracking the mild, difficult and fiendish codes within and downstream of the EGFR to link diagnostics and therapeutics. Biochem Soc Trans. 2007;35:1–6. doi: 10.1042/BST0350001. [DOI] [PubMed] [Google Scholar]
- 68.Wood ER, Truesdale AT, McDonald OB, Yuan D, Hassell A, et al. A unique structure for epidermal growth factor receptor bound to GW572016 (Lapatinib): relationships among protein conformation, inhibitor off-rate, and receptor activity in tumor cells. Cancer Res. 2004;64:6652–59. doi: 10.1158/0008-5472.CAN-04-1168. [DOI] [PubMed] [Google Scholar]
- 69.Yarden Y, Sliwkowski MX. Untangling the ErbB signalling network. Nat Rev Mol Cell Biol. 2001;2:127–37. doi: 10.1038/35052073. [DOI] [PubMed] [Google Scholar]
- 70.Yu X, Sharma KD, Takahashi T, Iwamoto R, Mekada E. Ligand-independent dimer formation of epidermal growth factor receptor (EGFR) is a step separable from ligand-induced EGFR signaling. Mol Biol Cell. 2002;13:2547–57. doi: 10.1091/mbc.01-08-0411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yun CH, Boggon TJ, Li Y, Woo MS, Greulich H, et al. Structures of lung cancer-derived EGFR mutants and inhibitor complexes: mechanism of activation and insights into differential inhibitor sensitivity. Cancer Cell. 2007;11:217–27. doi: 10.1016/j.ccr.2006.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yuzawa S, Opatowsky Y, Zhang Z, Mandiyan V, Lax I, Schlessinger J. Structural Basis for Activation of the Receptor Tyrosine Kinase KIT by Stem Cell Factor. Cell. 2007;130:323–34. doi: 10.1016/j.cell.2007.05.055. [DOI] [PubMed] [Google Scholar]
- 73.Zhang H, Berezov A, Wang Q, Zhang G, Drebin J, et al. ErbB receptors: from oncogenes to targeted cancer therapies. J Clin Invest. 2007;117:2051–8. doi: 10.1172/JCI32278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhang X, Gureasko J, Shen K, Cole PA, Kuriyan J. An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell. 2006;125:1137–49. doi: 10.1016/j.cell.2006.05.013. Confirms that the EGFR-TK is autoinhibited and proposes an elegant activation mechanism that involves kinase domain dimerization. Biophysical and biochemical data support this model that is also consistent with characteristics of receptor activation in larger ErbB family. [DOI] [PubMed] [Google Scholar]