

Abstract
There is much interest in the potential use of Cox-2 selective inhibitors in combination with other cancer therapeutics. Malignancies of hematopoietic and non-hematopoietic origin often have increased expression of cyclooxygenase-2 (Cox-2), a key modulator of inflammation. For example, hematological malignancies such as chronic lymphocytic leukemia, chronic myeloid leukemia, Hodgkin’s lymphoma, non-Hodgkin’s lymphoma and multiple myeloma often highly express Cox-2, which correlates with poor patient prognosis. Expression of Cox-2 enhances survival and proliferation of malignant cells, while negatively influencing anti-tumor immunity. Hematological malignancies expressing elevated levels of Cox-2 potentially avoid immune responses by producing factors that enhance angiogenesis and metastases. Cellular immune responses regulated by natural killer cells, cytotoxic T lymphocytes, and T regulatory cells are also influenced by Cox-2 expression. Therefore, Cox-2 selective inhibitors have promising therapeutic potential in patients suffering from certain hematological malignancies.
Introduction
Non-steroidal inflammatory drugs (NSAIDs) and cyclooxygenase-2 (Cox-2) selective inhibitors have shown potential as immunotherapeutics for malignancies. Clinical studies have employed Cox inhibitors in conjunction with chemotherapy or radiotherapy in order to attempt to eliminate metastatic disease. Cox-2 is typically not expressed in resting cells, but can be induced in response to cytokines, various growth factors and inflammatory stimuli [1]. Cox-2 is highly expressed in many types of cancers including breast, prostate, colon and lung [1]. More recently, hematological malignancies have also been shown to constitutively highly express Cox-2 compared to normal cells [2-4]. In many types of cancer, elevated Cox-2 expression correlates with poor response to therapy and decreased survival [5]. Interestingly, Cox-2 deficient mice are resistant to multiple forms of induced cancer [6]. Conversely, over-expression of Cox-2 caused tumorigenesis to occur spontaneously [6, 7]. This indicates that Cox-2 may act as an oncogene and its expression plays an important role in the pathogenesis of cancers of both non-hematological and hematological origin.
Cyclooxygenases are inflammatory regulators, responsible for the conversion of arachidonic acid to prostaglandin H2 (PGH2). The constitutively expressed isoform, Cox-1, maintains homeostatic levels of prostaglandins. Cox-2, which is inducible, mediates inflammation through increased production of prostaglandins, which are important bioactive mediators of inflammation and immunity. Prostaglandin synthases coordinate with cyclooxygenases to produce a spectrum of prostaglandins. Tumors that highly express Cox-2 produce high levels of prostaglandins, including prostaglandin E2 (PGE2) [8-10]. Cox-2 selective inhibitors, as well as NSAIDs that inhibit both Cox-1 and Cox-2, have been shown to improve survival in patients with breast, colon, or prostate cancer [11].
As high Cox-2 expression in solid tumors has become more appreciated, there has been heightened interest in Cox-2 in cells of hematopoietic origin. Our laboratory has shown that while normal resting B cells constitutively express Cox-1, they do not express Cox-2 [12, 13]. However, after activation with polyclonal stimuli that trigger receptors such as CD40, TLR9 and the B cell receptor, human B lymphocytes were found to highly express Cox-2 [12, 13]. Cox-2 was determined to be enzymatically active as evidenced by increased PGE2 production. We and other groups have demonstrated that malignant B cells, namely chronic lymphocytic leukemia (CLL), highly express Cox-2, which confers increased survival [2, 14]. Conversely, Cox-2 selective inhibitors increased apoptosis in B-CLL cells, indicating their potential to act as anti-malignant tumor therapeutic agents [2, 14].
Tumors evade the immune system and sustain their growth in part by controlling the tumor microenvironment. Control features include the release of factors that promote tumorigenesis, angiogenesis, metastasis, inhibit regulatory T cell (Treg) function or antitumor cytotoxicity. It is important to keep in mind that small molecules, such as Cox-2 selective inhibitors, not only target malignant cells, but also cells within the tumor microenvironment. As many types of hematological malignancies express elevated levels of Cox-2, Cox-2 selective inhibitors have promise as therapeutic treatments against these diseases by influencing not only the tumor itself, but the surrounding microenvironment as well (Fig. 1). This review will focus on what is known about Cox-2 expression in hematological malignancies and will discuss the use Cox-2 selective inhibitors in treatments.
Fig. 1. Cox-2 selective inhibitors promote immunity to and reduce survival of hematological malignancies.
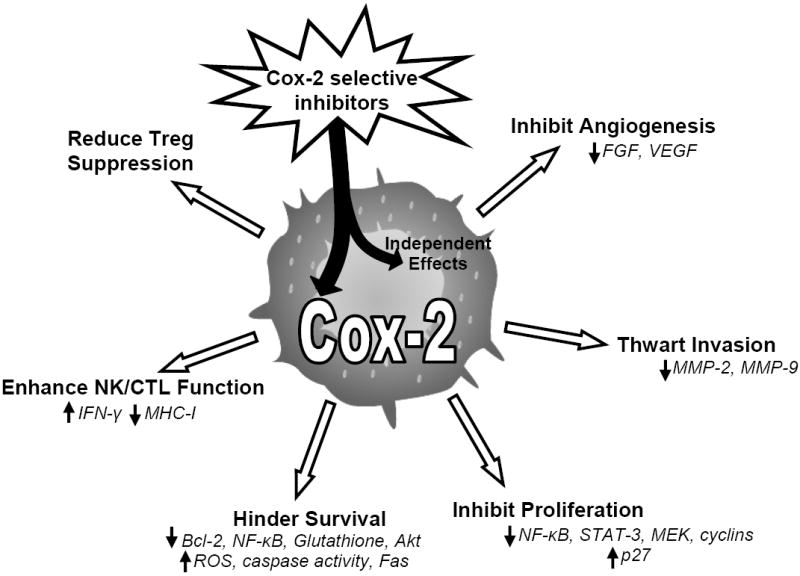
Tumors of hematopoietic origin can express elevated levels of Cox-2, which can enhance cell growth and survival. Cox-2 expression influences growth of new blood vessels surrounding the tumor and promotes metastasis. Activity of regulatory T cells, CTLs, and NK cells, which play important roles in antitumor immunity, are negative influenced by Cox-2 and subsequent prostaglandin production. For this reason, Cox-2 selective inhibitors have the potential to affect many avenues of tumor evasion, acting as promising therapeutic agents for many hematological malignancies. Drug effects independent of Cox-2 activity may also play a role in enhancing anti-tumor immunity, demonstrating that study of new derivative drugs will be important.
Chronic lymphocytic leukemia
Chronic lymphocytic leukemia (CLL) is a malignancy of mature, antigen-experienced B lymphocytes that are highly proliferative [15]. Due to their uncontrolled growth, B-CLL cells have increased resistance to chemotherapeutic drugs. Investigators have hypothesized that Cox-2 enhances tumorigenesis, because its expression promotes resistance to apoptosis [16]. Secchiero et al. analyzed B-CLL samples from patients by cDNA microarray and reported that Cox-2 steady-state mRNA levels were consistently upregulated compared to normal B lymphocytes [14]. In agreement, Cox-2 protein was also elevated, as determined by western blot and immunohistochemistry. Apoptosis was induced in B-CLL cells treated with NS-398 (a small molecule Cox-2 selective inhibitor), but not in normal lymphocytes. This suggests that B-CLL survival is dependent upon Cox-2 expression. The authors also demonstrated the ability of NS-398 to enhance the cytotoxic chemotherapeutic potential of chlorambucil (a commonly used B-CLL therapy). NS-398 significantly increased the B-CLL cytotoxic activity of chlorambucil, effectively increasing cell death by ~50% [14]. Patients with early stage B-CLL were enrolled in a small preliminary clinical study (13 patients) investigating a role for the Cox-2 selective inhibitor celecoxib, at a dose of 400 mg/day [17]. Although, not significant due to the small size of the study, three patients showed a greater than 10% decrease in peripheral CLL load, suggesting that celecoxib could prove useful as a therapeutic agent. Together, these studies suggest that Cox-2 selective inhibitors could enhance chemotherapeutic treatments in patients suffering from B-CLL, however, more clinical investigation is needed.
