

Abstract
Interpenetrating polymer networks (IPNs) have been the subject of extensive study since their advent in the 1960s. Hydrogel IPN systems have garnered significant attention in the last two decades due to their usefulness in biomedical applications. Of particular interest are the mechanical enhancements observed in “double network” IPN systems which exhibit nonlinear increases in fracture properties despite being composed of otherwise weak polymers. We have built upon pioneering work in this field as well as in responsive IPN systems to develop an IPN system based on end-linked poly-(ethylene glycol) (PEG) and loosely crosslinked poly(acrylic acid) (PAA) with hydrogen bond-reinforced strain-hardening behavior in water and high initial Young’s moduli under physiologic buffer conditions through osmotically induced pre-stress. Uniaxial tensile tests and equilibrium swelling measurements were used to study PEG/PAA IPN hydrogels having second networks prepared with varying crosslinking and photoinitiator content, pH, solids content, and comonomers. Studies involving the addition of non-ionic comonomers and neutralization of the second network showed that template polymerization appears to be important in the formation of mechanically enhanced IPNs.
Keywords: hydrogels, double network, interpenetrating polymer networks
INTRODUCTION
Interpenetrating polymer networks (IPNs) are unique “alloys” of crosslinked polymers1 in which at least one network is synthesized and/or crosslinked in the presence of the other. IPNs are often created for the purpose of conferring key attributes of one of the components while maintaining the critical attributes of another. In some cases, entirely new, and sometimes surprising properties are exhibited by the IPN that are not observed in either of the two single networks alone.
From a synthetic standpoint, IPNs come in two varieties: (a) a sequential IPN in which one network is swollen and polymerized in the presence of the other, and (b) simultaneous IPN, in which both of the network precursors are synthesized at the same time by independent, non-interfering routes. When one of the two polymers is linear (uncrosslinked), a semi-IPN results and when both of the polymers are identical, a homo-IPN results.2 An important parameter that must be considered in the fabrication of IPNs is the mutual miscibility of the interpenetrating polymers; in general, polymers do not mix well with each other, resulting in the phase separation of the resultant blend.3,4 However, because crosslinking provides a way to “enforce” mixing between two otherwise immiscible materials, there are virtually endless combinations of polymers worthy of exploration as IPNs for a variety of applications.
This paper begins with an overview of pioneering work in the development and characterization of IPNs followed by a focused review of developments in hydrogel-based IPN systems, and ends with a presentation of recent work done in our laboratory to characterize the mechanical properties of PEG/PAA IPNs.
BACKGROUND
Pioneering work in IPN science
While the science of IPNs began with the work of Millar in 1960, the first publication on the subject came through a patent by Aylsworth in 1914.5 Since then, IPNs have been the subject of extensive study by investigators looking into the synthesis, morphology, properties, and applications of these materials. Pioneering work by Frisch et al.6 and Sperling and Friedman7 established that IPNs are characterized by topological interlocking of two or more crosslinked chains by two independent polymerization reactions.5 IPNs came to be defined as entanglements of polymer networks that are ideally held together only by permanent topological interactions.1,5,6 The inter-network entanglements are permanent by virtue of the fact that the two networks are themselves chemically crosslinked, and thus cannot be “pulled apart” from each other without fracturing one or both of the networks.
IPN formation has been shown by numerous investigators to be a useful way to enhance the performance of hydrogels. Hydrogels are water-swollen polymers that are useful in a variety of biomedical device applications due to their biocompatibility, high water content, and in some cases, responsiveness to stimuli. These properties make hydrogels excellent materials for drug delivery vehicles and tissue scaffolds, devices that are dependent on the controlled transport of small molecules. IPN hydrogels are typically produced by first synthesizing a hydrophilic polymer network, swelling it in a second aqueous monomer solution, and polymerizing the latter to form a water-swollen mesh of two different polymers. IPNs can also be prepared by synthesizing simultaneously two polymers by different polymerization techniques such as condensation and free radical polymerization. Semi-IPNs are usually synthesized by polymerizing a monomer or prepolymer around pre-existing polymer chains; alternatively, they are made by diffusing polymer chains into a pre-formed polymer network.
The hydrophilicity/hydrophobicity and stimuli-responsiveness of IPNs can be controlled by proper selection of the two polymers and by varying their composition. This is a useful strategy that has been employed by a number of investigators to create novel IPN systems for commercial and biomedical applications that are responsive to stimuli in a controlled and predicable fashion. A review of such systems is described in the ensuing section.
Responsive IPN hydrogel systems
Numerous IPN hydrogels have been described in the literature that utilize different combinations of neutral and ionic components. Ilmain et al. described an IPN system based on poly(acrylamide) and poly(acrylic acid) (PAAm/PAA) that exhibited large volume transitions driven by hydrogen bonding between the two networks.8 Other notable examples of IPNs described in the literature include but are not limited to the poly(vinyl alcohol)/PAA system developed by Gudeman and Peppas,9 the chitosan/PAA systems developed by Lee et al.,10 and the temperature-responsive poly(vinyl alcohol)/poly(N-isopropylacrylamide) semi-IPNs of Zhang et al.11
One of the more extensively studied systems—and the subject of this paper—is an IPN of poly(ethylene glycol) (PEG) and PAA. Like the previously mentioned example of the PAAm/PAA network of Ilmain et al.,8 this system belongs to a subclass of IPNs known for their capacity for inter-network “complexation” via non-covalent interactions. Investigators have previously applied the concept of complex formation to IPNs and hydrogels. Nishi et al.12–14 created IPNs of equimolar quantities of crosslinked PEG and crosslinked PAA. By characterizing the swelling and mechanical properties of these IPNs, they demonstrated that the IPN undergoes elongation and contraction as the pH of the swelling liquid was changed. They had the idea of using this system as an ultrafiltration membrane, demonstrating that the molecular weight (MW) cut-off of the membrane could be changed reversibly by altering the pH of the system.12–14
Pioneering work by Osada and Takeuchi15 based on interpolymer complexation of PEG/PMAA and PEG/PAA systems showed that large, pH-dependent dilations and contractions took place when a PMAA membrane was treated with PEG. With PEG treatment, PMAA was able to lift and lower large loads and also serve as environmentally sensitive membranes. They showed that the permeability of the PMAA membranes to hemoglobin and albumin could be altered by reversible treatment with PEG chains. They also showed that longer PEG chains (≥2000 Da) were necessary for sufficient contraction of the membrane and formation of pores for permeation. Osada has written a review on this concept of chemical valves and converting chemical energy into mechanical work.12 A thermodynamic theory of this complexation behavior was proposed by Khokhlov and Kramarenko,16 a theory that was based primarily on Flory–Huggins theory but also included a free energy contribution due to complex formation.