Our laboratory previously showed that B-CLL cells constitutively express Cox-2 and that its expression enhances their survival [2]. In contrast to naive human B cells, B-CLL cells constitutively express both Cox-1 and Cox-2, which promoted synthesis of PGE2, PGF2α and TXA2. Expression of Cox-2 in B-CLL cells was further elevated by stimulation with CD40 ligand. We observed that B-CLL cells from patients that carried un-mutated variable heavy chains and elevated CD38 expression (indicators of poor prognosis) correlated with higher levels of Cox-2. This provides evidence that Cox-2 could be an important prognostic indicator in B-CLL. Treatment with indomethacin (non-selective Cox-1/Cox-2 inhibitor) or SC-58125 (Cox-2 selective inhibitor) reduced B-CLL viability in a concentration dependent manner. Relatively high doses of indomethacin or SC-58125 were required to enhance cell death, suggesting that a Cox-2-independent mechanism plays a role in inhibiting B-CLL proliferation and survival. Overall, these data demonstrate potential for the therapeutic use of Cox-2 inhibitors in B-CLL patients.
As Cox-2 selective inhibitors may have unwanted secondary effects on the cardiovascular system, a new generation of derivatives has been developed. A third generation derivative of celecoxib, called OSU03012, induced apoptosis in prostate cancer cell lines [18]. Johnson et al. investigated the effect of OSU03012 on B-CLL cells in vitro and found that the celecoxib derivative induced cytotoxicity in B-CLL at low concentrations (<10 μM) [19]. They concluded that OSU03012 mediated cell death by a caspase-independent mechanism in contrast to celecoxib, which is believed to induce apoptosis via a caspase-dependent mechanism [19, 20]. More recently, our laboratory has examined the effects of both celecoxib and OSU03012 on B-CLL, as well as B cell lymphomas [21]. Treatment with either drug significantly attenuated glutathione levels. Glutathione controls damaging reactive oxygen species (ROS) production and low levels of glutathione were associated with decreased malignant B cell viability. These observations suggest that Cox-2 selective inhibitors may reduce B-CLL cell survival by modulating oxidative stress.
Chronic myeloid leukemia
Chronic myeloid leukemia (CML) is characterized by a chromosomal translocation and fusion of the BCR and ABL genes, which results in unrestrained proliferation. CML treatment therapies include allogeneic bone marrow transplants, IFN-γ immunotherapy and chemotherapy which includes the use of imatinib, a drug that specifically inhibits the activity of the BCR/ABL fusion protein [22]. New therapies are needed as imatinib is not efficacious in later stages of the disease and some patients become resistant to the drug due to new mutations in the fusion protein [22].
Giles et al. observed that primary CML cells expressed high levels of Cox-2 protein compared to healthy cells, which correlated with a reduction in patient survival [23]. Cox-2 expression was also found to be elevated in the bone marrow of patients with CML and similarly correlated with reduced survival [23]. These observations have raised interest in studying the effect of Cox inhibitors on CML cells. Indomethacin attenuated proliferation and induced apoptosis in the K562 CML cell line and primary CML cells [24]. Investigators observed that indomethacin-induced cell death occurred through a decline in Bcl-2 levels. Whether or not this was mediated via cyclooxygenases is uncertain, as relatively high doses of drug (400 μM) were used. Celecoxib inhibited proliferation and induced apoptosis in the K562 cell line in a dose-dependent manner, however doses upwards of 50 μM were most effective [25, 26]. The pro-apoptotic and anti-proliferative effects on the CML cell line was determined to be a result of reduced Bcl-2 expression, cytochrome c release, and inhibition of NF-κB [25]. Other highly selective inhibitors of Cox-2, such as DuP-697 or nabumetone, also inhibited proliferation of K562 cells, as well as primary CML cells and induced apoptosis [27, 28]. This further supports a role for Cox-2 expression in CML survival and resistance to apoptosis, indicating that selective inhibitors have promise in myeloid leukemia therapies. It is, however, important to keep in mind that Cox-2 independent effects of inhibitors may also be responsible for anti-proliferative effects.
Hodgkin’s lymphoma
Hodgkin’s lymphoma, also known as Hodgkin’s disease, is one of the most commonly diagnosed lymphomas. It is characterized by the presence of Hodgkin’s lymphoma cells and giant cells called Reed-Sternberg cells (HRS) [29]. A majority (90-95%) of Hodgkin’s patients respond well to current treatments which consist of radiation and chemotherapy, however there can be recurrence [29]. As a result, new therapies are needed for those who respond poorly to treatments and to reduce recurrence of the disease.
Macrophages in patients with Hodgkin’s lymphoma highly produce PGE2, which can suppress cell-mediated immunity [30, 31]. In a small study, Hazar et al. demonstrated that in 70% of Hodgkin’s patients’ HRS cells expressed Cox-2 [32]. Another study showed that HRS cells from nearly half of patients screened expressed Cox-2 and this was associated with increased proliferation and malignant cell survival [3]. Cox-2 expression was positively correlated with increased microvessel density, signifying an increase in angiogenesis. Investigators also observed higher levels of Ki-67 in the nucleus of Cox-2 positive HRS cells, which indicates active proliferation. These studies demonstrate that Cox-2 expression in Hodgkin’s lymphoma cells could play a role in tumor cell proliferation and survival. Patients with Hodgkin’s lymphoma may benefit from Cox-2 inhibitor treatment, as a majority of lymphoma cells expressed Cox-2 and Cox-2 was positively correlated with increased growth of the cancer. In support of this, one group reported a reduced risk of Hodgkin’s lymphoma in individuals taking low dose aspirin [33], which acts as a non-selective Cox inhibitor. Consequently, Cox-2 inhibitors have the potential to enhance cellular immunity, reduce angiogenesis, and inhibit proliferation of malignant cells in patients with Hodgkin’s lymphoma.
Non-Hodgkin’s lymphoma
Non-Hodgkin’s lymphoma is a diverse assemblage of hematological cancers derived from B, T and NK cells. There is an expanding need for new therapies to combat the various types of non-Hodgkin’s lymphomas. Early investigation revealed that a majority of non-Hodgkin’s lymphoma patients, as well as Hodgkin’s lymphoma patients had elevated levels of plasma PGE2, however, at the time no clinical correlation was made [34]. More recent studies have shown that patients whose lymphomas expressed Cox-2 were more likely to have aggressive tumor growth. In one study, 57% of patients expressed Cox-2 in their non-Hodgkin’s lymphoma cells [32]. Cox-2 expression was determined through immunohistochemical analysis and was further correlated with poor response to chemotherapy. However, no correlation was found between Cox-2 and overall survival or tumor grade. Gastric mucosa-associated lymphoid tissue (MALT) lymphoma is a non-Hodgkin’s lymphoma of B cell origin. Li et al. demonstrated that Cox-2 levels were elevated in ~69% of primary MALT lymphomas screened [35]. Cox-2 expression was found to positively correlate with Ki-67 expression, enhanced proliferation, and accumulation of p53 in lymphoma cells. In a broader screening study, including 177 patients diagnosed with various subtypes of non-Hodgkin’s lymphoma, 60% of patients expressed Cox-2 in their tumor cells [3]. Though not quite reaching statistical significance, Cox-2 negative or low expressing lymphomas were found in patients with better survival outcomes. Patients with extranodal NK/T-cell lymphoma, characterized by aggressive cancer in the mid-facial region, showed an inverse correlation with Cox-2 expression and survival [36]. Systemic disease recurrence was not observed in patients negative for Cox-2, while there was a 40% recurrence in patients with lymphomas expressing Cox-2. Five year overall survival was 70% in patients who lacked Cox-2 and 32% for those who were positive for Cox-2. These studies demonstrate that Cox-2 is associated with tumor pathogenesis and that inhibitor therapy may benefit patients suffering from any one of the multiple subtypes of non-Hodgkin’s lymphoma.
Burkitt’s lymphoma, a B cell derived non-Hodgkin’s lymphoma, is particularly aggressive. Wun et al. first described elevated expression of Cox-2 in the Burkitt’s lymphoma cell lines Daudi, Namalwa, Raji and Ramos [37]. Inhibition of Cox-2 with 50 μM celecoxib reduced proliferation and induced apoptosis in more than 85% of Burkitt’s lymphoma cells. Akt (an anti-apoptotic factor) expression was also found to be decreased following inhibition of Cox-2. Kardosh et al. provide supporting evidence that celecoxib and a structural analog, dimethyl-celecoxib (DMC), which lacks Cox-2 inhibitory activity, inhibited proliferation and in vivo tumor growth of Burkitt’s lymphoma [38]. Both drugs inhibited proliferation by negatively influencing expression of cyclin A and B and increased p27Kip1 levels. It is important to note that both groups used relatively high doses of drug (>25 μM) to mediate these effects. This demonstrates that Cox-2 inhibitors can induce apoptosis in Burkitt’s lymphoma. However, at high doses the effects seen may not be dependent upon Cox-2, but rather are off-target effects. Further, DMC which lacks Cox-2 inhibitory activity was also effective in Burkitt’s lymphoma. Kobayashi et al. investigated the effects of etodolac, a Cox-2 selective inhibitor, on Daudi and Raji Burkitt’s lymphoma cell lines (although they state that these cells did not express Cox-2) [39]. Treatment with etodolac induced apoptosis through a mechanism where Bcl-2 levels were decreased. An in vivo model of primary central nervous system lymphoma (PCNSL) utilized intra-cranially injected Raji Burkitt’s lymphoma cells to study the effects of Cox-2 selective inhibitors on tumor growth [40]. Mice administered celecoxib in their food supply displayed increased apoptosis within tumors and a decrease in the levels of Bcl-2, which prevents apoptosis. Celecoxib treatment enhanced the overall survival of mice with PCNSL. Collectively these studies indicate significant promise for the use of certain Cox-2 selective inhibitors against non-Hodgkin’s lymphomas.
Multiple myeloma
Multiple myeloma (MM) is a plasma cell malignancy residing within the bone marrow. In the face of extensive therapies such as radiation, autologous stem cell transplantation and chemotherapy, MM remains an essentially incurable disease with relatively poor patient survival [41]. Therefore, new therapeutics are urgently required.
Cox-2 expression in MM was first reported by Ladetto et al. to be a poor prognostic indicator [4]. Investigators screened Cox-2 expression by western blot in over one hundred tumor samples collected from MM patients undergoing various treatment modalities. They demonstrated that Cox-2 was highly expressed in more than 30% of malignant cells of primary diagnoses of MM and in nearly half of patients who had relapsed. In a later study the same group showed that 54% of MM patients expressed Cox-2, although expression was not correlated with staging of the disease [42]. Investigators observed that patients with MM positive for Cox-2 were disease free on average 18 months, as opposed to 36 months for those who were negative for Cox-2 [4]. Survival was also significantly reduced for patients highly expressing Cox-2 (28 months), compared to those who were negative for Cox-2 (52 months) [4]. These data demonstrate that Cox-2 was an indicator of earlier relapse and poor prognosis for MM patients. Similarly, Cetin et al. observed weak to high Cox-2 expression in 70% of MM cells isolated from patients and that overall patient survival correlated with Cox-2 expression levels [43].
As malignant plasma cells have elevated levels of Cox-2, it is pertinent to ask whether Cox-2 inhibition would have any repercussion on MM. Zhang et al. demonstrated that the MM cell lines PCM6 and RPMI8226, as well as primary MM cells constitutively expressed Cox-2 [44]. The MM cell line U266 however lacked Cox-2 expression. NS-398, a Cox-2 selective inhibitor, inhibited proliferation of PCM6, RPMI8226, and primary MM cells, but not U266 cells, supporting that proliferation was dependent upon Cox-2 expression. NS-398 induced apoptosis at concentrations >25 μM by way of increased cellular Fas expression and caspase 8 and 9 activation [44]. The anti-proliferative and apoptotic effects of dexamethasone or thalidomide, two chemotherapeutic drugs used in the treatment of MM, were enhanced when used together with NS-398. These observations suggest that Cox-2 selective inhibitors might boost the efficacy of chemotherapeutic treatments already in clinical use. In contrast, investigators performing a similar study showed that ARH-77 and IM-9 MM cell lines expressed Cox-2 mRNA, while RPMI8226, U266 were negative for Cox-2 [45]. ARH-77 cells contained enzymatically active Cox-2 and produced PGE2 and PGF2α, whose production was inhibited by Cox-2 selective inhibitors. Proliferation of ARH-77 cells was attenuated by indomethacin, NS-398 or SC-58125, however, only at supra-pharmacologic concentrations (100 μM). In this study, however, proliferation of cells stated to lack Cox-2 (RPMI8226 and U266) was also inhibited by SC-58125, suggesting that the effects of the Cox-2 selective inhibitor were independent of Cox-2. It is possible that the effects of Cox-2 selective inhibitors may only be partially Cox-2 dependent as many in vitro studies use celecoxib at doses higher than necessary to blunt Cox-2 activity. These studies, while raising the issue of drugs acting in a Cox-2 dependent or independent manner, still demonstrate promise for use in patients suffering from MM.
Patients with MM often become resistant to chemotherapeutic drugs and are in need of better therapies [46]. RPMI8226 and chemotherapeutic drug-resistant variant cell lines were used to investigate whether Cox-2 selective inhibitors and derivative drugs could benefit MM patients who had developed resistance to therapy [46]. The authors observed that both celecoxib and DMC, a structural analog lacking Cox-2 inhibitory activity, inhibited proliferation and induced cell death in both drug-sensitive and drug-resistant cell lines. This was achieved through the inhibition of STAT-3, MEK, and NF-kB signaling, as well as through increased caspase-3 activity [46]. Comparatively, DMC induced stronger anti-proliferative and apoptotic effects compared to celecoxib, which demonstrates that DMC may be superior to other Cox-2 selective inhibitors, as it lacks the Cox-2 associated side-effects.
Thrombotic complications can arise in MM patients undergoing chemotherapy [47]. Baz et al. investigated the use of low-dose aspirin therapy in MM patients enduring multiple forms of chemotherapy in order to lower the risk of thrombotic events [47, 48]. Investigators observed increased overall survival of patients receiving low-dose aspirin therapy, along with a significant reduction in thrombotic complications. Another phase II clinical trial investigated the use of celecoxib in combination with thalidomide chemotherapy for treatment of patients suffering from relapsed and refractory MM [49]. Patients enrolled in the trial were administered thalidomide with celecoxib at doses ranging from 200 to 800 mg/day. Investigators compared patients taking less than or equal to 400 mg/day to those taking doses greater than 400 mg/day. Progression-free survival was significantly increased in MM patients who received the larger dose of celecoxib (12.7 vs. 4.6 months). Overall survival was also significantly increased for patients who received the larger dose of celecoxib (29.6 vs. 18.9 months). Unfortunately there were adverse side effects in some patients, including peripheral edema and renal complications. However, no cardiovascular complications were observed. This clinical trial demonstrated that celecoxib has the potential to improve MM therapies, although some patients may develop complications, most likely related to doses higher than necessary to inhibit Cox-2. Therefore, it will be necessary to test the efficacy of lower doses of celecoxib, as well as the use of other Cox inhibitors.
Cox-2 Promotes Progression to Malignancy
Epidemiological studies have provided insight into the role of cyclooxygenases in tumor occurrence and in the progression of healthy cells to malignancy. Celecoxib is approved for use in patients with familial adenomatous polyposis, to reduce progression of this pre-malignant neoplasm to colorectal cancer. Cox-2 selective inhibitors may also reduce the risk of lung, gastric and breast cancers [50-52]. A few epidemiological studies have attempted to determine if NSAIDs with cyclooxygenase inhibitory activity lower the risk of hematological malignancies. There was a reported reduced risk of leukemia with regular aspirin use [53], while decreased risk of MM was not associated with aspirin consumption [54]. Another study found a 60% lower incidence of non-Hodgkin’s lymphoma in females with long-term analgesic use [55]. Moderate aspirin use in women (3 to 9 years of use) reduced the chance of developing non-Hodgkin’s lymphoma by 50% [56]. Conversely, two separate studies have claimed no correlation between the development of non-Hodgkin’s lymphoma and NSAID use [57, 58]. One investigation of women taking Cox-2 selective inhibitors for as many as four years, showed a two-fold higher risk of developing non-Hodgkin’s lymphoma [59]. No significant correlations were found in men taking either aspirin or Cox-2 selective inhibitors. A limitation of this study was that patient data was not separated based upon subtype of non-Hodgkin’s malignancy. Overall, these studies suggest that NSAIDs and Cox inhibitors may prevent the onset of certain hematological malignancies and that larger, more defined studies are necessary to draw accurate conclusions.