Mechanically enhanced IPN hydrogel systems
Double networks
Unfortunately, the mechanical fragility of most hydrogels poses a formidable obstacle to their application as substitutes for load-bearing tissues, which have exceptional mechanical properties despite high water content (>75%). Although a number of strategies—such as high crosslinking density, fiber-reinforcement, and copolymerization—can be used to improve the strength of hydrogels, the enhancement afforded by these often involves some compromise in the desired characteristics of the original material such as hydrophilicity, transparency, or permeability.17 “Double networks” are a unique type of IPN first described by Gong et al. in 2003, which exhibits a nonlinear enhancement of mechanical strength. These mechanically enhanced IPN hydrogels, containing 60–90% water, exhibit high resistance to wear and high fracture strength, up to 17 times that of its component networks. They have been heralded as the beginning of a new frontier for tissue replacement, particularly for load-bearing structures.17
In Gong et al.’s most extensively studied DN system, a densely crosslinked, ionizable first network consisting of poly(2-acrylamido-2-methylpropanesulfonic acid) (PAMPS) is interpenetrated with a flexible, loosely crosslinked, neutral second network consisting of PAAm. In recent work, they have characterized this material through compression tests,17 dynamic light scattering,18 and tear-testing to determine fracture energy,19,20 and have shed light on the role of the neutral PAAm second network in retarding crack propagation within “voids” present in the PAMPS first network.20
Mechanically enhanced PEG/PAA IPNs
In recent work, we have reported results on an IPN system that is, in effect, the “inverse” of the double networks prepared by Gong et al.: a neutral crosslinked polymer in the first network and an ionizable crosslinked polymer in the second.21–24 Specifically, the first network is composed of end-linked PEG-diacrylate macromonomers with defined average MW. The second network is, in contrast, a loosely crosslinked, ionizable network of PAA. PEG-DA and PAA networks are both relatively fragile materials, so neither would be expected to make the sole contribution to mechanical enhancement. As described in the previous section, interpenetrating networks of PEG and PAA have been explored as vehicles for drug delivery and as chemomechanical systems due to their reversible, pH-dependent swelling behavior.14,25,26 The two polymers form complexes through hydrogen bonds between the ether groups on PEG-DA and the carboxyl groups on PAA.14 This interpolymer hydrogen bonding enhances their mutual miscibility in aqueous solution, which, in turn, yields optically clear polymer blends.
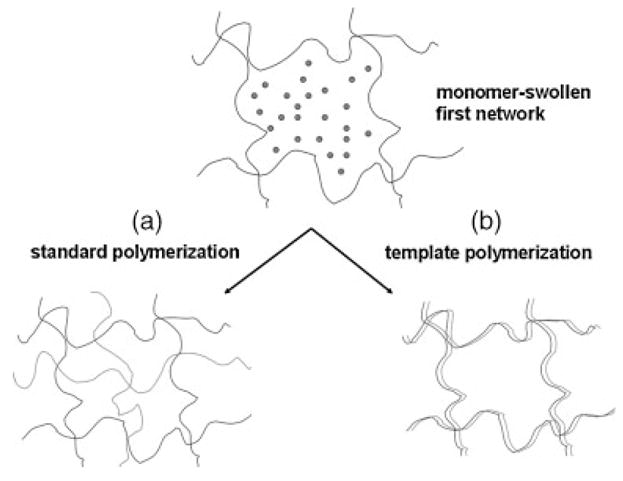
We have shown that strain-hardening behavior in the PEG/PAA system is particularly pronounced under conditions where hydrogen bonding is possible (low pH and pure water). On the other hand, the IPNs strain harden and, in turn, become “pre-stressed” with high values for initial Young’s moduli when swollen in buffers of physiologic pH and salt concentrations (i.e. phosphate buffered saline (PBS)). The strain hardening under these conditions is the result of the constraining effect that the tightly crosslinked, neutral PEG network has on the swelling of the ionized PAA network. This constraining effect leads to additional physical crosslinks between the two networks and manifests as an increase in the initial Young’s modulus of the IPN. In our previous work,27 we paid specific attention to the effect of the PEG macromonomer MW and the PAA solids content as strength-determining parameters in the IPN. In this paper, we present recent investigations focused on various aspects of the second network. A number of investigators have shown that acrylic acid monomers undergo “template polymerization” when formed in the presence of PEG macromonomers,28 a process illustrated in Fig. 1.
Figure 1.

Schematic representation of chain template polymerization “type II” (pick-up mechanism), adapted from Polowinski, Progress in Polymer Science 2002.28 During polymerization, (a) the monomers (gray circles) begin to polymerize by a free-radical chain reaction. Once the chain has reached a certain length, it (b) anneals to the existing template chain (black line), while the chain reaction continues (arrow). The result is a second polymer that is polymerized along the length of the template chain.
In this figure, an oligomer of a forming PAA chain complexes with an adjacent, pre-existing PEG chain. Polymerization of the PAA chain continues as a templated process, with each successive AA monomer simultaneously adding to the PAA polymer and hydrogen bonding to an ethylene oxide repeat unit. Extensive work has been carried out on the subject of template polymerization, which is related to the well-known example of template-based synthetic reactions that take place in biological systems such as DNA and RNA. The organization of the monomer unit can be accomplished by a number of means such as hydrogen bonds, electrostatic forces, or by covalent bonds. The criteria for classifying a process as a templated process is not clear-cut, and usually is the result of a “template effect” in which either the kinetics, MW, or tacticity of a polymer is altered by the presence of another.28
We hypothesized that template polymerization may be an important processing element in the formation of mechanically strong PEG/PAA IPNs. To investigate this, we systematically varied the second network through (1) changes in the concentration of AA at the time of polymerization, and (2) concentrations of the crosslinker and photoinitiator, (3) concentration of non-ionic comonomers, and (4) neutralization of the AA monomer solution prior to polymerization.
MATERIALS AND METHODS
Chemicals
PEG (MW 3400, 4600, and 8000), acryloyl chloride, acrylic acid (AA), 2-hydroxy-2-methyl-propiophenone, 2-hydroxyethylacrylate (HEA), tetrahydrofuran, and triethylene glycol dimethacrylate (TEGDMA) were purchased from Sigma-Aldrich Chemical Company (Milwaukee, WI). PBS was purchased from Invitrogen (Carlsbad, CA).
IPN synthesis
IPN hydrogels were synthesized by a (two-step) sequential network formation technique based on UV-initiated free radical polymerization. The first hydrogel network was prepared from PEG-DA synthesized by the reaction between PEG and acryloyl chloride. Briefly, PEG-DA was synthesized by first dissolving the PEG macromonomer in anhydrous tetrahydrofuran at 50°C. Next, a molar excess of acryloyl chloride was added to the PEG solution and allowed to react for 5 hr under a nitrogen atmosphere. The solution was allowed to cool to room temperature and then was recrystallized at 4°C. The PEG-DA was then purified by a second recrystallization step in fresh anhydrous tetrahydrofuran. Purified PEG-DA was then dissolved in deionized water. The photoinitiator, 2-hydroxy-2-methyl propiophenone (Sigma) was added to the PEG-DA solution at a concentration of 1% by volume with respect to the macromonomer. This precursor solution was cast within a Teflon spacer (250 μm thick, 2 cm inner diameter) positioned on a glass plate (1.0 mm thick), sandwiched with a second glass plate, and then reacted under a 75 W xenon ultraviolet (UV) light source (Oriel Instruments, Mountain View, CA) with a broad range of wavelengths (200–2500 nm) for 10 min. Upon exposure to UV light, the precursor solution underwent free radical-induced gelation and became insoluble in water.