Presence of Cox-2 in pre-malignant cells indicates that Cox-2 may promote the progression of healthy cells to a malignant phenotype. Many forms of non-malignant neoplasms express Cox-2 [60]. Plasmacytoma is a plasma cell neoplasm that can progress to MM. In a mouse model of plasmacytoma, indomethacin was administered through drinking water [61]. Continuous treatment of mice with indomethacin reduced plasmacytoma formation and increased survival, indicating that cyclooxygenase activity may be responsible for tumor survival and progression. More recently, a mouse model of lymphoma induced via dioxin (TCDD, an aryl hydrocarbon receptor ligand) treatment, demonstrated that induction of lymphoma was associated with elevated levels of Cox-2 mRNA in lymph nodes [62]. Large numbers of highly proliferating cells and increased Bcl-2 expression was correlated with increased Cox-2 and development of lymphoma. These studies provide evidence that Cox-2 could be associated with progression of normal to malignant cells.
Cox-2 expression in the microenvironment surrounding a potential neoplasm can also promote the progression to malignancy. Chronic inflammation can activate stromal fibroblasts leading to enhanced Cox-2 expression and inflammatory prostaglandins. This can be necessary for promoting progression of other cells to malignancy [8]. Therefore, hematopoeitic neoplasms in contact with stromal cells expressing Cox-2 and secreting prostaglandins could be pushed towards malignancy. The tumor microenvironment plays a role in initial tumorigenesis, but can also be regulated by malignant cells that express Cox-2 to promote further growth and survival.
Cox-2/PGE2 Promotes Angiogenesis and Metastasis
Formation of new blood vessels can be driven by malignant cells in order to support their further proliferation, survival and metastasis. Increased angiogenesis was recently found to play a positive role in the growth of multiple types of hematological malignancies and is correlated with poor disease outcome [63]. Accumulating data show that angiogenesis occurs in the bone marrow of patients with MM [64] and that there is a direct correlation between increased angiogenesis and disease progression [65]. Pro-angiogenic factors include vascular endothelial growth factor (VEGF) and fibroblast growth factor (FGF). Hematological malignancies are associated with high levels of VEGF and FGF which correlates with advancement of disease stage [66, 67]. B-CLL patients were found to have high levels of serum VEGF, which correlated with decreased survival [68].
Interestingly, Cox inhibitors, including celecoxib, rofecoxib, indomethacin, and roscovitine, significantly reduced angiogenesis in an ulcer healing model [69], indicating that Cox-2 may be important for new growth of blood vessels. VEGF and FGF production are also dependent upon Cox-2 expression [70]. Cox-2 activity is known to control fibroblast VEGF and FGF expression, as treatment with Cox-2 inhibitors significantly attenuated growth factor production [71, 72]. PGE2 can modulate VEGF synthesis through the EP3 receptor [73]. Cox-2 expression is positively correlated with high VEGF levels in patients with multiple types of cancer and is associated with increased angiogenesis and metastasis [74, 75]. Therefore, use of Cox-2 selective inhibitors could potentially reduce growth factor levels, thereby reducing angiogenesis. Administration of sulindac or celecoxib to mice with gastric cancer xenografts decreased levels of both FGF and VEGF, effectively reducing microvessel density within the tumor [76]. Celecoxib in conjunction with chemotherapy reduced VEGF in mice implanted with a colon cancer cell line and also increased IFN-gamma, which is important for antitumor immunity [77]. Zhou et al. reported that post-operative gastric cancer patients given celecoxib had reduced VEGF levels in cancerous tissue compared to those that received surgical intervention alone [78]. As previously mentioned, Cox-2 expression in Hodgkin’s lymphoma correlated with a greater degree of angiogenesis [3]. In a phase II clinical trial, Buckstein, et al., gave relapsed or refractory diffuse large B cell lymphoma patients celecoxib with cyclophosphamide chemotherapy in an attempt to reduce angiogenic factors [79]. Indicators of angiogenesis were decreased in responding patients, including a reduction in serum VEGF levels and fewer circulating endothelial cells and endothelial cell precursors. Celecoxib and cyclophosphamide treatment was well tolerated in these patients and 37% responded positively to the combined treatment. These studies provide evidence that Cox-2 selective inhibitors can reduce expression of angiogenic factors, VEGF and FGF, in tumors and therefore, show rationale that these drugs may prove therapeutic in hematological malignancies.
Enhanced angiogenesis creates a means for tumor dissemination to distant tissues. Matrix metalloproteinases (MMP), including MMP-9, are responsible for increased invasion and metastasis of cancerous cells. Investigators have shown that MMP-9 can be expressed by adult T cell leukemia cells and that high levels correlated with increased metastases [80]. B-CLL cells expressed significant amounts of MMP-9, which was associated with increased invasion and lower patient survival rates [81, 82]. One group has even developed a pro-drug that is activated by MMP-9 for MM therapy, as MM cells are known to overexpress MMP-9 [83]. Since levels of MMP-9, a mediator of tumor invasion, are increased in multiple types of hematological malignancies, it will be important to develop drugs that target MMPs.
Cox-2 selective inhibitors have anti-metastatic potential, as there is evidence that MMP-9 production can be dependent upon Cox-2 activity. Macrophages increased MMP-9 production after treatment with PGE2 and expression was mediated through the PGE2 receptor, EP4 [84]. Following transfer of Cox-2 cDNA into a carcinoma cell line, Takaoka et al. reported an up-regulation of MMP-2 and MMP-9 [85]. Upon administration of these malignant cells to nude mice they observed increased tumor aggressiveness and invasion, demonstrating that Cox-2 was responsible for MMP expression and its downstream control of metastasis. The Cox-2 selective inhibitor NS-398 reduced MMP-2 and MMP-9 expression and invasion of prostate cancer cells into a gel matrix [86]. Kwak et al. also showed that celecoxib reduced MMP-2 and MMP-9 in a squamous cell carcinoma and inhibited migration through a collagen matrix [87]. In a mouse model of lung adenocarcinoma both indomethacin and celecoxib significantly reduced MMP-9 activity and migration/invasion of malignant cells [88]. Since Cox-2 activity appears to have some control over MMP expression and invasion of tumor cells, it is hopeful that Cox inhibitors can curb metastasis of hematological malignancies.
Cox-2 Expression and T regulatory cells
Anti-tumor immune responses can be controlled by regulatory T cells (Treg) [89]. Depletion of Tregs is often necessary for vaccines to be effective against solid tumors [90, 91]. In a mouse model of lung cancer, Sharma et al. demonstrated that tumors highly expressing Cox-2 and producing significant amounts of PGE2, augmented Treg numbers and suppressive activity [92]. PGE2 increased expression of FoxP3, a transcription factor expressed exclusively by Treg cells, while inhibition of Cox-2 reduced FoxP3 expression in the spleen. Supporting data shows that Cox-2 inhibition decreased tumor burden and increased survival of mice. This indicates that Tregs may be dependent on Cox-2 to suppress anti-tumor immunity. Yaqub et al. have generated some interesting results in patients with colorectal cancer [93]. Investigators found that Tregs expressed Cox-2 and produced PGE2. Elevated numbers of Tregs were also found in the draining lymph nodes of patients. Depletion of Tregs or treatment of cells in vitro with indomethacin both increased the ability of peripheral blood mononuclear cells to produce inflammatory cytokines such as IFN-γ and TNF-α. This further suggests that FoxP3+ Tregs can control antitumor immunity and that Cox-2 activity is important for Treg function. The use of Cox-2 inhibitors therefore, may act to control the generation and activity of FoxP3+ Treg cells.
Regulatory T cells can control anti-tumor immunity in solid tumors. However, there are conflicting results for their role in the control of hematologic malignancies. Increased levels of FoxP3 in tumor infiltrating cells of diffuse large B cell lymphoma patients were correlated with reduced survival, indicating that Tregs may be suppressing anti-tumor immunity [94]. On the other hand, investigators also discovered that Hodgkin’s lymphoma and follicular lymphoma patients with higher FoxP3 expression had improved prognosis [94]. Lysates from patient MM cells induced FoxP3 in CD4+CD25- cells, which were capable of suppressing anti-tumor immunity [95]. Kelley et al. reported that increased FoxP3 levels in patients with follicular lymphoma correlated with poor disease outcome [96]. Other reports show that in patients diagnosed with T cell lymphoma, diffuse large B cell lymphoma, or follicular lymphoma, increased numbers of Tregs correlated with increased patient survival [97-99]. Although there are conflicting reports for the role of Tregs in hematological malignancies, it appears in some cases that Tregs may negatively influence hematological disease outcome. The dependency of Tregs on Cox-2 expression and activity suggests that treating some of these patients with Cox-2 inhibitors will reduce Treg numbers and suppressive activity in the tumor microenvironment and therefore, allow a stronger anti-tumor immune response. Conversely, in malignancies where Treg presence is protective, Cox-2 selective inhibitors may suppress their function, ultimately relieving their ability to control disease progression. These results provide some insight into how Cox-2 inhibitors may affect Treg generation and function in the tumor microenvironment. However, more disease-specific investigations must be performed to determine how important FoxP3 Treg function is in various types of hematological malignancies before the benefit of Cox inhibitors can be properly evaluated.
Cox-2 Influences Cytotoxic Activity
Cells with cytotoxic capabilities such as NK cells and cytotoxic T lymphocytes (CTLs) are key effectors against hematological malignancies [100]. The cyclooxygenase product, PGE2, has known immunosuppressive effects on NK and CTLs [101-103]. This provides therapeutic potential for the use of Cox-2 selective inhibitors, which could enhance cell-mediated cytotoxic immunity to tumors of hematological origin. PGE2 administered in vivo reduced NK cell and CTL numbers, as well as cytotoxic activity, promoting further tumor growth and metastasis [102, 104]. NK cell cytolytic activity was attenuated against squamous cell carcinoma capable of producing PGE2, and indomethacin restored killing ability [103]. This indicates that the cytotoxic function of the NK cells was dependent upon cyclooxygenase activity. Specht et al. demonstrated that mouse plasmacytoma CTL responses were impaired during late tumor stages [105], typically when high levels of Cox-2 and PGE2 were observed. Indomethacin treatment, which reduced serum PGE2 levels, restored cytotoxic activity and increased IFN-γ production from CD8+ T cells [105]. Indomethacin and celecoxib treated mice had increased cytotoxic activity against tumors, through increased sensitivity of NK cells and increased IFN-γ production [106]. Clearly Cox-2 and PGE2 play a positive role in dampening tumor-specific killing by reducing IFN-γ production, which is important for CTL responses and NK cell activity.
Another possible mechanism by which Cox-2 inhibitors influence NK cell activity is by modulating the balance of inhibitory and activating receptors. NK cells can become activated under conditions where activating receptors gain interactions and inhibitory receptors lose interactions, effectively offsetting the balance of receptor-ligand relationships. Reducing ligand interactions for inhibitory receptors, such as Ly49, which associates with MHC class I molecules, will activate NK cells, causing them to become cytotoxic [107]. Kundu et al. demonstrated that indomethacin or celecoxib treatment both down-regulated MHC class I expression on the surface of cells, offsetting the balance of inhibitory receptors, thereby activating the NK cells to kill tumor cells [108]. By increasing IFN-γ production, reducing MHC class I levels and attenuating inhibitory PGE2, Cox-2 selective inhibitors have the potential to activate more cells with cytolytic activity, enhance anti-tumor immunity and more effectively combat hematological tumors.
Conclusions
New therapies for the treatment of hematological malignancies are needed as many current therapies are insufficient and patients can develop resistance to therapeutics. It can be advantageous to use commonly prescribed or approved drugs as adjuvants with either radiation or chemotherapy. Cox-2 is expressed in many types of hematological malignancies and its high expression is often an indicator of poor prognosis. B-CLL, CML, Hodgkin’s lymphoma, non-Hodgkin’s lymphoma, and MM all highly express Cox-2 and therefore, may be suitable candidates for the use of Cox-2 selective inhibitor therapy. Further, Cox-2 promotes malignancy, angiogenesis, metastasis, influences regulatory T cell function and even affects activity of cells with cytotoxic function, all which regulate the ability of malignant cells to survive. Elevated Cox-2 expression is often correlated with decreased survival of patients with hematological malignancies. Through the use of NSAIDs and Cox-2 selective inhibitors in vitro, many investigators have shown that proliferation and survival of hematological malignancies is dependent upon Cox-2 activity. However, future clinical trials are essential to test the efficacy of Cox-2 selective inhibitors and their derivatives in patients suffering from hematological malignancies
One must also acknowledge that in many cases, Cox-2 selective drugs have anti-proliferative and pro-apoptotic effects at doses higher than necessary to inhibit Cox-2. In addition derivatives of Cox-2 selective inhibitors, lacking Cox-2 inhibitory function, have similar effects, often at lower, less toxic doses. Further study of both Cox-2 selective inhibitors and their associated derivatives will be necessary to determine the efficacy of each in certain hematological malignancies. New drug targets may be developed as a result of determining the specific Cox-2-dependent and independent targets in malignant cells. Regardless, there is growing evidence that Cox-2 expression in hematological malignancies perpetuates their growth and survival and acts a poor prognostic indicator. For this reason, Cox-2 inhibitors and their derivatives have exciting promise as therapeutics for patients afflicted with a hematological malignancy.
Acknowledgments
This work was supported by the National Institutes of Health Grants DE011390, AI071064, HL075432, HL088325 and the Training Program in Oral Sciences T32-DE007202.