To incorporate the second network, the PEG-DA hydrogel was removed from the mold and immersed in an acrylic acid monomer solution containing 1% v/v photoinitiator solution (with respect to the monomer) and 1% v/v TEGDMA (with respect to the monomer) as a cross-linking agent for 24 hr. For some samples, we varied the photoinitiator and crosslinker concentrations to 0.1% and 10% by volume with respect to the AA monomer. The swollen gel was exposed to the same UV source for 5 min and a second network, PAA, was polymerized and crosslinked in the presence of the PEG-DA network to form the interpenetrated polymer network structure. The resultant IPN was washed extensively for a minimum of 3 days in deionized water or PBS to remove any unreacted components and to reach equilibrium swelling in deionized water.
PEG/P(AA-co-HEA) IPNs were prepared by mixing HEA monomers into the second network precursor solution while keeping the photoinitiator and crosslinker concentration constant. After washing, the water content and mechanical strength were investigated. IPNs with neutralized PAA networks were synthesized by the addition of NaOH to AA solutions until the pH reached 5.5. (From this point onward, the PEG-DA networks will be referred to simply as PEG networks, and will be distinguished on the basis of the MW of the PEG macromonomer. Single networks and IPNs will be designated PEG(X) and PEG(X)/PAA, respectively, where X is the MW of the PEG network, while the copolymer will be designated as PEG-co-PAA) and the IPN copolymer containing HEA monomers will be called PEG/P(AA-co-HEA). In the case where the polymer content in the second network is changed, the IPN will be designated as PEG(X)/PAA[Y], where Y is the volume fraction of aqueous acrylic acid at the time of polymerization. The hydrogels were then equilibrated in 500 ml of either water or PBS and stored at room temperature until further use.
Swelling studies
The water content of the hydrogels was evaluated in terms of the swelling ratio. Swelling studies were carried out by comparing the swollen and dry weights of the hydrogels in either deionized water or electrolyte buffers. The swollen gels were removed from the molds, patted dry, and weighed at regular intervals until equilibrium was reached. The percentage water content (WC) was calculated from the swollen and dry weight of the hydrogel:
| (1) |
where Ws and Wd are the weights of swollen and dry hydrogel, respectively.
The equilibrium swelling ratio (q) of the hydrogels was determined by the equation:
| (2) |
Uniaxial tensile testing
The hydrogels were allowed to reach equilibrium in deionized water or pH buffers overnight prior to testing. The specimens were cut into dumbbell shapes that conform to ASTM D638-V standards using a dumbbell cutter (Ontario Die, Canada). Dumbbell-shaped specimens (gauge length of 9.5 mm, width 3.18 mm, and thickness 250–750 μm) of the hydrogels were tested using an Instron 5844 materials testing apparatus equipped with a 10 N load cell and BioPuls bath, submersible pneumatic grips, and standard video extensometer system (Instron Corp., Norwood, MA). Thicknesses were measured by gently clamping a digital caliper (VWR International, Westchester, PA) over the samples sandwiched between 150 μm thick glass coverslips in order not to compress or damage the hydrogels.
The lack of observed stress relaxation after stretching samples to a fixed strain indicated that the stress values obtained were equilibrium values. For the uniaxial tensile tests, the crosshead speed was set at 15 mm/min for all samples unless otherwise specified. Tensile tests were conducted under various pH and ionic strength conditions by submersing the gripped samples in 3.0 L of the swelling liquid of interest. Load and extension measurements that take into account the thinning of the samples by extension were collected automatically by the load cell and video extensometer, respectively, and were used to obtain the true stress and strain values for each sample. Each material was tested in triplicate, and average values for Young’s moduli, true stress-at-break (σtrue) and true strain-at-break (εbreak) of PEG, PAA, and PEG/PAA IPNs were calculated.
RESULTS
Variation of PAA solids content in the second network
Effect of PAA content on IPN swelling in pure water
PEG(4600) single networks were prepared and imbibed with varying concentrations of AA in the second network in the presence of the photoinitiator and crosslinker. IPNs based on these AA-swollen PEG networks were then formed by UV-initiated polymerization. The IPNs were then removed from their molds, immersed in deionized water, and allowed to reach equilibrium. The volume of the IPNs relative to the PEG single networks were then measured and compared. The results are plotted in Fig. 2.
Figure 2.
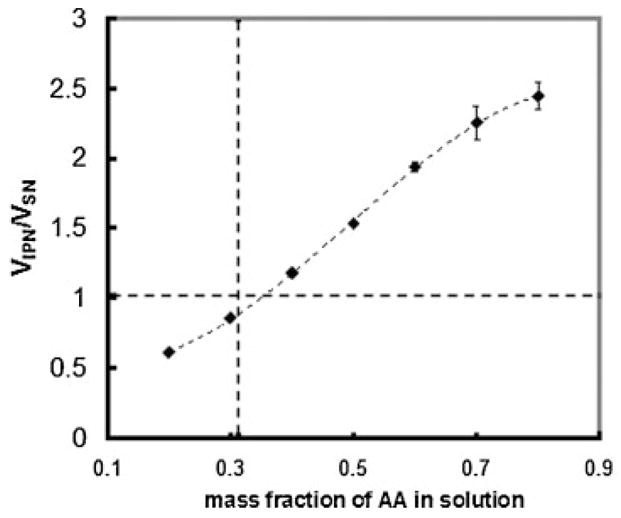
Effect of the mass fraction of AA monomer in the second network precursor solution on the volume change in the resultant IPN. The vertical dotted line indicates the point of equimolar amounts of AA and ethylene glycol (EG) monomer units in the IPN, while the horizontal dotted line indicates where the PEG network and the PEG/PAA IPN have the same volume.
Figure 2 shows that the volume of the IPN is increased with higher amounts of AA monomer in the second network. This is consistent with the understanding that PAA absorbs water, and therefore increased PAA content in the IPN should lead to increased water absorption. Of note, however, is the fact that the IPN deswells relative to the PEG single network when the AA/EG monomer ratio is less than unity, and swells relative to the PEG network when AA is in excess to EG monomers.
Effect of PAA content on IPN mechanical properties in pure water
The same PEG/PAA IPNs of varying AA monomer content were tested by unaxial tensile measurements. The results are shown in Fig. 3. In this figure, both the fracture stress and Young’s modulus are plotted as functions of AA mass fraction at the time of polymerization. Young’s modulus exhibited a modest monotonic increase as the AA concentration increased. In contrast, the fracture stress exhibited a dramatic increase in magnitude when the AA/EG ratio was increased beyond unity. As the AA monomer concentration increased, however, the fracture stress exhibited a monotonic decline.
Figure 3.
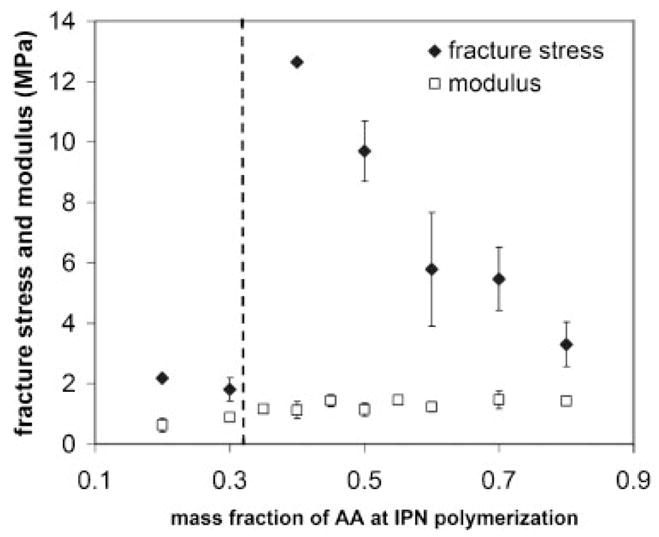
Dependence of the fracture stress and Young’s modulus of the PEG/PAA IPN on the mass fraction of AA in the IPN.
Effect of photoinitiator and crosslinker concentrations
A series of PEG(3.4k)/PAA IPNs were synthesized with varying concentrations of photoinitiator and crosslinking agents and swollen to equilibrium in either deionized water or PBS. The results are shown in Table 1.
Table 1.
Effect of crosslinker and photoinitiator concentrations on the mechanical properties of PEG/PAA hydrogels*
| Sample | Swelling Medium | Crosslinker (vol.%) | Photoinitiator (vol.%) | E0 (MPa) | σmax | εmax |
|---|---|---|---|---|---|---|
| 1 | dH2O | 0.1 | 1.0 | 1.0±30.1 | 3.9±31.2 | 0.63±30.07 |
| 2 | dH2O | 1.0 | 1.0 | 1.4±30.3 | 9.7±30.4 | 0.91±30.53 |
| 3 | dH2O | 10.0 | 1.0 | 0.8±30.0 | 5.6±33.7 | 1.07±30.41 |
| 4 | PBS, pH 7.4, I = 0.15 | 0.1 | 1.0 | 5.3±30.3 | 0.5±30.2 | 0.12±30.03 |
| 5 | PBS, pH 7.4, I = 0.15 | 1.0 | 1.0 | 8.4±30.5 | 4.3±30.8 | 0.44±30.03 |
| 6 | PBS, pH 7.4, I = 0.15 | 10.0 | 1.0 | 6.9±30.7 | 1.1±30.2 | 0.20±30.03 |
| 7 | dH2O | 1.0 | 0.1 | 0.9±30.2 | 5.2±32.4 | 1.11±30.08 |
| 8 | dH2O | 1.0 | 1.0 | 1.4±30.3 | 9.7±30.4 | 0.91±30.53 |
| 9 | dH2O | 1.0 | 10.0 | 0.9±30.0 | 4.2±30.0 | 0.67±30.00 |
| 10 | PBS, pH 7.4, I = 0.15 | 1.0 | 0.1 | 8.8±30.0 | 3.3±31.1 | 0.35±30.10 |
| 11 | PBS, pH 7.4, I = 0.15 | 1.0 | 1.0 | 8.4±30.5 | 4.3±30.8 | 0.44±30.03 |
| 12 | PBS, pH 7.4, I = 0.15 | 1.0 | 10.0 | 7.8±30.2 | 1.9±30.6 | 0.34±30.06 |
Samples 2 & 8 and 5 & 11 provided repeated data to aid visual comparison between experimental conditions.
In Samples 1–3, the water-swollen IPNs were prepared in which concentration of the crosslinking agent (TEGDMA) was varied between 0.1, 1.0, and 10% by volume with respect to the AA monomer while keeping the photoinitiator concentration constant. The most significant result of these experiments was the fact that in deionized water the IPN with the intermediate degree of crosslinking in the second network has substantially higher stress-at-break and strain-at-break values than its counterparts with higher and lower amounts of crosslinker. This is not the case, however, when these same samples are swollen in PBS (Samples 4–6) as well where the PAA network is fully ionized. Under these conditions, the IPN with intermediate concentration (1%) also exhibits the highest stress-at-break and strain at break values. IPNs with varying photoinitiator concentration in both water (Samples 7–9) and PBS (Samples 10–12) did not appear to show any significant mechanical enhancements relative to each other. Of note, Sample 12, which was prepared from the highest concentration of photoinitiator, exhibited the lowest stress-at-break value at the same crosslinker concentration.
Effect of P(AA-co-HEA) copolymerization on IPN mechanical properties in pure water
The AA monomers in the second network were mixed in three different concentrations relative to the HEA monomers: 10:1, 3:1, and 1:1. Uniaxial tensile testing experiments (Fig. 4) of the hydrogels swollen in deionized water showed that the PEG/P(AA-co-HEA) IPNs with the highest ratio of AA/HEA in the second network exhibited enhanced mechanical strength in terms of its stress-at-break and strain-at-break, while the IPNs with higher relative HEA content exhibited reduced strain-hardening behavior.
Figure 4.
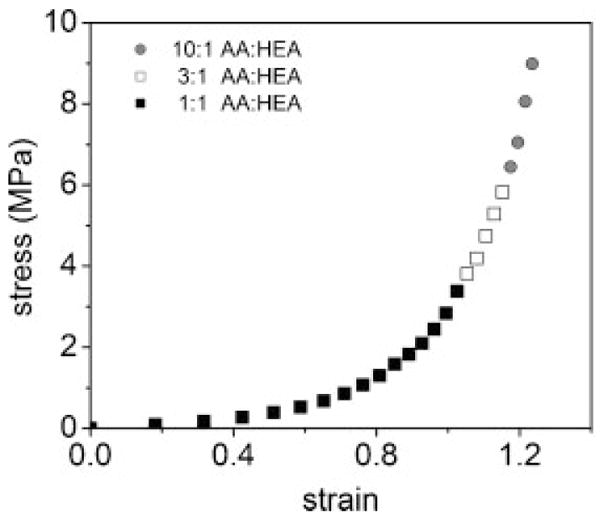
Effect of varying copolymerizing HEA into the second network of PEG(8000)/PAA IPNs on stress–strain behavior.
This result demonstrates that complexation between PEG and the PAA networks is necessary for mechanical enhancement of the fracture properties in this system. It is also strongly suggestive that a templated (rather than a randomly polymerized) second network is important for this enhancement, since PHEA is not known to complex with or template-polymerize on PEG.
Effect of AA neutralization on IPN mechanical properties
In this set of experiments, PEG networks were immersed in AA solutions (containing photoinitiator and crosslinker) that were partially neutralized to pH 5.5 by titration with sodium hydroxide. The monomer-swollen PEG networks were then exposed to UV light to form a partially neutralized PAA network within the PEG network. These “pre-neutralized” PEG/PAA IPNs were then washed in PBS and subjected to unaxial tensile tests.
Figure 5 shows that neutralizing the AA solution prior to polymerization and then forming the second network leads to an IPN with the same elastic modulus, but dramatically reduced fracture strength. The stress-at-break is reduced from nearly 4 MPa—in the case of the IPNs prepared under acidic conditions and then neutralized in PBS buffer—to roughly 0.5 MPa.
Figure 5.