References
- 1.Zha S, Yegnasubramanian V, Nelson WG, Isaacs WB, De Marzo AM. Cancer Lett. 2004;215:1–20. doi: 10.1016/j.canlet.2004.06.014. [DOI] [PubMed] [Google Scholar]
- 2.Ryan EP, Pollock SJ, Kaur K, Felgar RE, Bernstein SH, Chiorazzi N, Phipps RP. Clin Immunol. 2006;120:76–90. doi: 10.1016/j.clim.2005.12.012. [DOI] [PubMed] [Google Scholar]
- 3.Ohsawa M, Fukushima H, Ikura Y, Inoue T, Shirai N, Sugama Y, Suekane T, Kitabayashi C, Nakamae H, Hino M, Ueda M. Leuk Lymphoma. 2006;47:1863–71. doi: 10.1080/10428190600685442. [DOI] [PubMed] [Google Scholar]
- 4.Ladetto M, Vallet S, Trojan A, Dell’Aquila M, Monitillo L, Rosato R, Santo L, Drandi D, Bertola A, Falco P, Cavallo F, Ricca I, De Marco F, Mantoan B, Bode-Lesniewska B, Pagliano G, Francese R, Rocci A, Astolfi M, Compagno M, Mariani S, Godio L, Marino L, Ruggeri M, Omede P, Palumbo A, Boccadoro M. Blood. 2005;105:4784–91. doi: 10.1182/blood-2004-11-4201. [DOI] [PubMed] [Google Scholar]
- 5.Subbaramaiah K, Dannenberg AJ. Trends Pharmacol Sci. 2003;24:96–102. doi: 10.1016/S0165-6147(02)00043-3. [DOI] [PubMed] [Google Scholar]
- 6.Muller-Decker K, Furstenberger G. Mol Carcinog. 2007;46:705–10. doi: 10.1002/mc.20326. [DOI] [PubMed] [Google Scholar]
- 7.Liu CH, Chang SH, Narko K, Trifan OC, Wu MT, Smith E, Haudenschild C, Lane TF, Hla T. J Biol Chem. 2001;276:18563–9. doi: 10.1074/jbc.M010787200. [DOI] [PubMed] [Google Scholar]
- 8.Baglole CJ, Ray DM, Bernstein SH, Feldon SE, Smith TJ, Sime PJ, Phipps RP. Immunol Invest. 2006;35:297–325. doi: 10.1080/08820130600754960. [DOI] [PubMed] [Google Scholar]
- 9.Wang D, Dubois RN. Gut. 2006;55:115–22. doi: 10.1136/gut.2004.047100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Harris SG, Padilla J, Koumas L, Ray D, Phipps RP. Trends Immunol. 2002;23:144–50. doi: 10.1016/s1471-4906(01)02154-8. [DOI] [PubMed] [Google Scholar]
- 11.Ulrich CM, Bigler J, Potter JD. Nat Rev Cancer. 2006;6:130–40. doi: 10.1038/nrc1801. [DOI] [PubMed] [Google Scholar]
- 12.Ryan EP, Pollock SJ, Murant TI, Bernstein SH, Felgar RE, Phipps RP. J Immunol. 2005;174:2619–26. doi: 10.4049/jimmunol.174.5.2619. [DOI] [PubMed] [Google Scholar]
- 13.Bernard MP, Phipps RP. Clin Immunol. 2007;125:138–48. doi: 10.1016/j.clim.2007.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Secchiero P, Barbarotto E, Gonelli A, Tiribelli M, Zerbinati C, Celeghini C, Agostinelli C, Pileri SA, Zauli G. Am J Pathol. 2005;167:1599–607. doi: 10.1016/S0002-9440(10)61244-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chiorazzi N, Rai KR, Ferrarini M. N Engl J Med. 2005;352:804–15. doi: 10.1056/NEJMra041720. [DOI] [PubMed] [Google Scholar]
- 16.Tsujii M, DuBois RN. Cell. 1995;83:493–501. doi: 10.1016/0092-8674(95)90127-2. [DOI] [PubMed] [Google Scholar]
- 17.Kara IO, Sahin B. Leuk Lymphoma. 2004;45:1495–6. doi: 10.1080/10428190410001663608. [DOI] [PubMed] [Google Scholar]
- 18.Zhu J, Huang JW, Tseng PH, Yang YT, Fowble J, Shiau CW, Shaw YJ, Kulp SK, Chen CS. Cancer Res. 2004;64:4309–18. doi: 10.1158/0008-5472.CAN-03-4063. [DOI] [PubMed] [Google Scholar]
- 19.Johnson AJ, Smith LL, Zhu J, Heerema NA, Jefferson S, Mone A, Grever M, Chen CS, Byrd JC. Blood. 2005;105:2504–9. doi: 10.1182/blood-2004-05-1957. [DOI] [PubMed] [Google Scholar]
- 20.Hsu AL, Ching TT, Wang DS, Song X, Rangnekar VM, Chen CS. J Biol Chem. 2000;275:11397–403. doi: 10.1074/jbc.275.15.11397. [DOI] [PubMed] [Google Scholar]
- 21.Ryan EP, Bushnell TP, Friedman AE, Rahman I, Phipps RP. Cancer Immunol Immunother. 2008;57:347–58. doi: 10.1007/s00262-007-0374-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fausel C. Am J Health Syst Pharm. 2007;64:S9–15. doi: 10.2146/ajhp070482. [DOI] [PubMed] [Google Scholar]
- 23.Giles FJ, Kantarjian HM, Bekele BN, Cortes JE, Faderl S, Thomas DA, Manshouri T, Rogers A, Keating MJ, Talpaz M, O’Brien S, Albitar M. Br J Haematol. 2002;119:38–45. doi: 10.1046/j.1365-2141.2002.03784.x. [DOI] [PubMed] [Google Scholar]
- 24.Zhang G, Tu C, Zhang G, Zhou G, Zheng W. Leuk Res. 2000;24:385–92. doi: 10.1016/s0145-2126(99)00198-8. [DOI] [PubMed] [Google Scholar]
- 25.Subhashini J, Mahipal SV, Reddanna P. Cancer Lett. 2005;224:31–43. doi: 10.1016/j.canlet.2004.11.002. [DOI] [PubMed] [Google Scholar]
- 26.Zhang GS, Liu DS, Dai CW, Li RJ. Am J Hematol. 2006;81:242–55. doi: 10.1002/ajh.20542. [DOI] [PubMed] [Google Scholar]
- 27.Peng HL, Zhang GS, Liu JH, Gong FJ, Li RJ. Ann Hematol. 2008;87:121–9. doi: 10.1007/s00277-007-0385-4. [DOI] [PubMed] [Google Scholar]
- 28.Vural F, Ozcan MA, Ozsan GH, Ates H, Demirkan F, Piskin O, Undar B. Leuk Lymphoma. 2005;46:753–6. doi: 10.1080/10428190400027860. [DOI] [PubMed] [Google Scholar]
- 29.Kuppers R, Yahalom J, Josting A. Biol Blood Marrow Transplant. 2006;12:66–76. doi: 10.1016/j.bbmt.2005.10.016. [DOI] [PubMed] [Google Scholar]
- 30.Passwell J, Levanon M, Davidsohn J, Ramot B. Clin Exp Immunol. 1983;51:61–8. [PMC free article] [PubMed] [Google Scholar]
- 31.Chemnitz JM, Driesen J, Classen S, Riley JL, Debey S, Beyer M, Popov A, Zander T, Schultze JL. Cancer Res. 2006;66:1114–22. doi: 10.1158/0008-5472.CAN-05-3252. [DOI] [PubMed] [Google Scholar]
- 32.Hazar B, Ergin M, Seyrek E, Erdogan S, Tuncer I, Hakverdi S. Leuk Lymphoma. 2004;45:1395–9. doi: 10.1080/10428190310001654032. [DOI] [PubMed] [Google Scholar]
- 33.Chang ET, Zheng T, Weir EG, Borowitz M, Mann RB, Spiegelman D, Mueller NE. J Natl Cancer Inst. 2004;96:305–15. doi: 10.1093/jnci/djh038. [DOI] [PubMed] [Google Scholar]
- 34.Sebahoun G, Maraninchi D, Carcassonne Y. Acta Haematol. 1985;74:132–6. doi: 10.1159/000206188. [DOI] [PubMed] [Google Scholar]
- 35.Li HL, Sun BZ, Ma FC. World J Gastroenterol. 2004;10:1862–6. doi: 10.3748/wjg.v10.i13.1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shim SJ, Yang WI, Shin E, Koom WS, Kim YB, Cho JH, Suh CO, Kim JH, Kim GE. Int J Radiat Oncol Biol Phys. 2007;67:31–8. doi: 10.1016/j.ijrobp.2006.07.1387. [DOI] [PubMed] [Google Scholar]
- 37.Wun T, McKnight H, Tuscano JM. Leuk Res. 2004;28:179–90. doi: 10.1016/s0145-2126(03)00183-8. [DOI] [PubMed] [Google Scholar]
- 38.Kardosh A, Wang W, Uddin J, Petasis NA, Hofman FM, Chen TC, Schonthal AH. Cancer Biol Ther. 2005;4:571–82. doi: 10.4161/cbt.4.5.1699. [DOI] [PubMed] [Google Scholar]
- 39.Kobayashi M, Nakamura S, Shibata K, Sahara N, Shigeno K, Shinjo K, Naito K, Ohnishi K. Eur J Haematol. 2005;75:212–20. doi: 10.1111/j.1600-0609.2005.00498.x. [DOI] [PubMed] [Google Scholar]
- 40.Wang W, Kardosh A, Su YS, Schonthal AH, Chen TC. Neurosurg Focus. 2006;21:E14. doi: 10.3171/foc.2006.21.5.15. [DOI] [PubMed] [Google Scholar]
- 41.Merchionne F, Perosa F, Dammacco F. Clin Exp Med. 2007;7:83–97. doi: 10.1007/s10238-007-0134-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Trojan A, Tinguely M, Vallet S, Seifert B, Jenni B, Zippelius A, Witzens-Harig M, Mechtersheimer G, Ho A, Goldschmidt H, Jager D, Boccadoro M, Ladetto M. Swiss Med Wkly. 2006;136:400–3. doi: 10.4414/smw.2006.11467. [DOI] [PubMed] [Google Scholar]
- 43.Cetin M, Buyukberber S, Demir M, Sari I, Sari I, Deniz K, Eser B, Altuntas F, Camci C, Ozturk A, Turgut B, Vural O, Unal A. Am J Hematol. 2005;80:169–73. doi: 10.1002/ajh.20460. [DOI] [PubMed] [Google Scholar]
- 44.Zhang M, Abe Y, Matsushima T, Nishimura J, Nawata H, Muta K. Leuk Lymphoma. 2005;46:425–33. doi: 10.1080/10428190400015691. [DOI] [PubMed] [Google Scholar]
- 45.Ding J, Tsuboi K, Hoshikawa H, Goto R, Mori N, Katsukawa M, Hiraki E, Yamamoto S, Abe M, Ueda N. Mol Carcinog. 2006;45:250–9. doi: 10.1002/mc.20175. [DOI] [PubMed] [Google Scholar]
- 46.Kardosh A, Soriano N, Liu YT, Uddin J, Petasis NA, Hofman FM, Chen TC, Schonthal AH. Blood. 2005;106:4330–8. doi: 10.1182/blood-2005-07-2819. [DOI] [PubMed] [Google Scholar]
- 47.Baz R, Li L, Kottke-Marchant K, Srkalovic G, McGowan B, Yiannaki E, Karam MA, Faiman B, Jawde RA, Andresen S, Zeldis J, Hussein MA. Mayo Clin Proc. 2005;80:1568–74. doi: 10.4065/80.12.1568. [DOI] [PubMed] [Google Scholar]
- 48.Baz R, Hussein MA. Br J Haematol. 2006;134:349–50. doi: 10.1111/j.1365-2141.2006.06195.x. [DOI] [PubMed] [Google Scholar]
- 49.Prince HM, Mileshkin L, Roberts A, Ganju V, Underhill C, Catalano J, Bell R, Seymour JF, Westerman D, Simmons PJ, Lillie K, Milner AD, Iulio JD, Zeldis JB, Ramsay R. Clin Cancer Res. 2005;11:5504–14. doi: 10.1158/1078-0432.CCR-05-0213. [DOI] [PubMed] [Google Scholar]
- 50.Harris RE, Beebe-Donk J, Alshafie GA. Int J Biol Sci. 2007;3:328–34. doi: 10.7150/ijbs.3.328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Harris RE, Beebe-Donk J, Alshafie GA. BMC Cancer. 2006;6:27. doi: 10.1186/1471-2407-6-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Futagami S, Suzuki K, Hiratsuka T, Shindo T, Hamamoto T, Ueki N, Kusunoki M, Miyake K, Gudis K, Tsukui T, Sakamoto C. Inflammopharmacology. 2007;15:1–4. doi: 10.1007/s10787-006-1541-5. [DOI] [PubMed] [Google Scholar]
- 53.Kasum CM, Blair CK, Folsom AR, Ross JA. Cancer Epidemiol Biomarkers Prev. 2003;12:534–7. [PubMed] [Google Scholar]
- 54.Moysich KB, Bonner MR, Beehler GP, Marshall JR, Menezes RJ, Baker JA, Weiss JR, Chanan-Khan A. Leuk Res. 2007;31:547–51. doi: 10.1016/j.leukres.2006.07.027. [DOI] [PubMed] [Google Scholar]
- 55.Beiderbeck AB, Holly EA, Sturkenboom MC, Coebergh JW, Stricker BH, Leufkens HG. Am J Epidemiol. 2003;157:510–6. doi: 10.1093/aje/kwg004. [DOI] [PubMed] [Google Scholar]
- 56.Flick ED, Chan KA, Bracci PM, Holly EA. Am J Epidemiol. 2006;164:497–504. doi: 10.1093/aje/kwj223. [DOI] [PubMed] [Google Scholar]
- 57.Bernatsky S, Lee JL, Rahme E. Rheumatology (Oxford) 2007;46:690–4. doi: 10.1093/rheumatology/kel396. [DOI] [PubMed] [Google Scholar]
- 58.Zhang Y, Coogan PF, Palmer JR, Strom BL, Rosenberg L. Cancer Detect Prev. 2006;30:99–101. doi: 10.1016/j.cdp.2005.12.002. [DOI] [PubMed] [Google Scholar]
- 59.Cerhan JR, Anderson KE, Janney CA, Vachon CM, Witzig TE, Habermann TM. Int J Cancer. 2003;106:784–8. doi: 10.1002/ijc.11311. [DOI] [PubMed] [Google Scholar]
- 60.Harris RE. Subcell Biochem. 2007;42:93–126. doi: 10.1007/1-4020-5688-5_4. [DOI] [PubMed] [Google Scholar]
- 61.Potter M, Wax J, Jones GM. Blood. 1997;90:260–9. [PubMed] [Google Scholar]
- 62.Vogel CF, Li W, Sciullo E, Newman J, Hammock B, Reader JR, Tuscano J, Matsumura F. Am J Pathol. 2007;171:1538–48. doi: 10.2353/ajpath.2007.070406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ribatti D, Scavelli C, Roccaro AM, Crivellato E, Nico B, Vacca A. Stem Cells Dev. 2004;13:484–95. doi: 10.1089/scd.2004.13.484. [DOI] [PubMed] [Google Scholar]
- 64.Rajkumar SV, Leong T, Roche PC, Fonseca R, Dispenzieri A, Lacy MQ, Lust JA, Witzig TE, Kyle RA, Gertz MA, Greipp PR. Clin Cancer Res. 2000;6:3111–6. [PubMed] [Google Scholar]
- 65.Pruneri G, Ponzoni M, Ferreri AJ, Decarli N, Tresoldi M, Raggi F, Baldessari C, Freschi M, Baldini L, Goldaniga M, Neri A, Carboni N, Bertolini F, Viale G. Br J Haematol. 2002;118:817–20. doi: 10.1046/j.1365-2141.2002.03654.x. [DOI] [PubMed] [Google Scholar]
- 66.Ribatti D, Vacca A, Rusnati M, Presta M. Cytokine Growth Factor Rev. 2007;18:327–34. doi: 10.1016/j.cytogfr.2007.04.011. [DOI] [PubMed] [Google Scholar]
- 67.Jakob C, Sterz J, Zavrski I, Heider U, Kleeberg L, Fleissner C, Kaiser M, Sezer O. Eur J Cancer. 2006;42:1581–90. doi: 10.1016/j.ejca.2006.02.017. [DOI] [PubMed] [Google Scholar]
- 68.Molica S, Vitelli G, Levato D, Gandolfo GM, Liso V. Br J Haematol. 1999;107:605–10. doi: 10.1046/j.1365-2141.1999.01752.x. [DOI] [PubMed] [Google Scholar]
- 69.Jones MK, Wang H, Peskar BM, Levin E, Itani RM, Sarfeh IJ, Tarnawski AS. Nat Med. 1999;5:1418–23. doi: 10.1038/70995. [DOI] [PubMed] [Google Scholar]
- 70.Gately S, Li WW. Semin Oncol. 2004;31:2–11. doi: 10.1053/j.seminoncol.2004.03.040. [DOI] [PubMed] [Google Scholar]
- 71.Williams CS, Tsujii M, Reese J, Dey SK, DuBois RN. J Clin Invest. 2000;105:1589–94. doi: 10.1172/JCI9621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Baguma-Nibasheka M, Barclay C, Li AW, Geldenhuys L, Porter GA, Blay J, Casson AG, Murphy PR. Mol Carcinog. 2007;46:971–80. doi: 10.1002/mc.20339. [DOI] [PubMed] [Google Scholar]