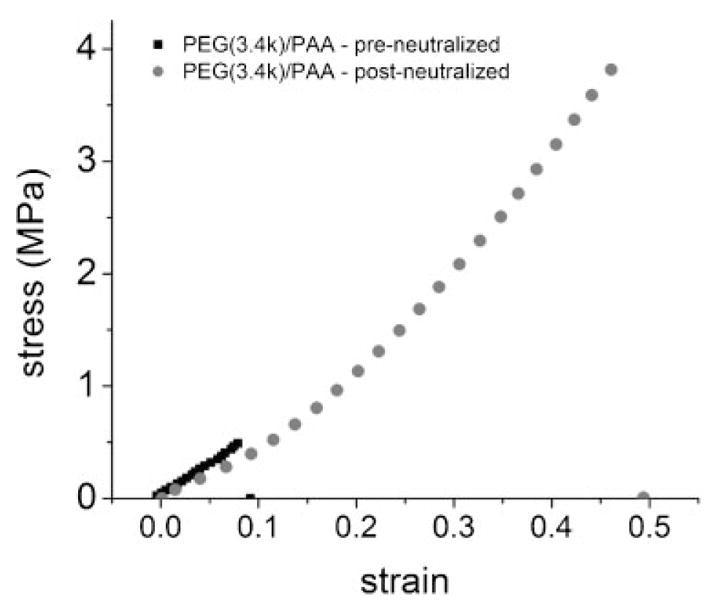
Effect of neutralizing the AA monomer solution prior to polymerization (“pre-neutralized”) on the stress–strain behavior of a PEG(3.4k)/PAA IPN (black) compared to a PEG(3.4k)/PAA IPN prepared under acidic conditions and neutralized after polymerization (“post-neutralized”).
To further investigate the effect of neutralizing the AA monomer solution prior to polymerization, PAA alone was prepared from AA monomer solutions at varying pH and constant salt concentration. Immediately after polymerization, the PAA hydrogels were placed in PBS. The swelling ratio was then measured from these samples. Figure 6 shows that that by increasing the pH of the monomer solution above the pKa of PAA (pH 4.7) leads to a significant increase in its swelling. In contrast, the swelling ratio of PAA when polymerized in pure water and then equilibrated in PBS is only 22.0.27
Figure 6.
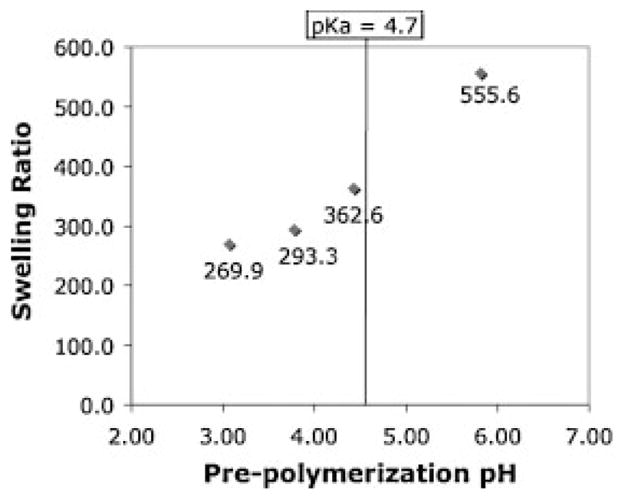
Effect of the pre-polymerization pH on the swelling of PAA at a constant ionic strength of 2.05.
Effect of unaxial extension rate on IPN mechanical properties
To determine whether this large decrease in fracture strength is due to rapid swelling and in turn, the production of defects in the PEG network, the post-neutralized PEG(3.4k)/PAA IPN was tested under different extension rates. The extension rate was varied from 15 mm/min to 500 mm/min, and the results are shown in Fig. 7. The results show that there is essentially no difference in the stress–strain behavior of the IPN due to changes in the extension rate. Both the modulus and the fracture strength are within experimental error and show no dependence on the extension rate.
Figure 7.
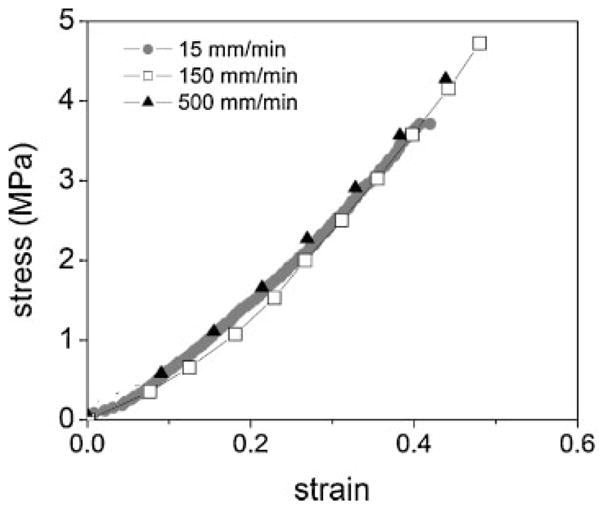
Effect of the extension rate on the stress–strain behavior of post-neutralized PEG(3.4k)/PAA IPNs.
DISCUSSION
In our previous work, we demonstrated that hydrogen bonding is an important factor for increasing the fracture strength and strain of PEG/PAA IPNs. We also showed that hydrogen bonding was not required for mechanical enhancement by virtue of the fact that the PEG/PAA system exhibits large enhancements in Young’s modulus at physiologic pH and ionic strength conditions in which hydrogen bonding does not occur. In this paper, we explored the notion that hydrogen bonding may still be an important determinant of the IPNs mechanical properties as it relates to template polymerization.
Hydrogen bonding takes place when an electron-deficient hydrogen interacts with a region of high electron density. Hydrogen bonding between macromolecules is observed in many biological systems. There are certain prerequisites for complexation to take place in synthetic systems. For instance, there typically is a critical chain length below which complexation does not occur, so therefore polymer chains must be sufficiently long. The complexes also tend to take place in aqueous solution under a small range of pH, ionic strength, and solvent composition. The complex stability is impacted by temperature, polymer concentration, structure, and hydrophobic stabilization.29 PEG is well known to form interpolymer complexes with both PAA and its more hydrophobic counterpart, poly(methacrylic acid) (PMAA). PEG is a proton acceptor while PAA and PMAA are polycarboxylic acids that are proton donors. The protons on the carboxylic acid groups form hydrogen bonds with the ether oxygens on the PEG chains. Since complexation in these systems occurs between hydrophilic acid groups of the PAA or PMAA with the oxygens on the PEG chain, the resultant interpolymer complex is more hydrophobic than the individual polymers alone.29 The complexation between PEG and PAA has been verified experimentally by viscometry and potentiometry experiments,13,14,30–32 excimer fluorescence,33,34 photon correlation spectroscopy,35 solution micro-calorimetry,36 differential scanning calorimetry, Fourier-transform infrared spectroscopy,37 and other fluorescence-based techniques.31,38
The results shown in Fig. 2—in which the IPNs at or below equimolar amounts of AA and EG monomers are contracted relative to the original PEG network—are consistent with this scenario. If PAA is template polymerized along PEG, the annealing between the two complexing polymers would lead to a contracted/compacted IPN structure relative to the PEG network. The template-polymerized IPN illustrated in the right-hand scheme of Fig. 8 would be expected to be contracted relative to the monomer-swollen first (single) network due to compact association between the networks.
Figure 8.
Schematic illustrating the differences in IPN structure after (a) standard polymerization and (b) template polymerization of a second network in the presence of a preexisting first network, at equimolar concentrations of acrylic acid and ethylene glycol monomer units.
With regard to the PAA network itself, it has been proposed that it undergoes “self-template” polymerization. Chapiro et al.39–42 studied acrylic acid bulk polymerization in different solvents and under different temperatures. Based on IR spectrometry data, they found that in solutions of acrylic acid, two types of associations between AA monomers exist: cyclic dimers and linear oligomers. While in hydrocarbon and chlorinated derivatives, cyclic dimers predominate; in water, methanol, or dioxane, linear oligomers are stabilized. This led them to the assumption that PAA formed during the course of polymerization with itself as a template. Linear oligomers of AA monomers associate with the PAA template and polymerization proceeds as a template process.
However, the situation becomes more complex when equimolarity is exceeded. The primary PEG/PAA system explored in this paper contains a significant excess of PAA relative to the PEG network by dry weight. This means that there are substantially more AA monomer units than there are EG monomer units. In this case, polymerization of PAA within PEG can proceed in one of two ways (Fig. 9). Standard polymerization would proceed as illustrated in the pathway shown in Fig. 9(A), where the second network is formed in a random configuration within the PEG meshwork. In another scenario (Fig. 9(B)), some of the PAA can be formed by template polymerization (an equimolar portion relative to PEG) while the excess is polymerized in a random distribution within the aqueous solution. The excess PAA may self-template to some degree (as double stranded PAA chains) since it has been shown that AA-monomers dimerize, but these self-templated chains would still adopt a random coil configuration within the free volume of the PEG network.
Figure 9.

Schematic illustrating the differences in IPN structure after (A) standard polymerization and (B) template polymerization of a second network in the presence of a preexisting first network, at excess concentrations of acrylic acid relative to ethylene glycol monomer units.
The phenomenon shown in Fig. 2 is supportive of the mechanism shown in Figs 8(b) and 9(B). In the equimolar scenario, PAA template polymerizes with PEG, and the IPN contracts. In the excess AA scenario, the equimolar portion of the PAA template polymerizes, while the rest of the AA polymerizes randomly. While the double stranded PEG/PAA chains would like to contract, the thermodynamically favorable thing for the excess PAA chains to do is to absorb water, leading to swelling of the IPN. At the same time, the excess PAA chains also enhance the fracture properties of the IPN (Fig. 3). In this case, the mechanism proposed by Gong et al. may be applicable, in which the second network macromolecular chains act as molecular “crackstoppers;” in addition, strain-dependent interpolymer hydrogen bonding reinforces the network, enhancing the fracture strength of the IPN. On the other hand, the observed decrease in fracture strength at high mass fractions of AA monomers (Fig. 3) is not intuitive. This may be due to two factors: (1) introduction of more network inhomogeneities in the second network due to the large amount of solid matter at the time of polymerization, or, more likely, (2) the greater degree of swelling exhibited by the IPNs based on greater AA mass fractions. The polymer-diluting effect of swelling in the higher AA-content systems would lead to a reduction in fracture strength of these IPNs.
The complexation between PEG and PAA was disrupted in two ways. First, the second network was copolymerized with increasing amounts of HEA monomers to create a P(AA-co-HEA) copolymer. This was done because PHEA does not complex or template polymerize with PEG, and therefore a P(AA-co-HEA) copolymer would be expected to have reduced complexation behavior with PEG, and in turn, reduced fracture strength. Figure 10 illustrates how a copolymer with one interacting species and one noninteracting species (like AA and HEA, respectively) would be expected to polymerize in the presence of a complementary polymer (like PEG). The final two-stranded structure would be the “partially templated” or “partially annealed” complex shown in Fig. 10.
Figure 10.

Investigators have proposed that template copolymerization with complexing and non-complexing species would proceed by the mechanism shown here, where the first network polymer (black line) associates strongly with monomer A (gray circles) and does not interact with monomer B (white circles), yielding a partially templated structure after polymerization. In the case of a PEG/P(AA-co-HEA) IPN, the PEG is the black line, AA monomers are the pink circles, and HEA monomers are the white circles (adapted from Polowinski, Progress in Polymer Science, 200228).
If inter-network complexation between PEG and PAA were indeed important for its strength enhancement, one would expect that a P(AA-co-HEA) network would have compromised fracture properties. As shown in Fig. 4, this is indeed the case. While the IPNs with mostly AA in the second network (a 10:1 AA to HEA ratio) had enhanced mechanical strength, those with a higher ratio of AA to HEA (3:1 and 1:1) show progressively reduced fracture stress and fracture strain. This is supportive of the concept we have proposed that hydrogen bonding between the PAA and PEG networks serves to reinforce the network and prevent cracks from propagating, leading to an IPN with increased resistance to fracture at high strains. This is consistent with the work of Krupers et al.32 who first showed in solutions that the nature of the AA-based copolymer is important for complexation with PEG.
To further explore the impact of template polymerization, we neutralized the AA monomer solution prior to polymerization. This causes two things to happen. First, it changes the chain configuration of the polymerized PAA chain because the negative charges on the carboxylic acid will cause it to assume a self-avoiding walk and “stretch out.” This manifests itself as rapid swelling of the network after placement in solution. Second, it will eliminate any template polymerization of PAA on PEG because charged PAA chains cannot hydrogen bond to PEG ether oxygens. The result of this experiment, shown in Fig. 5, demonstrates that pre-neutralization of AA has a devastating effect on the IPN’s fracture properties. The pre-neutralized IPN has an eightfold reduced stress-at-break relative to the IPNs prepared in the standard fashion under acidic conditions. Of note, the moduli of the two materials have nearly the same value (~4 MPa). This indicates that the number of entanglements between the two networks is roughly the same. However, the reduction of fracture strength may be due to a number of factors. It is possible that polymerization of pre-neutralized AA leads to more fragmented, shorter PAA chains. If this is the case, then the fracture strength would be expected to be lower, since Gong et al. have already demonstrated that long chains in the second network are necessary for increased fracture properties. The lack of template polymerization may be directly related to this; having a PEG template on which PAA may polymerize could result in longer PAA chains. The tacticity of the chain may also be affected—since templated PAA is syndiotactic and non-templated PAA is atactic—but the impact of this on the mechanical properties of the IPN is unproven.
Another possibility is that the pre-neutralized PAA swells so much and so rapidly after polymerization that as it becomes more entangled with the PEG network, it also over-strains and creates defects in it. Figure 6 demonstrates how neutralization of AA monomers impacts the equilibrium swelling of the crosslinked polymer. The osmotic pressure built up by this rapid swelling (which takes place in a matter of minutes) may be one reason that the pre-neutralized IPN is so susceptible to crack formation and failure. In the case of the IPN prepared under acidic conditions and then neutralized, however, the PAA network swells slowly as it gradually loses protons and becomes charged. This slow process of swelling, which takes place over a period of hours, could allow the two networks to mutually accommodate each other through chain rearrangement, also leading to a highly entangled network, but one that is relatively defect free.
On the other hand, if the swelling rate after polymerization were indeed the cause of the reduced fracture strength, then the post-neutralized PEG/PAA IPN network should also exhibit some strain dependence to its mechanical properties. The data shown in Fig. 7 demonstrates that this is not the case. Regardless of whether the IPN is extended at 15, 150, or 500 mm/min, its stress–strain profile remains effectively unchanged. The lack of dependence of the stress–strain curves on the extension rate casts doubt on the idea that the rapid swelling after polymerizing a pre-neutralized PAA network is the cause of network defects. In turn, it strengthens the hypothesis that template polymerization is important to the formation of the mechanically enhanced PEG/PAA IPN, most likely because it facilitates the polymerization of long PAA chains that are intimately entangled with PEG and with other PAA chains.