- 73.Amano H, Hayashi I, Endo H, Kitasato H, Yamashina S, Maruyama T, Kobayashi M, Satoh K, Narita M, Sugimoto Y, Murata T, Yoshimura H, Narumiya S, Majima M. J Exp Med. 2003;197:221–32. doi: 10.1084/jem.20021408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhang XH, Huang DP, Guo GL, Chen GR, Zhang HX, Wan L, Chen SY. BMC Cancer. 2008;8:4. doi: 10.1186/1471-2407-8-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tang H, Wang J, Bai F, Zhai H, Gao J, Hong L, Xie H, Zhang F, Lan M, Yao W, Liu J, Wu K, Fan D. Cancer Invest. 2008;26:60–7. doi: 10.1080/07357900701519279. [DOI] [PubMed] [Google Scholar]
- 76.Wu YL, Fu SL, Zhang YP, Qiao MM, Chen Y. Biomed Pharmacother. 2005;59(Suppl 2):S289–92. doi: 10.1016/s0753-3322(05)80048-4. [DOI] [PubMed] [Google Scholar]
- 77.Irie T, Tsujii M, Tsuji S, Yoshio T, Ishii S, Shinzaki S, Egawa S, Kakiuchi Y, Nishida T, Yasumaru M, Iijima H, Murata H, Takehara T, Kawano S, Hayashi N. Int J Cancer. 2007;121:878–83. doi: 10.1002/ijc.22720. [DOI] [PubMed] [Google Scholar]
- 78.Zhou Y, Ran J, Tang C, Wu J, Honghua L, Xingwen L, Ning C, Qiao L. Cancer Biol Ther. 2007;6:269–75. doi: 10.4161/cbt.6.2.3629. [DOI] [PubMed] [Google Scholar]
- 79.Buckstein R, Kerbel RS, Shaked Y, Nayar R, Foden C, Turner R, Lee CR, Taylor D, Zhang L, Man S, Baruchel S, Stempak D, Bertolini F, Crump M. Clin Cancer Res. 2006;12:5190–8. doi: 10.1158/1078-0432.CCR-06-0474. [DOI] [PubMed] [Google Scholar]
- 80.Hayashibara T, Yamada Y, Onimaru Y, Tsutsumi C, Nakayama S, Mori N, Miyanishi T, Kamihira S, Tomonaga M, Maita T. Br J Haematol. 2002;116:94–102. doi: 10.1046/j.1365-2141.2002.03255.x. [DOI] [PubMed] [Google Scholar]
- 81.Bauvois B, Dumont J, Mathiot C, Kolb JP. Leukemia. 2002;16:791–8. doi: 10.1038/sj.leu.2402472. [DOI] [PubMed] [Google Scholar]
- 82.Kamiguti AS, Lee ES, Till KJ, Harris RJ, Glenn MA, Lin K, Chen HJ, Zuzel M, Cawley JC. Br J Haematol. 2004;125:128–40. doi: 10.1111/j.1365-2141.2004.04877.x. [DOI] [PubMed] [Google Scholar]
- 83.Van Valckenborgh E, Mincher D, Di Salvo A, Van Riet I, Young L, Van Camp B, Vanderkerken K. Leukemia. 2005;19:1628–33. doi: 10.1038/sj.leu.2403866. [DOI] [PubMed] [Google Scholar]
- 84.Pavlovic S, Du B, Sakamoto K, Khan KM, Natarajan C, Breyer RM, Dannenberg AJ, Falcone DJ. J Biol Chem. 2006;281:3321–8. doi: 10.1074/jbc.M506846200. [DOI] [PubMed] [Google Scholar]
- 85.Takaoka K, Kishimoto H, Segawa E, Hashitani S, Zushi Y, Noguchi K, Sakurai K, Urade M. Int J Oncol. 2006;29:1095–101. [PubMed] [Google Scholar]
- 86.Attiga FA, Fernandez PM, Weeraratna AT, Manyak MJ, Patierno SR. Cancer Res. 2000;60:4629–37. [PubMed] [Google Scholar]
- 87.Kwak YE, Jeon NK, Kim J, Lee EJ. Ann N Y Acad Sci. 2007;1095:99–112. doi: 10.1196/annals.1397.014. [DOI] [PubMed] [Google Scholar]
- 88.Peluffo GD, Stillitani I, Rodriguez VA, Diament MJ, Klein SM. Int J Cancer. 2004;110:825–30. doi: 10.1002/ijc.20226. [DOI] [PubMed] [Google Scholar]
- 89.Wang HY, Wang RF. Curr Opin Immunol. 2007;19:217–23. doi: 10.1016/j.coi.2007.02.004. [DOI] [PubMed] [Google Scholar]
- 90.Dannull J, Su Z, Rizzieri D, Yang BK, Coleman D, Yancey D, Zhang A, Dahm P, Chao N, Gilboa E, Vieweg J. J Clin Invest. 2005;115:3623–33. doi: 10.1172/JCI25947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Grauer OM, Nierkens S, Bennink E, Toonen LW, Boon L, Wesseling P, Sutmuller RP, Adema GJ. Int J Cancer. 2007;121:95–105. doi: 10.1002/ijc.22607. [DOI] [PubMed] [Google Scholar]
- 92.Sharma S, Yang SC, Zhu L, Reckamp K, Gardner B, Baratelli F, Huang M, Batra RK, Dubinett SM. Cancer Res. 2005;65:5211–20. doi: 10.1158/0008-5472.CAN-05-0141. [DOI] [PubMed] [Google Scholar]
- 93.Yaqub S, Henjum K, Mahic M, Jahnsen FL, Aandahl EM, Bjornbeth BA, Tasken K. Cancer Immunol Immunother. 2007 doi: 10.1007/s00262-007-0417-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Tzankov A, Meier C, Hirschmann P, Went P, Pileri SA, Dirnhofer S. Haematologica. 2008;93:193–200. doi: 10.3324/haematol.11702. [DOI] [PubMed] [Google Scholar]
- 95.Han S, Wang B, Cotter MJ, Yang LJ, Zucali J, Moreb JS, Chang LJ. Mol Ther. 2008;16:269–79. doi: 10.1038/sj.mt.6300369. [DOI] [PubMed] [Google Scholar]
- 96.Kelley T, Beck R, Absi A, Jin T, Pohlman B, Hsi E. Leuk Lymphoma. 2007;48:2403–11. doi: 10.1080/10428190701665954. [DOI] [PubMed] [Google Scholar]
- 97.Gjerdrum LM, Woetmann A, Odum N, Burton CM, Rossen K, Skovgaard GL, Ryder LP, Ralfkiaer E. Leukemia. 2007;21:2512–8. doi: 10.1038/sj.leu.2404913. [DOI] [PubMed] [Google Scholar]
- 98.Carreras J, Lopez-Guillermo A, Fox BC, Colomo L, Martinez A, Roncador G, Montserrat E, Campo E, Banham AH. Blood. 2006;108:2957–64. doi: 10.1182/blood-2006-04-018218. [DOI] [PubMed] [Google Scholar]
- 99.Lee NR, Song EK, Jang KY, Choi HN, Moon WS, Kwon K, Lee JH, Yim CY, Kwak JY. Leuk Lymphoma. 2008;49:247–56. doi: 10.1080/10428190701824536. [DOI] [PubMed] [Google Scholar]
- 100.Costello RT, Fauriat C, Sivori S, Marcenaro E, Olive D. Trends Immunol. 2004;25:328–33. doi: 10.1016/j.it.2004.04.005. [DOI] [PubMed] [Google Scholar]
- 101.Ting CC, Hargrove ME. J Immunol. 1983;131:1734–41. [PubMed] [Google Scholar]
- 102.Yakar I, Melamed R, Shakhar G, Shakhar K, Rosenne E, Abudarham N, Page GG, Ben-Eliyahu S. Ann Surg Oncol. 2003;10:469–79. doi: 10.1245/aso.2003.08.017. [DOI] [PubMed] [Google Scholar]
- 103.Kim CD, Sung MW, Lee SJ, Heo DS, Yoon SJ, Kim KH. Anticancer Res. 1999;19:455–9. [PubMed] [Google Scholar]
- 104.Okuno K, Jinnai H, Lee YS, Nakamura K, Hirohata T, Shigeoka H, Yasutomi M. Surg Today. 1995;25:954–8. doi: 10.1007/BF00312380. [DOI] [PubMed] [Google Scholar]
- 105.Specht C, Bexten S, Kolsch E, Pauels HG. Int J Cancer. 2001;91:705–12. doi: 10.1002/1097-0215(200002)9999:9999<::aid-ijc1066>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 106.Okuno T, Takagaki Y, Pluznik DH, Djeu JY. J Immunol. 1986;136:4652–8. [PubMed] [Google Scholar]
- 107.Malarkannan S. Semin Immunol. 2006;18:186–92. doi: 10.1016/j.smim.2006.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Kundu N, Walser TC, Ma X, Fulton AM. Cancer Immunol Immunother. 2005;54:981–7. doi: 10.1007/s00262-005-0669-2. [DOI] [PMC free article] [PubMed] [Google Scholar]