The mechanical properties of PEG/PAA and other strength-enhanced IPNs are distinguished from conventional hydrogels in that they exhibit nonlinear enhancement in modulus and fracture properties despite being composed of approximately the same amount of water. Classical models of structure–property relations fail to predict this behavior. In recent work, investigators have attempted to develop equations to describe the “toughness” (in contrast to the stiffness, or modulus) of double networks. The work of Brown43,44 and Okumura45 are salient examples. Brown expands upon classic Lake–Thomas and DeGennes’ theory of polymer fracture, and has developed a simple model for accounting for the high toughness of Gong’s double networks. His model is based on the assumption that the first, more rigid network breaks up to form multiple cracks that are held together by the second network when the applied stress is above a defined value. A multiply cracked damage zone forms around the macroscopic crack in the material, causing energy dissipation and shielding by the second network. Brown developed an expression that predicts the toughness enhancement by this process to be approximately 40-fold, which is consistent with data presented by Gong et al.44 Okumura has discussed the Griffith criterion for heterogeneous materials consisting of a soft phase and a hard phase to propose a mechanism for the extremely large fracture energy and strength of the double network gels.45
The results from our studies of the PEG/PAA system indicate that the processing of these IPNs is an important determinant of the mechanical properties of these materials. It is not enough to have “pre-stress” in the hydrogel. On the contrary, our results suggest that at least two prerequisite processing elements may also be necessary: (1) that the pre-stress must be gradually built into the IPN through buffer-induced ionization of the PAA network after polymerization, and (2) that the second network is formed by a template polymerization process along the preformed first network. However, template polymerization does not appear to be sufficient for conferring mechanical strength, since an excess of ionizable second network material in the presence of a neutral first network is necessary to produce an increase in fracture properties. This observation is consistent with the findings of Gong et al., who has reported that excess neutral second network material in the presence of an ionizable first network improves fracture strength.17
CONCLUSIONS
IPNs have been the subject of extensive study since their advent in the 1960s. Hydrogel IPN systems have garnered significant attention in the last two decades due to their usefulness in biomedical applications. Of particular interest are the mechanical enhancements observed in “double network” IPN systems which exhibit nonlinear increases in fracture properties despite being composed of otherwise weak polymers. We have built upon pioneering work in this field as well as in responsive IPN systems to develop an IPN system based on end-linked PEG and loosely crosslinked PAA with hydrogen bond-reinforced strain-hardening behavior in water and high initial Young’s moduli under physiologic buffer conditions through swelling-induced pre-stress. Uniaxial tensile tests and equilibrium swelling measurements were used to study PEG/PAA IPN hydrogels with 2nd networks prepared with varying crosslinking and photoinitiator content, pH, solids content, and comonomers. Studies involving the addition of non-ionic comonomers and neutralization of the second network showed that hydrogen bonding template polymerization via inter-polymer hydrogen bonding may be an important process element in the formation of mechanically enhanced IPNs.
Acknowledgments
This research was supported by the Bio-X Interdisciplinary Initiatives Program and Bio-X Graduate Student Fellowship at Stanford University and the Office of Technology Licensing at Stanford University. Additional external support was also received from the National Institutes of Health (NIH Grant Number 1R01EY016987), the Signapore Eye Research Institute (SERI), the Fight for Sight Foundation, and VISX, Incorporated (now VISX Technology).
References
- 1.Suthar B, Xiao HX, Klempner D, Frisch KC. A review of kinetic studies on the formation of interpenetrating polymer networks. Polym Adv Technol. 1996;7:221–233. [Google Scholar]
- 2.Gupta N, Srivastava AK. Interpenetrating polymer networks: a review on synthesis and properties. Polym Int. 1994;35:109–118. [Google Scholar]
- 3.Flory PJ. Principles of Polymer Chemistry. Ithaca: Cornell University Press; 1953. [Google Scholar]
- 4.Olabisi O, Robeson LM, Shaw MT. Polymer-Polymer Miscibility. New York: Academic Press; 1979. [Google Scholar]
- 5.Sperling LH. Recent advances in interpenetrating polymer networks. Polym Eng Sci. 1985;25(9):517–520. [Google Scholar]
- 6.Frisch HL, Klempner D, Frisch KC. A topologically interpenetrating elastomeric network. J Polym Sci B. 1969;7:775. [Google Scholar]
- 7.Sperling LH, Friedman DW. Synthesis and mechanical behavior of interpenetrating polymer networks: poly(ethyl acrylate) and polystyrene. J Polym Sci A. 1969;7(2):425–427. [Google Scholar]
- 8.Ilmain F, Tanaka T, Kokufuta E. Volume transition in a gel driven by hydrogen bonding. Nature. 1991;349:400–401. [Google Scholar]
- 9.Gudeman LF, Peppas NA. Preparation and characterization of pH-sensitive, interpenetrating networks of poly(vinyl alcohol) and poly(acrylic acid) J Appl Polym Sci. 1995;55 :919–928. [Google Scholar]
- 10.Lee JW, Kim SY, Kim SS, Lee YM, Lee KH, Kim SJ. Synthesis and characteristics of interpenetrating polymer network hydrogel composed of chitosan and poly(acrylic acid) J Appl Polym Sci. 1999;73(1):113–120. [Google Scholar]
- 11.Zhang JT, Cheng SX, Zhuo RX. Poly(vinyl alcohol)/poly-(N-isopropylacrylamide) semi-interpenetrating polymer network hydrogels with rapid response to temperature changes. Colloid Polym Sci. 2003;281(6):580–583. [Google Scholar]
- 12.Osada Y. Conversion of chemical energy into mechanical energy by synthetic polymers (chemomechanical systems) Adv Polym Sci. 1987;82:2–46. [Google Scholar]
- 13.Nishi S, Kotaka T. Complex-forming poly(oxyethylene): poly(acrylic acid) interpenetrating polymer networks. 1. Preparation, structure, and viscoelastic properties. Macromolecules. 1985;18(8):1519–1525. [Google Scholar]
- 14.Nishi S, Kotaka T. Complex-forming polyoxyethylene: poly(acrylic acid) interpenetrating polymer networks III. Swelling and mechanochemical behavior. Polymer. 1989;21 (5):393–402. [Google Scholar]
- 15.Osada Y, Takeuchi Y. Water and protein permeation through polymeric membrane having mechanochemically expanding and contracting pores. Function of chemical valve. I. J Polym Sci Polym Lett Ed. 1981;19:303–308. [Google Scholar]
- 16.Khokhlov AR, Kramarenko EY, Makhaeva EE, Starodubtzev SG. Collapse of polyelectrolyte networks induced by their interaction with oppositely charged surfactants. Macromolecules. 1991;25:4779–4783. [Google Scholar]
- 17.Gong JP, Katsuyama Y, Kurokawa T, Osada Y. Double-network hydrogels with extremely high mechanical strength. Adv Mater. 2003;15(14):1155–1158. [Google Scholar]
- 18.Na YH, Kurokawa T, Katsuyama Y, Tsukeshiba H, Gong JP, Osada Y, Okabe S, Karino T, Shibayama M. Structural characteristics of double network gels with extremely high mechanical strength. Macromolecules. 2004;37(14):5370–5374. [Google Scholar]
- 19.Tanaka Y, Kurwabara R, Na YH, Kurokawa T, Gong JP, Osada Y. Determination of fracture energy of high strength double network hydrogels. J Phys Chem B. 2005;109:11559–11562. doi: 10.1021/jp0500790. [DOI] [PubMed] [Google Scholar]
- 20.Tsukeshiba H, Huang M, Na YH, Kurokawa T, Kuwabara R, Tanaka Y, Furukawa H, Osada Y, Gong JP. Effect of polymer entanglement on the toughening of double network hydrogels. J Phys Chem B. 2005;109:16304–16309. doi: 10.1021/jp052419n. [DOI] [PubMed] [Google Scholar]
- 21.Bakri A, Farooqui N, Myung D, Koh WG, Noolandi J, Carrasco M, Frank C, Ta CN. Biocompatibility of a hydrogel corneal inlay in vivo. Invest Ophthalmol Vis Sci. 2006;47 E-Abstract 3592. [Google Scholar]
- 22.Myung D, Koh W, Ko J, Noolandi J, Carrasco M, Smith A, Frank C, Ta C. Characterization of poly(ethylene glycol) and poly(acrylic acid) double networks for corneal implant applications. Invest Ophthalmol Vis Sci. 2005;46 E-Abstract 5003. [Google Scholar]
- 23.Koh WG, Myung D, Ko J, Noolandi J, Carrasco M, Smith A, Frank C, Ta C. Synthesis and surface modification of double network hydrogel from poly(ethylene glycol) and poly-(acrylic acid) Invest Ophthalmol Vis Sci. 2005;46 E-Abstract 4994. [Google Scholar]
- 24.Myung D, Koh W, Bakri A, Zhang F, Marshall A, Ko J, Noolandi J, Carrasco M, Cochran JR, Frank CW, Ta CN. Design and fabrication of an artificial cornea based on a photolithographically patterned hydrogel construct. Biomed Microdevices. 2007;9(6):911–922. doi: 10.1007/s10544-006-9040-4. [DOI] [PubMed] [Google Scholar]
- 25.Choi HK, Kim OJ, Chung CK, Cho CS. A novel mucoadhesive polymer prepared by template polymerization of acrylic acid in the presence of poly(ethylene glycol) J Appl Polym Sci. 1999;73:2740–2754. [Google Scholar]
- 26.Kim IS, Kim SH, Cho CS. Drug release from pH-sensitive interpenetrating polymer networks hydrogel based on poly (ethylene glycol) macromer and poly (acrylic acid) prepared by UV cured method. Arch Pharm Res. 1996;19(1):18–22. [Google Scholar]
- 27.Myung D, Koh W, Ko J, Hu Y, Carrasco M, Noolandi J, Ta CN, Frank CW. Biomimetic strain hardening in interpenetrating polymer network hydrogels. Polymer. 2007;48:5376–5387. [Google Scholar]
- 28.Polowinski S. Template polymerisation and co-polymerisation. Prog Polym Sci. 2002;27:537–577. [Google Scholar]
- 29.Bell CL, Peppas NA. Biomedical membranes from hydrogels and interpolymer complexes. In: Langer RS, Peppas NA, editors. Biopolymers II: Advances in Polymer Science. Springer-Verlag; Heidelberg: 1995. pp. 125–175. [Google Scholar]
- 30.Bokias G, Staikos G, Iliopoulos I, Audebert R. Interpolymer association between acrylic-acid copolymers and poly(ethylene glycol)—effects of the copolymer nature. Macromolecules. 1994;27(2):427–431. [Google Scholar]
- 31.Chen H-L, Morawetz H. Kinetics of polymer complex interchange in poly(acrylic acid)-poly(oxyethylene) solutions. Macromolecules. 1982;15:1445–1448. [Google Scholar]
- 32.Krupers MJ, VanderGaag FJ, Feijen J. Complexation of poly(ethylene oxide) with poly(acrylic acid-co-hydroxyethyl methacrylate)s. Eur Polym J. 1996;32(6):785–790. [Google Scholar]
- 33.Oyama HT, Tang WT, Frank CW. Complex formation between poly(acrylic acid) and pyrene-labeled poly(ethylene glycol) in aqueous solution. Macromolecules. 1987;20:474–480. [Google Scholar]
- 34.Oyama HT, Tang WT, Frank CW. Effect of the hydrophobic interaction in the poly(methacrylic acid)/pyrene end-labeled poly(ethylene glycol) complex. Macromolecules. 1987;20:1839–1847. [Google Scholar]
- 35.Hemker DJ, Frank CW. Dynamic light-scattering studies of the fractal aggregation of poly(methacrylic acid) and poly-(ethylene glycol) Macromolecules. 1990;23:4404–4410. [Google Scholar]
- 36.Daoust H, Darveau R, Laberge F. Microcalorimetric investigation on interaction between poly(acrylic acid) and oxyethylene oligomers in water. Polymer. 1990;31(10):1946–1949. [Google Scholar]
- 37.Jeon SH, Ree T. Characterization of poly(carboxylic acid)/poly(ethylene oxide) blends formed through hydrogen bonding by spectroscopic and calorimetric analyses. J Polym Sci A: Polym Chem. 1988;26:1419–1428. [Google Scholar]
- 38.Soutar I, Swanson L. Fluorescence anisotropy studies of polyelectrolyte mobility and interpolyelectrolyte complexation in aqueous solution. Macromolecules. 1990;23:5170–5172. [Google Scholar]
- 39.Ansarian M, Chapiro A, Mankowski Z. Sur une exaltation de l’ “effet de matrice” lors de la polymerisation de l’acide acrylique dans certains melanges ternaires. Eur Polym J. 1981;17(8):823–838. [Google Scholar]
- 40.Ali-Miraftab S, Chapiro A, Mankowski Z. Influence de la temperature sur les manifestations de l’ “effet de matrice” dans la polymerisation de l’acide acrylique en solution. Eur Polym J. 1981;17(3):259–273. [Google Scholar]
- 41.Chapiro A, Dulieu J. Influence of solvents on the molecular associations and on the radiation initiated polymerization of acrylic acid. Eur Polym J. 1977;13(7):563–577. [Google Scholar]
- 42.Chapiro A, Goldfeld-Freilich D, Perichon J. Influence de la temperature sur les associations moleculaires et sur la polymerisation de l’acide acrylique en solution. Eur Polym J. 1975;11(7):515–521. [Google Scholar]
- 43.Webber RE, Creton C, Brown HR, Gong JP. Large strain hysteresis and mullins effect of tough double-network hydrogels. Macromolecules. 2007;40:2919–2927. [Google Scholar]
- 44.Brown HR. A model of the fracture of double network gels. Macromolecules. 2007;40:3815–3818. [Google Scholar]
- 45.Okumura K. Toughness of double elastic networks. Euro-phys Lett. 2004;67(33):470–476. [Google Scholar]